Introduction
Syndactyly/polydactyly, a congenital anomaly of the
hands and feet, is one of the most common types of limb deformity,
and is characterized by fusion of the bone or soft tissue between
adjacent fingers or toes. Based on the differences in the fingers
or toes involved, non-syndromic syndactyly/polydactyly is
classified into five types according to Temtamy and McKusick’s
method (1). Syndactyly/polydactyly
type II is termed synpolydactyly with autosomal dominant
inheritance.
Synpolydactyly is the joint presentation of
syndactyly (fusion of fingers or toes) and polydactyly
(supplementary digits), characterized by incomplete penetrance and
inconsistent expressivity. In the current study, a synpolydactyly
kindred from Huaihua in China was examined. Based on the clinical
features and the degree of difficulty in deformity repair, a simple
and feasible method for functional classification was proposed.
Genetic linkage analysis was also performed at polymorphic
microsatellite loci and a candidate gene was sequenced in the
implicated chromosomal region.
Materials and methods
Family survey, functional classification
and specimen collection
Informed consent was obtained from all family
members prior to participation in the study. The kindred with
congenital synpolydactyly was from Huaihua (Hunan, China) and
consisted of three generations, totaling 39 members, including 19
affected members (4 males and 15 females) and 20 unaffected
members. The 19 affected members were medically examined, and
images and X-rays were obtained of their hands and feet.
Furthermore, a 5-ml peripheral blood sample was conventionally
extracted and placed in a vacuum heparinized tube. The present
study was approved by the ethics committee of the Family Planning
Institute of Hunan Province (Changsha, China).
As there were no marked foot dysfunctions involving
walking, standing or running, only the hand deformities were
grouped into mild, moderate or severe deformity categories,
according to the functional classification method established in
the present study. A mild deformity was classified as the possible
presence of supplementary fingers, a normal outline of the fingers
and joints, and no bone abnormalities or dysfunctions. A moderate
deformity was classified as the possible presence of supplementary
fingers and a normal shape and position of the metacarpals and
phalanges. There were no bone adhesions and the grip strength was
normal; however, webbed fingers or finger adhesions, as well as
abnormal joint flexion, extension or other joint abnormalities, may
have been present. A severe deformity was classified as the
possible presence of supplementary fingers, with marked deformities
or joint abnormalities, including metacarpal or phalangeal
adhesions, and an abnormal grip function.
Linkage analysis
DNA extraction and determination
A standard phenol-chloroform method was used to
extract the total genomic DNA, which was quantified using a UV
spectrophotometer (DU800, Beckman Coulter, Kraemer Boulevard Brea,
CA, USA), and subsequently stored at −20°C.
Selection of polymorphic
microsatellite DNA loci
Polymorphic microsatellite markers were selected
from the three identified loci for synpolydactyly at chromosomes
2q31, 22q13.31 and 14q11.2–q12. The markers and their corresponding
fluorescence-labeled primers (Sunny Biotech Co., Ltd., Shanghai,
China) are shown in Table I.
 | Table IPrimer sequences, fragment size and
chromosomal localization. |
Table I
Primer sequences, fragment size and
chromosomal localization.
| Genetic marker | Primer sequences | Fragment size
(bp) | Chromosomal
localization |
|---|
| D2S2314 | 5′
FAM-GGTGTCAGTGAGACCCTGT 3′ | 96–118 | |
| 5′
ATTTCTAGCGGCCCTAAAAC 3′ | | |
| D2S2310 | 5′
FAM-CGACTTGAGTAGACGCACTATTC 3′ | 244–260 | |
| 5′
GCATCTAAACTGTGAAATGAGC 3′ | | 2q31–32 |
| D22S444 | 5′
FAM-TTTGAACTAAGCCTTAAAAATGC 3′ | 123–131 | |
| 5′
TGTTTGGCTTGAAGAAGGAG 3′ | | |
| D22S1170 | 5′
FAM-ACCGTTGCCTATATCCA 3′ | 180–212 | |
| 5′ AGCCCACTCCACAATTT
3′ | | 22q13–14 |
| D14S264 | 5′
FAM-CCCCAAATATCACTCCAAAT 3′ | 216–234 | |
| 5′
GAGTTGGCAACCACTTCTGT 3′ | | |
| D14S283 | 5′
FAM-GGGACTATATCTCCCAGGC 3′ | 125–153 | |
| 5′
TGTTTTCCTAGTAACCGCA 3′ | | 14q11–12 |
Polymerase chain reaction (PCR)
amplification of the microsatellite sequences and genotyping
The microsatellites were amplified by multiplex PCR.
According to the length of the products, an appropriate
intramolecular marker (0.12 μl; GeneScan™ 500
TAMRA™; Applied Biosystems®; Invitrogen Life
Technologies, Foster City, CA, USA) was selected and mixed with
high-purity carboxamide (4 μl) and the PCR product (1 μl).
Following 3 min denaturation at 95°C, the mixture was cooled on ice
to maintain the single-stranded DNA. The mixture was subjected to
capillary electrophoresis on an ABI Prism® 310 Genetic
analyzer (Applied Biosystems®) for 2 h.
Data analysis
Data were collected using GeneScan 2.1 software
(Applied Biosystems®) and subjected to lane line
correction. The collected gel images were converted to digital
signals for internal calibration of the molecular weight and
analysis of the amplified fragment sizes.
HOXD13 gene mutation analysis
A proband and an unaffected relative were selected
from the family for mutation detection in the HOXD13 gene.
The online software, Primer3, was used to design primers to amplify
the exons and intron/exon boundaries of the gene (Table II). The PCR products obtained were
purified by agarose gel electrophoresis, quantified and sequenced
using the two primers. The obtained sequences were compared with
the reference gene sequence in GenBank (accession no. NM_000523).
The identified mutation was verified experimentally in all family
members.
| Table IIPrimers for HOXD13 mutation
analysis and polymerase chain reaction fragment sizes. |
Table II
Primers for HOXD13 mutation
analysis and polymerase chain reaction fragment sizes.
| Primer | Upstream | Downstream | Fragment size
(bp) |
|---|
| Hoxd13-1 | 5′
GAGAAAGGAGAGGAGGGAGGAG 3′ | 5′
AGGGCTCGTATAGCCCTGGT 3′ | 684 |
| Hoxd13-2 | 5′
GGCTCTAAATCAGCCGGACA 3′ | 5′
GGCAACTGCTGAGAGCTAATGA 3′ | 727 |
| Hoxd13-3 | 5′
CCGGCTATATCGACATGGTGT 3′ | 5′
CATGTCCGGCTGATTTAGAGC 3′ | 1008 |
Results
Genetic analysis and clinical phenotype
of the pedigree
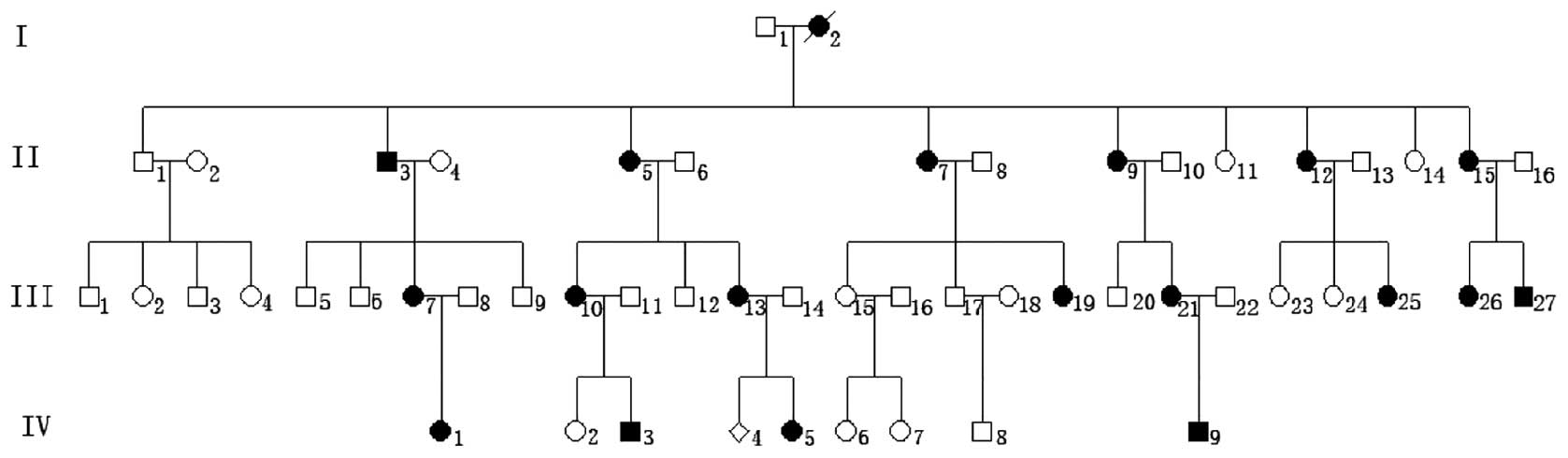
The clinical phenotypes of the kindred were
characterized as follows: i) Disease inheritance within the
kindred; ii) one parent of the proband is also affected; iii)
offspring of unaffected individuals are always unaffected; and iv)
equal opportunity for male and female members to be affected. The
disease was shown to be inherited in an autosomal dominant manner
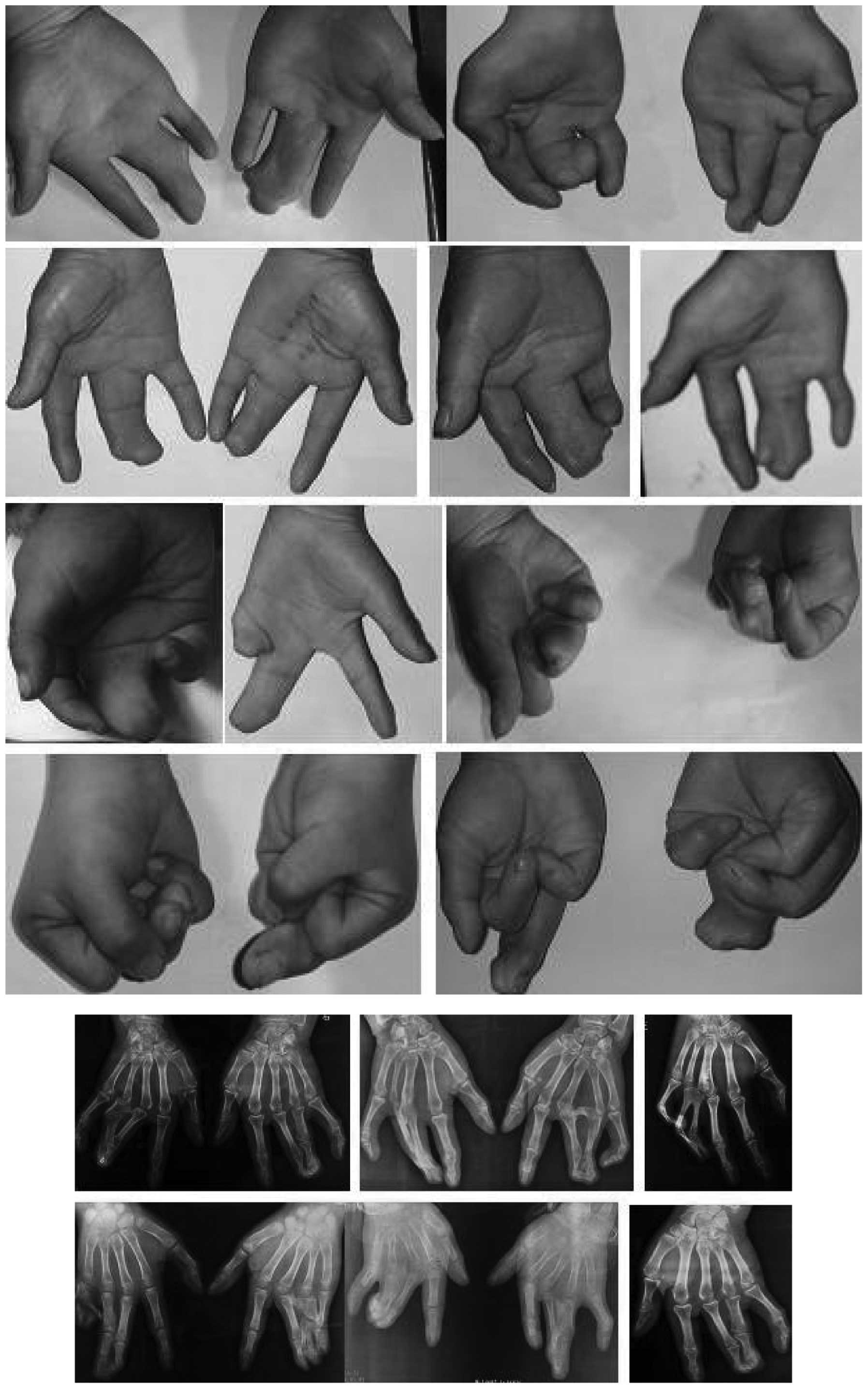
(Fig. 1). The patients revealed no
deformities other than those of the hands and feet; however, the
phenotypes of the affected members were complex, with variation in
the expressivity. There were significant phenotypic differences
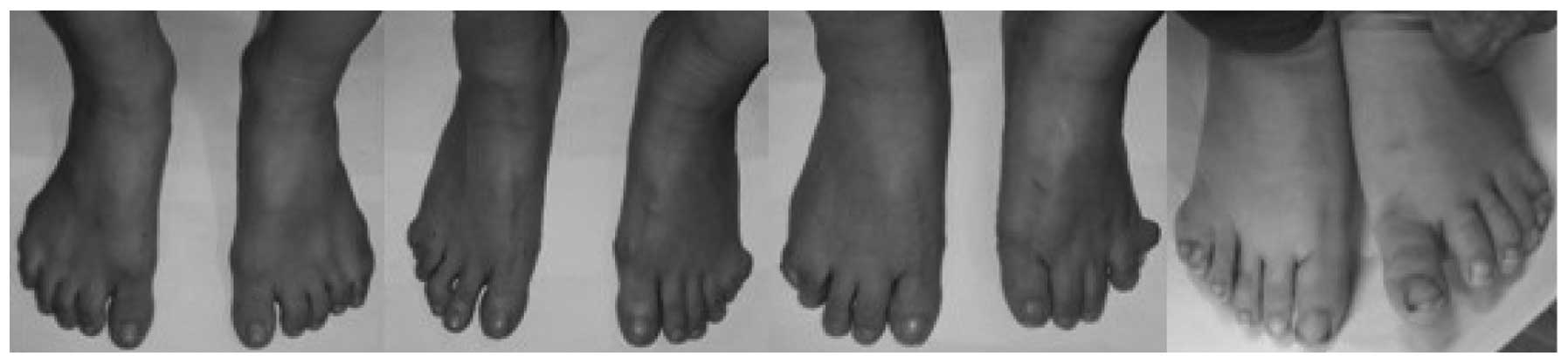
between the hands and feet of the affected members. No significant
dysfunction was observed in the toes that affected standing,
walking or running (Fig. 2). In
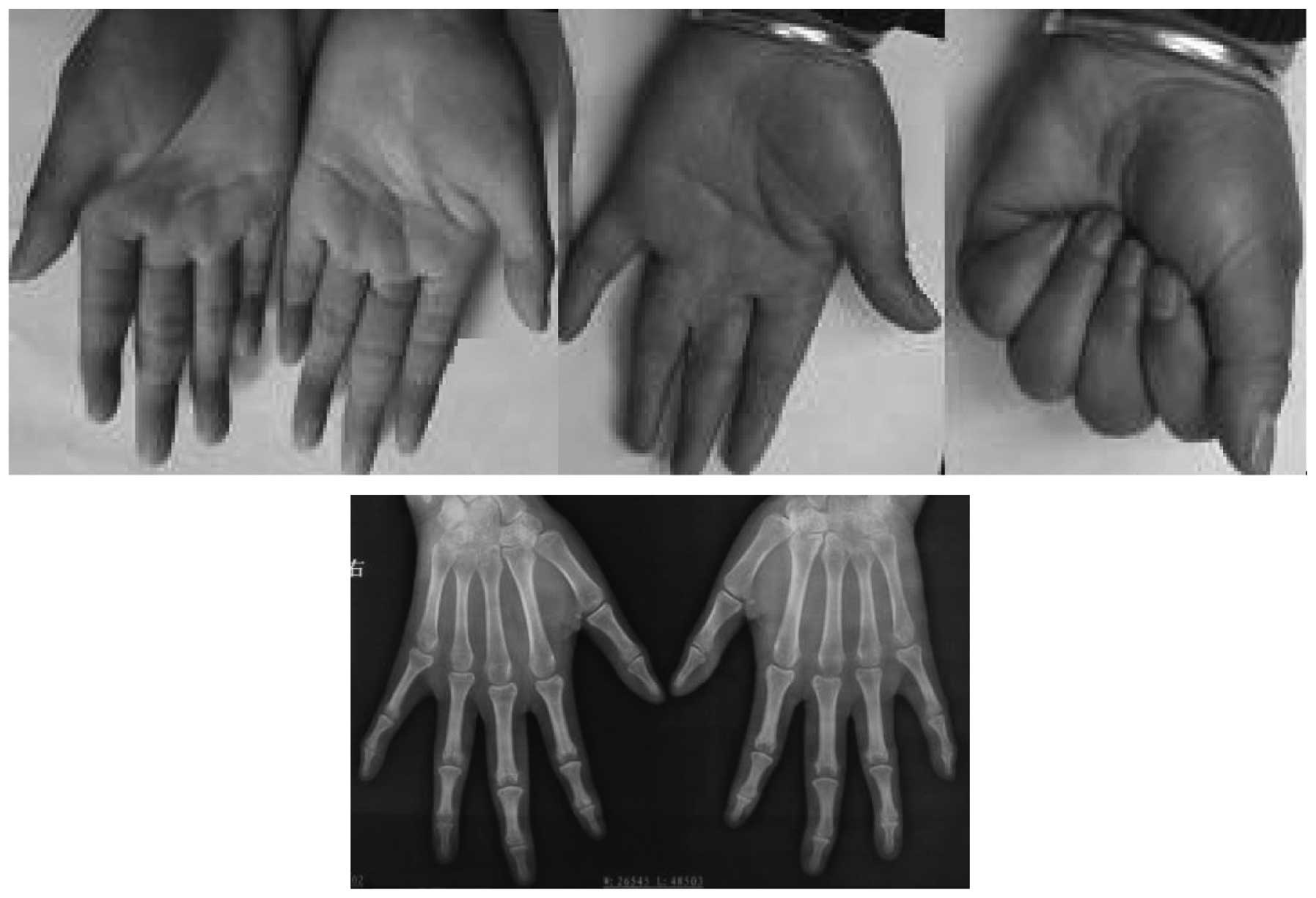
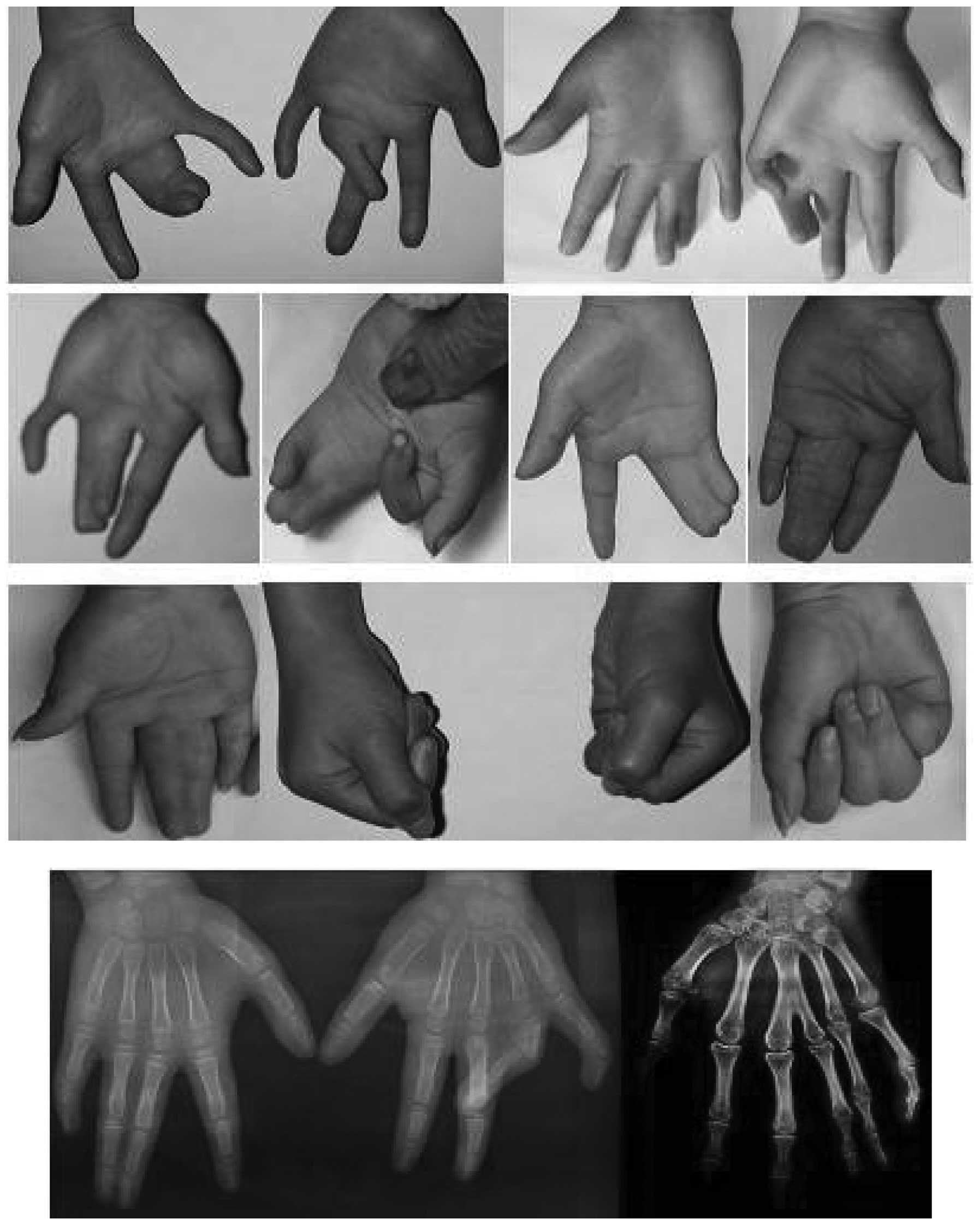
the 38 hands of the 19 affected patients, the number of hands with
a mild (Fig. 3), moderate
(Fig. 4) and severe (Fig. 5) deformity were 3, 17 and 18,
respectively. This demonstrates that the clinical features of the
feet and hands have significant differences in a pedigree with
individuals of an identical genotype.
Family linkage analysis
D2S2310 and D2S2314, genetic markers closely
associated with synpolydactyly type I, were genotyped in all the
affected members. D14S264, D14S283, D22S444 and D22S1170 revealed a
non-genetic linkage.
Mutation analysis of HOXD13
Sequencing of the HOXD13 PCR products
revealed that all the affected family members had a 27-bp insertion
encoding nine additional alanine residues. No other mutation was
identified in the coding region.
Discussion
Non-syndromic polydactyly is usually inherited in an
autosomal dominant manner; however, the condition has been reported
to demonstrate autosomal recessive inheritance in certain studies
(2). Currently, there are a number
of classification methods that are based on anatomical
classification (3). These include
Temtamy and McKusick’s method, which mainly emphasizes the
relevance between genotyping and the clinical phenotype of
syndactyly/polydactyly. This method has been accepted and utilized
by subsequent practitioners. However, this method has limitations
when selecting the clinical surgical method due to a complicated
and meticulous classification process that requires the promotion
of mutual communication between clinical staff. Therefore,
functional classification and sorting according to surgical
difficulty are recommended. In the current study, a method of
clinical classification, regardless of gene variation type, was
established. The classification was based only on clinical
phenotype and the severity of the syndactyly/polydactyly; thus, may
guide the selection of the clinical surgery method.
As there were no evident foot dysfunctions involving
walking, standing or running, the hand deformities only were
functionally grouped into mild, moderate or severe deformity
categories. Patients with a joint deformity and bone abnormalities
in the hands exhibited marked dysfunction of the hands. For a mild
deformity, finger resection and corrective surgery are usually
sufficient for treatment. For a moderate deformity, syndactyly
separation, webbing enlargement and articular repair are
recommended. Surgical treatment for a severe deformity is complex,
including osteotomy, bone separation, skin grafting, flap surgery
and joint reconstruction. The type of classification method used in
the present study may also have its application extended to all
cases of syndactyly/polydactyly.
Synpolydactyly types I, II, and III have been mapped
to the 2q31, 22q13.31 and 14q11.2–q12 chromosomes, respectively
(4–6). HOX genes encode a highly conserved
family of transcription factors that control the formation of the
primary and secondary body axes during embryonic development
(7). The 39 Hox genes are
generally arranged into four clusters: HOXA, HOXB, HOXC and HOXD,
which are located at chromosomes 7p14, 17q21, 12q13 and 2q31,
respectively (8). Each gene
cluster is ~120 kb and contains 9–11 genes (9–10).
Mutations in the HOXD13 gene have been previously associated
with synpolydactyly (11–13). All HOX proteins are capable of
binding to specific DNA sequences via a DNA-binding motif
consisting of 60 amino acids. HOXD13 comprises two exons
with a coding region of 1,008 bp, and encodes a protein of 335
amino acids (14), containing a
homeobox at the 3′ end and two polyserine chains and one
polyalanine chain at the 5′ end. The polyalanine chain, which
usually has a length of 15 residues, has been found to be expanded
in cases of synpolydactyly (7,15–19),
possibly resulting from the unequal crossover of imperfect
trinucleotide repeats in the GCN codon and exon 1 of HOXD13
(20,21). An abnormally long polyalanine chain
results in the migration of the HOXD13 protein from the nucleus to
the cytoplasm, which is followed by abnormal protein aggregation
and altered transcription factor activity (23). Goodman et al (22) observed that the penetrance in 20
typical synpolydactyly pedigrees was positively associated with the
severity of limb deformities and the number of alanine residues.
Other HOXD13 mutation types have also been associated with
synpolydactyly, including nonsense (24,25)
and missense (12) mutations,
small deletions, frameshift mutations and splice acceptor mutations
(17,26,27).
From the genotyping results of the microsatellites in these
regions, the causative gene in the current pedigree was mapped to
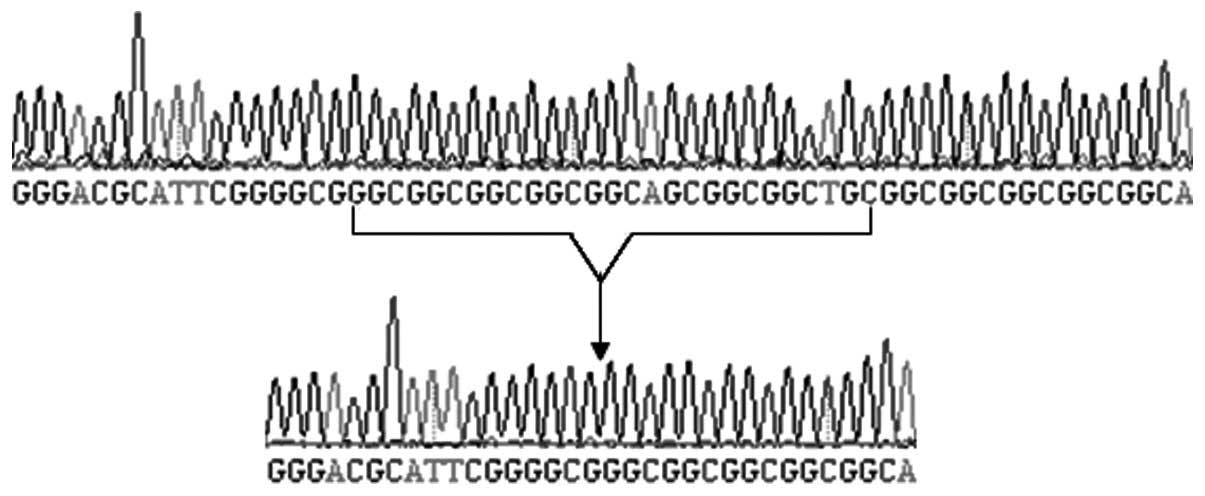
2q31 (type I). The first identified repetitive sequence of
c.168_194dup was GGCGGCGGCGGCGGCAGCGGCGGCTGC (27 bp; Fig 6); however, its encoded amino acid
sequence was the same as that reported by Goodman et al
(22).
In conclusion, the cause of synpolydactyly in the
Chinese family examined in the present study was identified to be a
polyalanine expansion in the HOXD13 gene. Penetrance in the
family was up to 100%; however, expressivity was inconsistent
between individuals and even between the hands and feet of the same
individual, with certain patients exhibiting different severity of
synpolydactyly between different phalangeal joints. The results of
the current study may be useful for prenatal genetic and molecular
diagnosis of synpolydactyly.
References
|
1
|
Temtamy SA and McKusick VA: The genetics
of hand malformations. Birth Defects Orig Artic Ser. 14:i–xviii.
1–619. 1978.PubMed/NCBI
|
|
2
|
Mollica F, Volti SL and Sorge G: Autosomal
recessive postaxial polydactyly type A in a Sicilian family. J Med
Genet. 15:212–216. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Al-Qattan MM: Type II familial
synpolydactyly: report on two families with an emphasis on
variations of expression. Eur J Hum Genet. 19:112–114. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sarfarazi M, Akarsu AN and Sayli BS:
Localization of the syndactyly type II (synpolydactyly) locus to
2q31 region and identification of tight linkage to HOXD8 intragenic
marker. Hum Mol Genet. 4:1453–1458. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Debeer P, Schoenmakers EF, Twal WO, et al:
The fibulin-1 gene (FBLN1) is disrupted in a t(12;22) associated
with a complex type of synpolydactyly. J Med Genet. 39:98–104.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Malik S, Abbasi AA, Ansar M, et al:
Genetic heterogeneity of synpolydactyly: a novel locus SPD3 maps to
chromosome 14q11.2–q12. Clin Genet. 69:518–524. 2006.PubMed/NCBI
|
|
7
|
Snajdr P, Grim M and Liska F: HOX genes
and the limb development in the clinical praxis and in the
experiment. Cas Lek Cesk. 149:4–9. 2010.(In Czech).
|
|
8
|
Apiou F, Flagiello D, Cillo C, et al: Fine
mapping of human HOX gene clusters. Cytogenet Cell Genet.
73:114–115. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Goodman FR and Scambler PJ: Human Hox gene
mutations. Clin Genet. 59:1–11. 2001. View Article : Google Scholar
|
|
10
|
Salsi V, Vigano MA, Cocchiarella F, et al:
Hoxd13 binds in vivo and regulates the expression of genes acting
in key pathways for early limb and skeletal patterning. Dev Biol.
317:497–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang B, Xu B, Cheng Z, et al: A novel
non-synonymous mutation in the homeodomain of HOXD-13 causes
synpolydactyly in a Chinese family. Clin Chim Acta. 413:1049–1052.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brison N, Debeer P, Fantini S, et al: An
N-terminal G11A mutation in HOXD13 causes synpolydactyly and
interferes with Gli3R function during limb pre-patterning. Hum Mol
Genet. 21:2464–2475. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kurban M, Wajid M, Petukhova L, et al: A
nonsense mutation in the HOXD13 gene underlies synpolydactyly with
incomplete penetrance. J Hum Genet. 56:701–706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
D’Esposito M, Morelli F, Acampora D, et
al: EVX2, a human homeobox gene homologous to the even-skipped
segmentation gene, is localized at the 5′ end of HOX4 locus on
chromosome 2. Genomics. 10:43–50. 1991.
|
|
15
|
Muragaki Y, Mundlos S, Upton J and Olsen
BR: Altered growth and branching patterns in synpolydactyly caused
by mutations in HOXD13. Science. 272:548–551. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gong L, Wang B, Wang J, et al: Polyalanine
repeat expansion mutation of the HOXD13 gene in a Chinese family
with unusual clinical manifestations of synpolydactyly. Eur J Med
Genet. 54:108–111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xin Q, Li L, Li J, et al: Eight-alanine
duplication in homeobox D13 in a Chinese family with
synpolydactyly. Gene. 499:48–51. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cocquempot O, Brault V, Babinet C and
Herault Y: Fork stalling and template switching as a mechanism for
polyalanine tract expansion affecting the DYC mutant of HOXD13, a
new murine model of synpolydactyly. Genetics. 183:23–30. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tüzel E, Samli H, Kuru I, et al:
Association of hypospadias with hypoplastic synpolydactyly and role
of HOXD13 gene mutations. Urology. 70:161–164. 2007.PubMed/NCBI
|
|
20
|
Warren ST: Polyalanine expansion in
synpolydactyly might result from unequal crossing-over of HOXD13.
Science. 275:408–409. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jin H, Lin PF, Wang QM, et al:
Synpolydactyly in a Chinese kindred: mutation detection, prenatal
ultrasonographic and molecular diagnosis. Zhonghua Yi Xue Yi Chuan
Xue Za Zhi. 28:601–605. 2011.(In Chinese).
|
|
22
|
Goodman FR, Mundlos S, Muragaki Y, et al:
Synpolydactyly phenotypes correlate with size of expansions in
HOXD13 polyalanine tract. Proc Natl Acad Sci USA. 94:7458–7463.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zákány J, Kmita M and Duboule D: A dual
role for Hox genes in limb anterior-posterior asymmetry. Science.
304:1669–1672. 2004.PubMed/NCBI
|
|
24
|
Kurban M, Wajid M, Petukhova L, et al: A
nonsense mutation in the HOXD13 gene underlies synpolydactyly with
incomplete penetrance. J Hum Genet. 56:701–706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Low KJ and Newbury-Ecob RA: Homozygous
nonsense mutation in HOXD13 underlies synpolydactyly with a cleft.
Clin Dysmorphol. 21:141–143. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brison N, Tylzanowski P and Debeer P: Limb
skeletal malformations - what the HOX is going on? Eur J Med Genet.
55:1–7. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wajid M, Ishii Y, Kurban M, et al:
Polyalanine repeat expansion mutations in the HOXD13 gene in
Pakistani families with synpolydactyly. Clin Genet. 76:300–302.
2009. View Article : Google Scholar : PubMed/NCBI
|