Introduction
Classical Guillain-Barré syndrome (GBS) is a
progressive symmetrical limb weakness, in which tendon reflexes
disappear. The disease is characterized by an acute onset and the
clinical symptoms often reach their peak at the 4th week. GBS is
manifested as multiple nerve root and peripheral nerve injury,
often with protein-cell separation in the cerebrospinal fluid. It
often presents a single-phase self-limiting course; intravenous
immunoglobulin and plasma exchange therapy are effective for the
treatment of GBS. Demyelination is the main electrophysiological
and pathological feature of this disease (1,2). In
the past 20 years, it has been recognized that there are extensive
subtypes of the condition, which include acute inflammatory
demyelinating polyneuropathy, acute motoraxonal neuropathy, acute
motor-sensory axonal neuropathy, Miller Fisher syndrome, acute
autonomic neuropathy and acute sensory neuropathy. Certain patients
with sensory neuropathy may actually exhibit sensory GBS. However,
case reports are rare (3,4). A case of sensory GBS treatment is
described in the present study.
Case report
A 58-year-old female patient was admitted to
hospital on August 3, 2012 having experienced limb numbness for one
month’. This study was conducted in accordance with the Declaration
of Helsinki and with approval from the Ethics Committee of the
Tianjin Third Central Hospital (Tianjin, China). Written informed
consent was obtained from the patient.
One month prior to hospitalization, the patient
suddenly felt numbness and pain at the fingertips of the hands
accompanied by palpitations. After four days, the symptoms
gradually involved the hands and feet and the patient was conscious
of lower limb weakness, although this was not accompanied by
posture or gait abnormalities. The patient additionally experienced
upper gastrointestinal discomfort, but without nausea or vomiting,
and zonesthesia from the double costal margin to the umbilical
level. After two weeks the patient exhibited numbness of the face,
mouth and the skin at the top of the temple. Following symptom
onset, she went to the clinic of the Tianjin Dagang Oilfield
Hospital (Tianjin, China), where a brain computed tomography (CT)
and magnetic resonance imaging examination of the brain and
cervical spinal cord showed no abnormalities. The local hospital
prescribed Mecobalamine as treatment, but the symptoms continued to
progress. During the illness, the patient exhibited no dry eye or
dry mouth symptoms, occasionally showed changes in bowel habit
(twice a day or once every two days) and exhibited a weight loss of
~6 kg compared with previously. Twenty days previously the patient
had also taken a health care product of an unknown name. The
patient’s blood glucose levels had increased for two years but,
following diet control, her fasting and postprandial blood glucose
levels could be maintained at ~7 mmol/l. The patient had no history
of habitation in an epidemic or rural environment and no history of
smoking or alcohol abuse. She was unaware of any familial
hereditary disease history or similar cases in her family.
Physical examination on admission indicated the
following characteristics: clear and co-operative mentality, with
normal advanced neural activity; prefrontal and bilateral facial
pain and a loss of heat sensation; no atrophy in the limb muscles;
normal muscle tension; muscle strength grade V; positive tendon
reflex of the upper limbs, with no tendon reflex of the lower
limbs; negative bilateral Hoffmann reflex, with the bilateral
Babinski reflex not being elicited; normal gait and no ataxia. The
patient experienced glove- and stocking-type sensations in her
hands and feet and a loss of pain and temperature sensation. The
patient’s diapason vibration sensation and joint position sense
were normal. In addition, her discriminative touch sense was
regular, and the internal medical examination revealed no
abnormalities. A routine blood test was conducted following
admission, as well as tests for liver and kidney function, five
types of hepatitis B, syphilis, human immunodeficiency virus,
thyroid function, tumor markers, immune components, vitamin
B12, folic acid, fasting blood glucose, mercury, lead,
manganese, chromium and other toxins. All results were normal. A
gastroscopy, chest CT, abdominal B-ultrasound, echocardiography and
thyroid B-ultrasound showed no evident abnormalities. On the second
day after admission, the electrophysiological examination was
performed. The results revealed that the bilateral median, ulnar,
right posterior tibial and peroneal nerves exhibited prolonged
distal motor latency, the amplitude was reduced, the proximal
amplitude was reduced with a normal speed, and the distal sensory
nerve did not elicit a positive waveform. The results of the needle
electromyography of the abductor digiti minimi and anterior tibial
muscles were normal. Following stimulation of the bilateral median,
ulnar and posterior tibial nerves there was a normal F wave, and
following stimulation of the bilateral posterior tibial nerve there
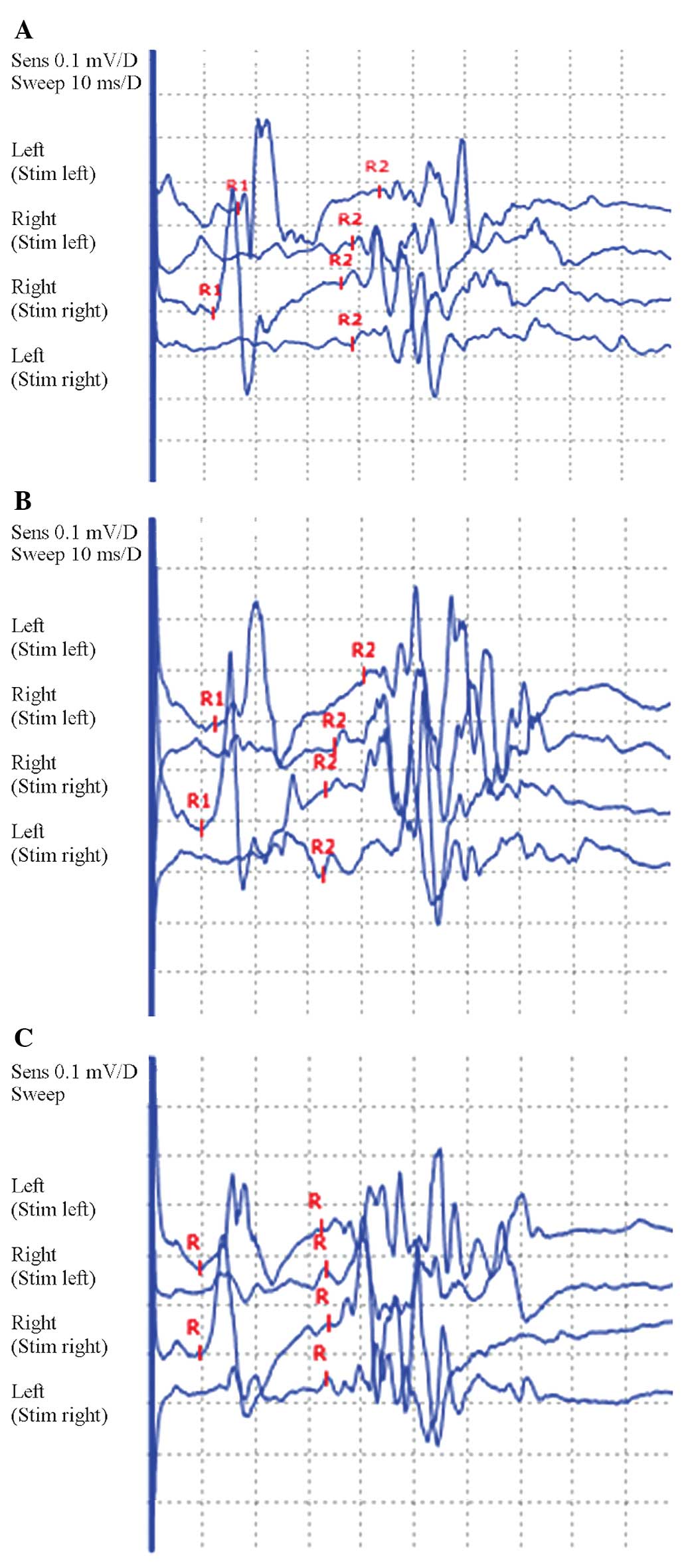
was no H reflex. In addition, the blink reflex showed prolongation
of the ipsilateral R1 and R2 and contralateral R2 latency. Finally,
the facial nerve motor conduction was normal, suggesting that the
damage may have been to the trigeminal primary afferent (Tables I–III and Fig. 1A). Examination of the cerebrospinal
fluid showed the number of cells and glucose and chloride levels to
be normal, while the protein levels were increased to 131.7 mg/dl
and the oligoclonal band was negative. Following admission, the
patient was diagnosed with sensory GBS, and was administered γ
globulin at a dosage of 400 mg/kg/day, intravenous immunoglobulin,
for five consecutive days, and heteropathy with vitamins
B1 and B12, and neurotropin. On August 16
(six weeks after the symptom onset), the review of the
neurophysiology showed that the peripheral nerve motor conduction
amplitude had recovered, and there were no clear changes in the
motor distal latency and sensory conduction results. The needle
electromyography results of the right abductor digiti minimi and
anterior tibial muscles showed much denervation potential. The
blink reflex was significantly improved (Tables I–III and Fig. 1B). The review of the lumbar
puncture showed protein levels to be 96.8 mg/dl on August 17. On
August 18, the clinical symptoms had completely remitted, and the
patient was discharged. Ten weeks after the onset of symptoms, the
review of the neurophysiological results showed that the amplitude
of the peripheral nerve motor conduction had further recovered and
the distal latency had improved. The sensory conduction results had
not changed significantly. The abductor digiti minimi and anterior
tibial muscles needle electromyography results returned to normal,
and the blink reflex was approximately normal (Tables I–III and Fig. 1C).
 | Table IRight median and ulnar nerve motor
conduction results at different times after onset. |
Table I
Right median and ulnar nerve motor
conduction results at different times after onset.
| Median nerve | Ulnar nerve |
|---|
|
|
|
|---|
| Time after onset
(weeks) | Distal latency (msec,
% change) | Amplitude (mV, %
change) | Speed, elbow-wrist
(m/sec) | Distal latency (msec,
% change) | Amplitude (mV, %
change) | Speed, elbow-wrist
(m/sec) |
|---|
| 4 | 15.2, ↑347 | 1.6, ↓89 | 52.6 | 4.27, ↑64 | 4.3, ↓75 | 67.1 |
| 6 | 15.5, ↑356 | 4.1, ↓74 | 57.1 | 4.53, ↑74 | 7.6 | 70.1 |
| 10 | 9.7, ↑185 | 4.7, ↓71 | 53.8 | 3.30, ↑27 | 7.8 | 70.4 |
| Table IIINeedle electromyography results of the
patient at different times after onset. |
Table III
Needle electromyography results of the
patient at different times after onset.
| Muscle | Time after onset
(weeks) | Rest | Minimal contraction
(MUP) | Maximal contraction
(interference phase, mV) |
|---|
|
|
|---|
| Denervation
potential | Duration (msec) | Amplitude (μV) |
|---|
| Right abductor minimi
muscle | | | | | |
| 4 | (−) | 12.9 | 478 | 2.87 |
| 6 | P++++F++ | 11.9 | 498 | 2.89 |
| 10 | (−) | 12.6 | 793 | 3.52 |
| Right anterior tibial
muscle | | | | | |
| 4 | (−) | 13.7 | 544 | 3.13 |
| 6 | P++++F++ | 11.6 | 529 | 3.08 |
| 10 | (−) | 11.8 | 580 | 3.46 |
Discussion
The clinical features exhibited by the patient
included numbness of the extremities, accompanied by the abatement
and disappearance of tendon reflexes and subjective fatigue.
Objective examination revealed muscle strength to be normal.
Symptoms reached their peak in four weeks. Analysis of
cerebrospinal fluid showed protein cell separation and the
electrophysiological examination showed primarily distal
sensorimotor fiber demyelination changes. Following treatment, the
clinical symptoms were alleviated from the sixth week after symptom
onset, and the protein levels in the cerebrospinal fluid reduced
from the levels of the fourth week. The clinical and laboratory
characteristics were consistent with classic GBS (5).
Oh et al (6)
proposed nine criteria for the diagnosis of sensory GBS in 2001: i)
Acute symmetrical sensory loss, ii) a peak in symptoms at four
weeks, iii) abating or disappearing tendon reflexes, iv) normal
muscle strength, v) at least two pieces of evidence for nerve
demyelination in the electrophysiological examination, vi)
single-phase course, vii) the exclusion of other neurological
diseases, viii) no family history, and ix) increases in protein
levels in the cerebrospinal fluid in the acute phase. As described,
the patient met all the aforementioned diagnostic criteria.
However, clinical case reports about sensory GBS remain rare, and
the understanding of this type of sensory GBS remains
superficial.
Firstly, clinical and electrophysiological
characteristics of sensory GBS show heterogeneity. Seneviratne and
Gunasekera (7) reported six cases
of sensory GBS with clinical manifestations of sensory impairment
to the extremities but no deep sensory abnormalities or ataxia. The
electrophysiological examinations were normal, and cerebrospinal
fluid examination showed isolated protein cells. The six patients
had a good prognosis, considering the effects of the small fiber
damage. Dawson et al (8)
described a case of sensory GBS in which the patient exhibited
abnormal sensation and joint position sense, vibratory sensory
abnormalities and ataxia. Certain patients may exhibit subjective
weak limb muscle strength, and electrophysiology tests can show
demyelination of the involved motor fiber, which is also considered
as a lesion in the large sensory fiber. Lee and Lee (9) believed that those patients who showed
only clinical sensory neuropathy, and who were indicated to have
motor and sensory fiber demyelination by electrophysiological
examination, or demyelination only involving the sensory fibers,
could be diagnosed with sensory type GBS.
In view of the clinical and electrophysiological
characteristic heterogeneity of sensory GBS, Uncini and Yuki
(10) suggested that, according to
the initial injury site and the diameter of the involved fibers,
sensory GBS could be divided into three categories: Acute sensory
demyelinating polyneuropathy, acute sensory large-fiber
axonopathy-ganglionopathy and acute sensory small-fiber
neuropathy-ganglionopathy. In the present case, the clinical
manifestations in the patient were sensory disturbance and mild
fatigue. The objective examination did not reveal loss of muscle
strength; however, the electrophysiological examination suggested
evidence of sensorimotor fiber demyelination; this was considered
to be a type of acute sensory fiber demyelinating polyneuropathy.
This type of sensory GBS is very similar to classical acute
inflammatory demyelinating polyneuropathy (AIDP); however, the
difference is primarily sensory injury. Of note, the patient
exhibited clinical manifestations of trigeminal nerve involvement,
and electrophysiology tests provided objective evidence of
trigeminal nerve involvement, which has not been reported in
previous cases. It is therefore confirmed that there can be sensory
cranial nerve fiber involvement in cases of sensory GBS, just as
AIDP can have motor cranial nerve fiber involvement. There are also
numerous sensory neuropathies with acute or subacute sensory
disturbances as the clinical onset, and the differential points
between these and sensory GBS remain unclear. Yee and Katz
(11) proposed that sensory
disturbance primarily exhibits a multi-asymmetric onset, but this
feature is not specific; the truly significant differential feature
is that the former exhibits continuous clinical symptoms, and the
latter is a one-way course. However, it can be observed from the
present case that while sensory disturbances only involve sensory
fibers, sensory GBS can exhibit the clinical involvement of motor
fibers. Furthermore, the one-way course of sensory GBS indicates a
clinical cure, but the electrophysiological examination manifests
the abnormality. For patients in the present study, the clinical
symptoms ceased, but the electrophysiological examination did not
reveal complete restoration, which is consistent with the studies
by Bannister and Sears (12) and
Sauron et al (13). It has
been suggested that myasthenia can no longer be considered to be
the core symptom, while a prodrome of GBS and cerebrospinal fluid
protein cell separation are not required conditions for the
diagnosis of GBS.
In conclusion, further understanding of the features
of sensory GBS would be greatly advantageous for improving the
treatment rate and enabling patients with GBS to receive timely and
effective treatment. Sensory GBS is an important type of GBS, which
may be associated with involvement of the cranial nerve.
References
|
1
|
Asbury AK: Diagnostic considerations in
Guillain-Barré syndrome. Ann Neurol. 9(Suppl): 1–5. 1981.
|
|
2
|
Asbury AK and Cornblath DR: Assessment of
current diagnostic criteria for Guillain-Barré syndrome. Ann
Neurol. 27:S21–S24. 1990.
|
|
3
|
No authors listed. Guillain-Barré syndrome
variants in Emilia-Romagna, Italy, 1992–3: incidence, clinical
features, and prognosis. Emilia-Romagna Study Group on Clinical and
Epidemiological Problems in Neurology. J Neurol Neurosurg
Psychiatry. 65:218–224. 1998.
|
|
4
|
Ropper AH: Unusual clinical variants and
signs in Guillain-Barré syndrome. Arch Neurol. 43:1150–1152.
1986.PubMed/NCBI
|
|
5
|
Ropper AH: The Guillain-Barré syndrome. N
Engl J Med. 326:1130–1136. 1992.
|
|
6
|
Oh SJ, LaGanke C and Claussen GC: Sensory
Guillain-Barré syndrome. Neurology. 56:82–86. 2001.
|
|
7
|
Seneviratne U and Gunasekera S: Acute
small fibre sensory neuropathy: another variant of Guillain-Barré
syndrome? J Neurol Neurosurg Psychiatry. 72:540–542.
2002.PubMed/NCBI
|
|
8
|
Dawson DM, Samuels MA and Morris J:
Sensory form of acute polyneuritis. Neurology. 38:1728–1731. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee SS and Lee SH: Does sensory
Guillain-Barré syndrome exist without any abnormalities in motor
nerve conduction? Neurology. 66:947–948. 2006.
|
|
10
|
Uncini A and Yuki N: Sensory
Guillain-Barré syndrome and related disorders: an attempt at
systematization. Muscle Nerve. 45:464–470. 2012.
|
|
11
|
Yee T and Katz JS: Acute sensory
neuropathy: a sensory form of Guillain-Barré syndrome? J Clin
Neuromuscul Dis. 2:135–138. 2001.PubMed/NCBI
|
|
12
|
Bannister RG and Sears TA: The changes in
nerve conduction in acute idiopathic polyneuritis. J Neurol
Neurosurg Psychiatry. 25:321–328. 1962. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sauron B, Bouche P, Cathala HP, Chain F
and Castaigne P: Miller Fisher syndrome: clinical and
electrophysiologic evidence of peripheral origin in 10 cases.
Neurology. 34:953–956. 1984. View Article : Google Scholar : PubMed/NCBI
|