Introduction
N-acetylcysteine (NAC) is an excellent source
of sulfhydryl groups and is rapidly absorbed following an oral
dose. The well-known role of NAC is as an antidote to acetaminophen
toxicity; it has been used for this purpose for >30 years
(1). It is also widely used to
treat chronic obstructive pulmonary disorder, pulmonary fibrosis,
and contrast-induced nephropathy. NAC also appears to have
beneficial effects in conditions such as liver injury, cancer,
heart disease, HIV infection and cigarette smoking (2,3).
In vitro, NAC that has been deacetylated by cytosolic
esterases in cells, causes an increase in the concentration of
dissociated cysteine and an increase in glutathione (GSH) synthesis
at a concentration of 5 mM (4,5).
In vivo, NAC is actively transported into hepatocytes and
converted into metabolites capable of stimulating GSH synthesis.
GSH, a tripeptide synthesized from glutamic acid, cysteine and
glycine via GSH synthetase and L-glutamate-cysteine ligase, plays
an pivotal physiological role in the maintenance of redox status.
During conditions of oxidative stress, GSH is depleted in cells,
and its biosynthesis is limited by the rate of cellular cysteine
uptake (6,7). Effective treatment with NAC provides
sufficient cysteine to promote detoxification and directly
eliminate reactive oxygen species (ROS). NAC also modulates the
inflammatory response through signaling pathways that control
pro-inflammatory nuclear factor (NF)-κB activation (8,9).
The chemotherapeutic agent cisplatin (CDDP) is a
potent drug that has successfully been used clinically to treat
cancers of the lungs, testis, ovary, cervix and genitourinary tract
(10,11). The biological activity of CDDP is
founded on the formation of covalent products with nucleic acids
that interrupt DNA replication, transcription and ultimately, cell
cleavage that finally leads to cell death. However, the clinical
use of CDDP in chemotherapy is hampered by its severe side-effects.
There is evidence that oxidative stress is involved in the liver
damage that occurs following the administration of CDDP. A number
of studies have demonstrated that CDDP-induced cell damage is
contributed to by several ROS (12–15).
Thus, the protective effect of free radical scavengers may prevent
the generation of ROS and block the downstream cascade that leads
to cell apoptosis.
Our previous study in vivo showed that NAC
raised the antioxidant capacity of hepatic tissue in rats
administered alcohol (16).
However, considering the conspicuous effects of NAC in vivo,
the protective roles observed for CDDP have not been completely
investigated. Therefore, the objective of the present study was to
investigate whether beneficial effects are observed in vitro
with NAC when cells are exposed to CDDP. Thus, CDDP was employed as
damage inducer, and the protective effects of NAC were evaluated
through a cell viability assay, apoptosis assay, single cell gel
electrophoresis (SCGE) assay and the measurement of biochemical
parameters related to redox status in HepG2 cells.
Materials and methods
Chemical products
NAC (CAS No. 616-91-1) and dimethyl sulfoxide (DMSO,
CAS No. 67-68-5) were purchased from Sigma-Aldrich (St. Louis, MO,
USA). CDDP (cis-diamminedichloroplatinum(II), CAS No.
15663-27-1) was purchased from Melone Pharmaceutical Co., Ltd.
(Dalian, China). All other chemicals were analytical grade
products.
Cell culture and treatments
HepG2 cells were provided by Keygen Biotech Co.,
Ltd. (Nanjing, China). The cells were stored in liquid nitrogen.
All experiments were performed within the third and fifth passages.
Cells were grown in a complete cell cycle in Dulbecco’s modified
Eagle’s medium (DMEM; Gibco Life Technologies, Carlsbad, CA, USA)
with 10% heat-inactivated fetal bovine serum (FBS; Hangzhou
Sijiqing Biological Engineering Materials Co., Ltd., Hangzhou,
China) and 1% penicillin/streptomycin antibiotic mixture (Gibco
Life Technologies) in culture flasks (25 cm2) in a
humidified incubator (Thermo Fisher Scientific, Waltham, MA, USA)
with an atmosphere of 95% air and 5% CO2 at 37°C. For
subcultivation, cells were passaged when they reached 80–90%
confluency in the flask. The cells were trypsinized, washed with
phosphate-buffered saline (PBS; pH 7.4) and centrifuged (450 × g, 5
min). Cell viability was determined by staining the cells with
trypan blue.
To evaluate the impact of the CDDP on cell
viability, the cells were exposed to CDDP (1.0, 2.0, 4.0, 8.0 μM)
for a period of 24, 48, 72 h and then were analyzed by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Cells incubated with culture medium alone were used as a
negative control. HepG2 cells were pre-treated for 3 h with various
concentrations of NAC (50.0, 100.0, 200.0 μM) prior to exposure to
CDDP (2.0 μM) for 48 h.
Cell lysates were formed with 0.1% sodium dodecyl
sulfate solution in PBS for determination of the biochemical
parameters. The cell suspensions were incubated for 30 min on ice
and centrifuged at 550 × g for 10 min (4°C). Subsequently, the
supernatants were used for the assessment of the biochemical
parameters associated with redox status. In all experiments,
solvent controls were included, and NAC and CDDP were dissolved in
DMEM.
Cell viability assessment
Cell viability was evaluated biochemically with the
MTT assay (17). The HepG2 cells
were seeded at ~1×105 cells/ml into the wells of a
96-well plate in complete DMEM, then allowed to attach and recover
for ≥24 h prior to assay. Different concentrations of NAC and CDDP
were added to each well. Following treatment, the medium was
carefully replaced with 200 μl DMEM without FBS for 1 h at 37°C.
Then, 20 μl 5 mg/ml MTT was added to each well and the cells were
incubated for a further 4 h. The MTT solution was removed and 150
μl DMSO was added to lyse the cells. The components of the wells
were mixed thoroughly with shaking until the crystals were
completely dissolved. The absorbance at 570 nm was measured using a
microenzyme-linked immunosorbent assay (ELISA) reader (model 680;
Bio-Rad Laboratories, Hercules, CA, USA). The reduction in
viability of the cells was expressed as a percentage compared with
the viability of the control group, which was designated a
viability of 100%. Each experiment was repeated three times in
triplicate samples.
Cell apoptosis assay
In order to determine the effects of CDDP on HepG2
cell apoptosis, annexin V/propidium iodide (PI) staining was
performed and analysis was conducted by flow cytometry
(FACSCalibur; BD Biosciences, San Jose, CA, USA), following the
instructions of the annexin V FITC apoptosis detection kit (BD
Biosciences). Briefly, cells were harvested, washed twice with
precooled PBS and resuspended in 500 μl binding buffer following
treatment with different concentrations of CDDP and NAC. Then, the
cells were stained with 5 μl annexin V-FITC and 5 μl PI for 15 min
at room temperature in the dark. The cell suspension was analyzed
by flow cytometry. A blue (488 nm) excitation laser was used for
both FITC and PI emission channels. The measured results were
analyzed using CellQuest version 3.3 software (BD Biosciences).
SCGE assay
The SCGE assay was performed according to the
procedure described by Singh et al (18). Following treatment with different
concentrations of CDDP and NAC, HepG2 cells were washed twice with
PBS, trypsinized for 3 min, and suspended in 1 ml PBS. Cell
suspensions were used for the determination of DNA damage. Frosted
glass slides were coated previously with 100 μl normal melting
agarose (NMA, 1%) and refrigerated to gel the agarose at 0°C for 5
min. For each slide, 75 μl low melting agarose (LMA, 0.65%) was
mixed with 5–10 μl single cell suspension at 37°C. All the slides
were covered with coverslips and refrigerated to gel the agarose in
an ice box. After ~10 min, the coverslips were removed. Then, the
slides were coated with 75 μl LMA (0.65%) and the agarose was
solidified. The slides were then placed in a closed box containing
freshly prepared cell lysis solution (2.5 M NaCl, 100 mM EDTA, 10
mM Tris HCl, 0.5% sodium N-lauroyl sarcosinate, adjusted to
pH 10.0 with NaOH, 1% Triton X-100 and 10% DMSO) at 4°C for 1 h.
Alkaline unwinding was carried out for 20 min in a horizontal
DYCP-38C electrophoresis chamber (Beijing Liuyi Instrument Factory,
Beijing, China), which was filled with freshly precooled alkaline
buffer (300 mM NaOH, 1 mM EDTA), and electrophoresis was performed
in the same buffer for 20 min at 300 mA, 20 V. The slides were then
neutralized three times with a neutralization buffer (0.4 M Tris,
pH 7.5) for 5 min. Then, the slides were immersed in methanol for 3
min and stained with GelRed (Biotium Inc., CA, USA).
Thereafter, the slides were covered with coverslips
and analyzed immediately. The following steps were carried out in
the dark. The SCGE results were examined at a magnification of 200
using a BX51 fluorescence microscope (Olympus Corporation, Tokyo,
Japan), which was equipped with an excitation filter at 590 nm.
Images of the comets were captured by an Olympus DP50 digital
camera. The CASPLab analysis system (http://mailbox.univie.ac.at/christoph.helma/comet/)
was employed to measure various comet parameters. The results were
expressed as tail length, tail moment and olive tail moment in 100
randomly selected comet cells.
Measurement of GSH, malondialdehyde (MDA)
and superoxide dismutase (SOD)
The GSH level was measured by the addition of
5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB). DTNB, a symmetric aryl
disulfide, reacts with thiols to yield a yellow colored
chromophore; this product can be quantified by its maximum
absorbance at 412 nm. Concentrations of GSH were determined from a
freshly prepared standard curve. The measurements were conducted
with a model UV-2550 spectrophotometer (Shimadzu Co., Ltd., Tokyo,
Japan). The protein concentrations of the supernatant were
determined using a bicinchoninic acid (BCA) protein assay kit
(Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Lipid peroxidation was evaluated by measuring MDA
concentrations according to the thiobarbituric acid (TBA) method.
This method involved the spectrophotometric measurement of the red
color produced during the condensation reaction of MDA with
thiobarbituric acid. The reactive products were quantified by the
absorbance at 532 nm. MDA concentrations were expressed in terms of
nmol/mg protein.
SOD activity was measured using a kit (Nanjing
Jiancheng Bioengineering Institute), based on the ability of SOD to
inhibit the conversion of WST-1 to a formazan dye by superoxide
anions produced from a xanthine-xanthine oxidase system. One unit
of SOD activity was defined as the amount that caused a 50%
reduction in the absorbance at 450 nm. The amount of SOD was
expressed in units of U/mg protein.
Statistical analysis
All data analyses were performed with SPSS version
16 software (SPSS, Inc., Chicago, IL, USA). All results are
expressed as mean ± standard deviation (SD). Statistical analyses
were evaluated using one way analysis of variance followed by
Dunnett’s test. P<0.05 was considered to indicate a
statistically significant result. All experiments were performed in
triplicate.
Results
Inhibitory effect of CDDP on HepG2 cell
growth
HepG2 cells were incubated with various
concentrations of CDDP for 24, 48 and 72 h. The viability of the
cells was determine by MTT assay. The viability of the negative
control was designated as 100%, and the viability of each
experimental culture was expressed as a percentage compared with
the negative control. The results obtained are shown in Fig. 1A. CDDP decreased the survival of
the cells in a concentration-dependent manner. Following exposure
to CDDP at concentrations of 1.0 μM or higher for 24 h, the
cellular viability decreased compared with that of the negative
control group (P<0.05). In addition, the viability was markedly
reduced following 48 and 72 h of treatment with concentrations of
CDDP from 1.0 to 8.0 μM compared with that of the control cells
(P<0.01).
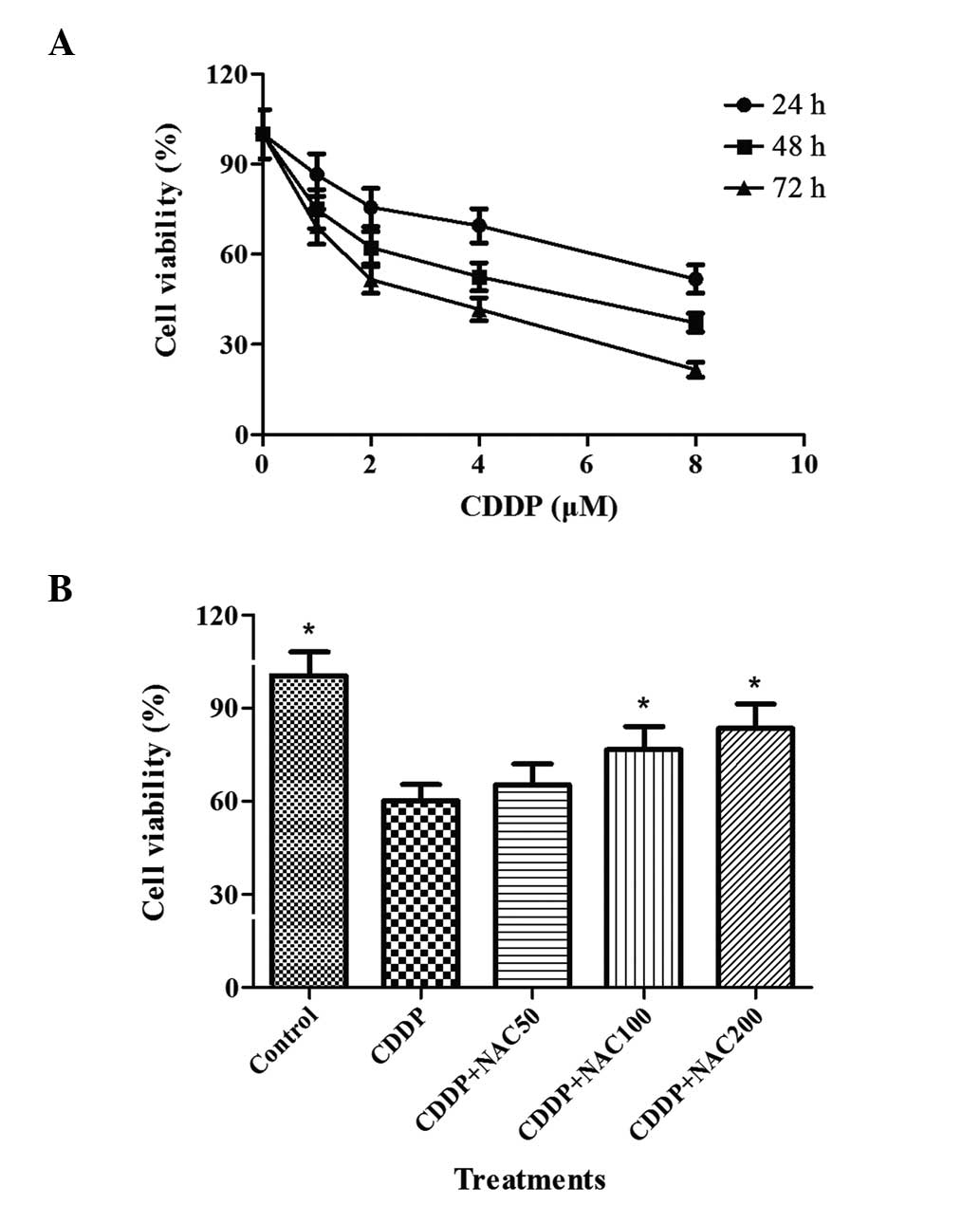 | Figure 1Impact of the combination of CDDP and
NAC on the viability of HepG2 cells. (A) HepG2 cells were incubated
with 0–8 μM CDDP for 24, 48 and 72 h, prior to the analysis of
viability using the MTT assay as described in Materials and
methods. For each experimental point, three cultures were prepared
in parallel in different 96-well microplates with eight replicates
per microplate. (B) Different concentrations of NAC were added for
3 h prior to treatment with 2 μM CDDP for 48 h. Bars indicate the
means ± standard deviation of results obtained from three
independent cultures per experimental point. Cell viability
assessment was as described in Materials and methods.
*P<0.05, vs. the CDDP group, as determined by one way
analysis of variance and Dunnett’s test. CDDP, cisplatin; NAC,
N-acetylcysteine; MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. |
Apoptosis induced by CDDP in HepG2 cells was
evaluated by flow cytometry. In the flow cytometric study, the
early apoptotic and late apoptotic cells were labeled by PI in red
or FITC in green. As shown in Fig.
2A, the apoptotic cell population, which was displayed on the
scatter plot in red or green or both colors, increased from 26.9 to
63.3% as the concentration of CDDP increased from 1.0 to 8.0 μM for
the 48-h treatment. Significant elevations were observed for the
apoptotic cell population as the CDDP concentration increased.
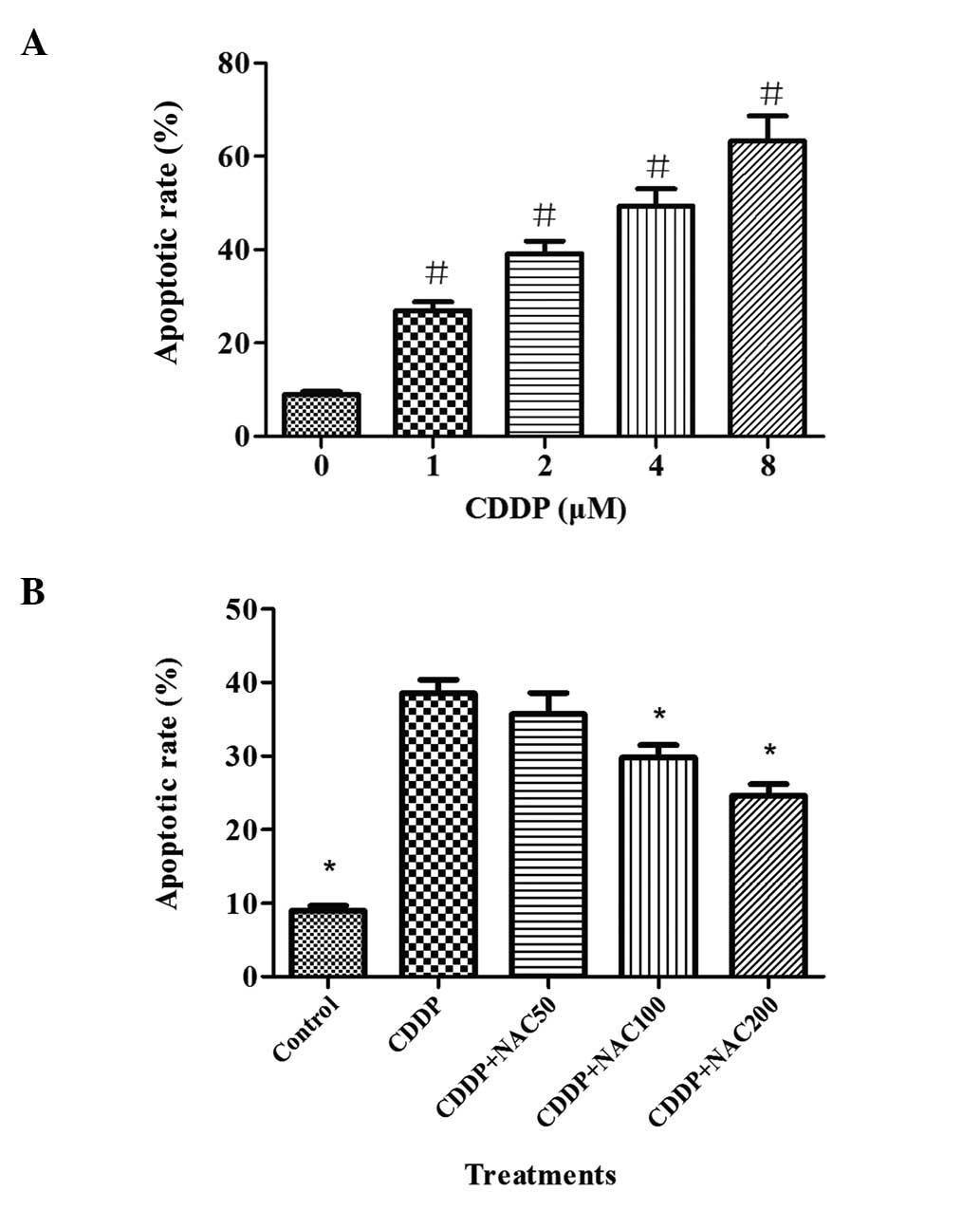 | Figure 2Effects of CDDP and NAC on HepG2 cell
apoptosis as evaluated by flow cytometry. Cells were exposed to 0,
1, 2, 4, 8 μM of CDDP for 48 h. Then, a flow cytometric assay was
carried out for the detection of apoptotic cells. NAC pretreatment
was conducted at concentrations of 50, 100 and 200 μM for 3 h prior
to exposure to 2 μM CDDP in HepG2 cells. (A) The apoptotic rate
after exposure to various concentrations of CDDP for 48 h. (B)
Apoptotic rate following NAC pre-treatment for 3 h and subsequent
incubation with 2 μM CDDP for 48 h. Bars indicate the means ± SD of
results obtained from three independent cultures per experimental
point. The apoptosis assay was conducted as described in Materials
and methods. #P<0.05, vs. the control group.
*P<0.05, vs. the CDDP group, as determined by one way
analysis of variance and Dunnett’s test. CDDP, cisplatin; NAC,
N-acetylcysteine. |
HepG2 cell apoptosis contributes to DNA
damage
The results concerning the impact of treatment with
CDDP on DNA stability in HepG2 cells are shown in Table I. The evident DNA damage induced by
CDDP, as indicated by the tail length, markedly increased even at a
low concentration (1.0 μM). Upon exposure to CDDP, the tail length,
tail moment and olive tail moment increased in a
concentration-dependent manner in the HepG2 cells compared with
control group. DNA damage appeared more frequently in cells treated
with higher concentrations of CDDP than in cells treated with lower
ones. Marked increases were observed in tail length, tail moment
and olive tail moment at higher concentrations compared with the
control and lower concentrations.
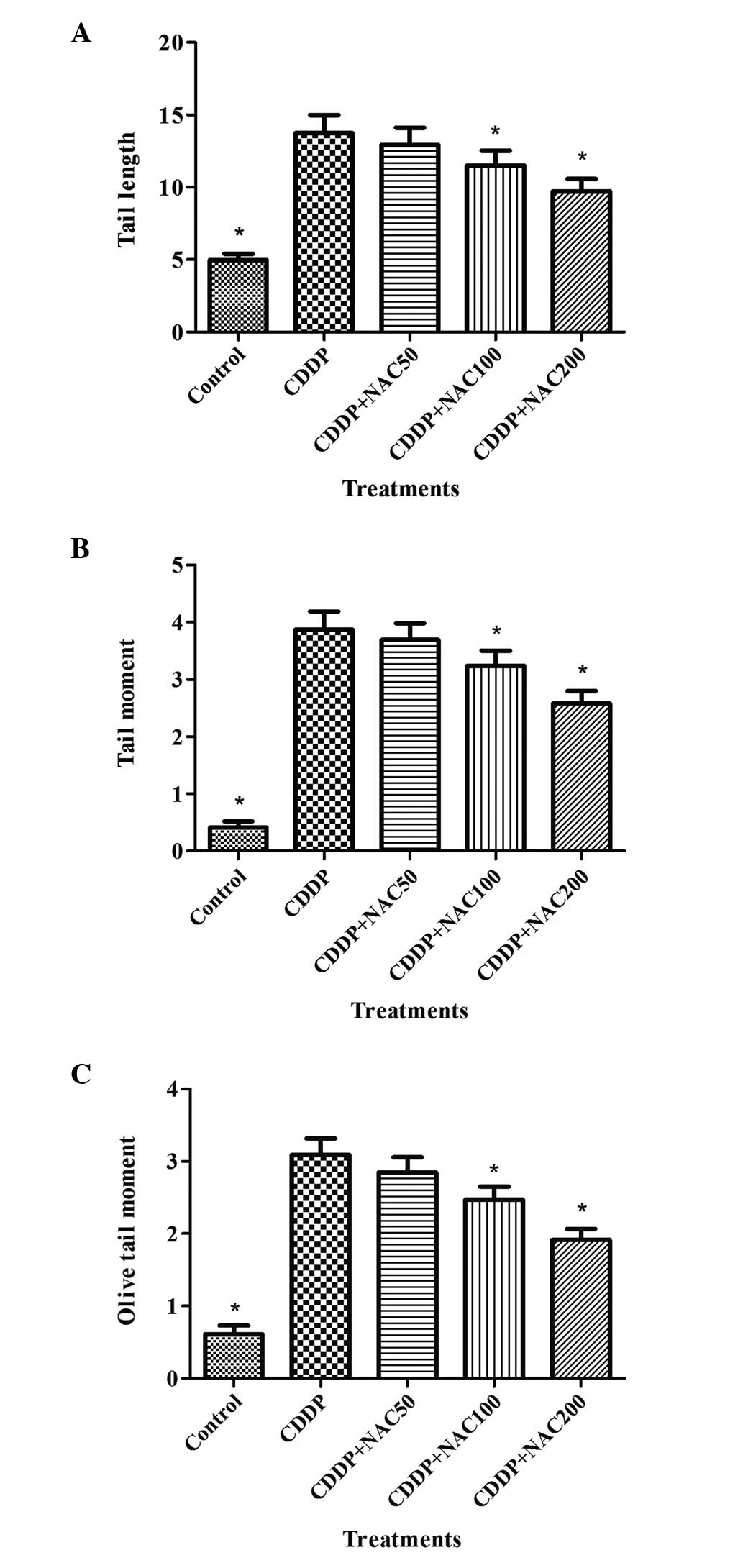
 | Table IResults of the single cell gel
electrophoresis assay after exposure of cultured HepG2 cells to
cisplatin for 48 h. |
Table I
Results of the single cell gel
electrophoresis assay after exposure of cultured HepG2 cells to
cisplatin for 48 h.
| Concentration
(μM) | Tail length | Tail moment | Olive tail
moment |
|---|
| 0 | 5.16±1.03 | 0.43±0.13 | 0.57±0.19 |
| 1 | 13.97±2.11a | 2.55±0.27a | 2.03±0.36a |
| 2 | 19.81±2.73a | 3.82±0.39a | 3.15±0.42a |
| 4 | 34.94±4.28a | 6.29±0.52a | 5.19±0.53a |
| 8 | 60.15±6.46a | 11.73±1.04a | 9.61±0.89a |
Oxidative stress damage is induced by
CDDP in HepG2 cells
To evaluate the effects of CDDP on oxidative stress
damage in HepG2 cells, the contents of MDA and GSH were determined,
as well as the activity of SOD. As shown in Fig. 3, different concentrations of CDDP
led to significant (P<0.05) reductions of GSH content and SOD
activity compared with that in the control group. The levels of GSH
and SOD activity were inversely associated with the concentration
of CDDP. When the cells were treated with various concentrations of
CDDP (1–8 μM) for 48 h, the MDA content increased from 1.9 to 4.9
nmol/mg protein. These results suggest that CDDP induces oxidative
damage in HepG2 cells.
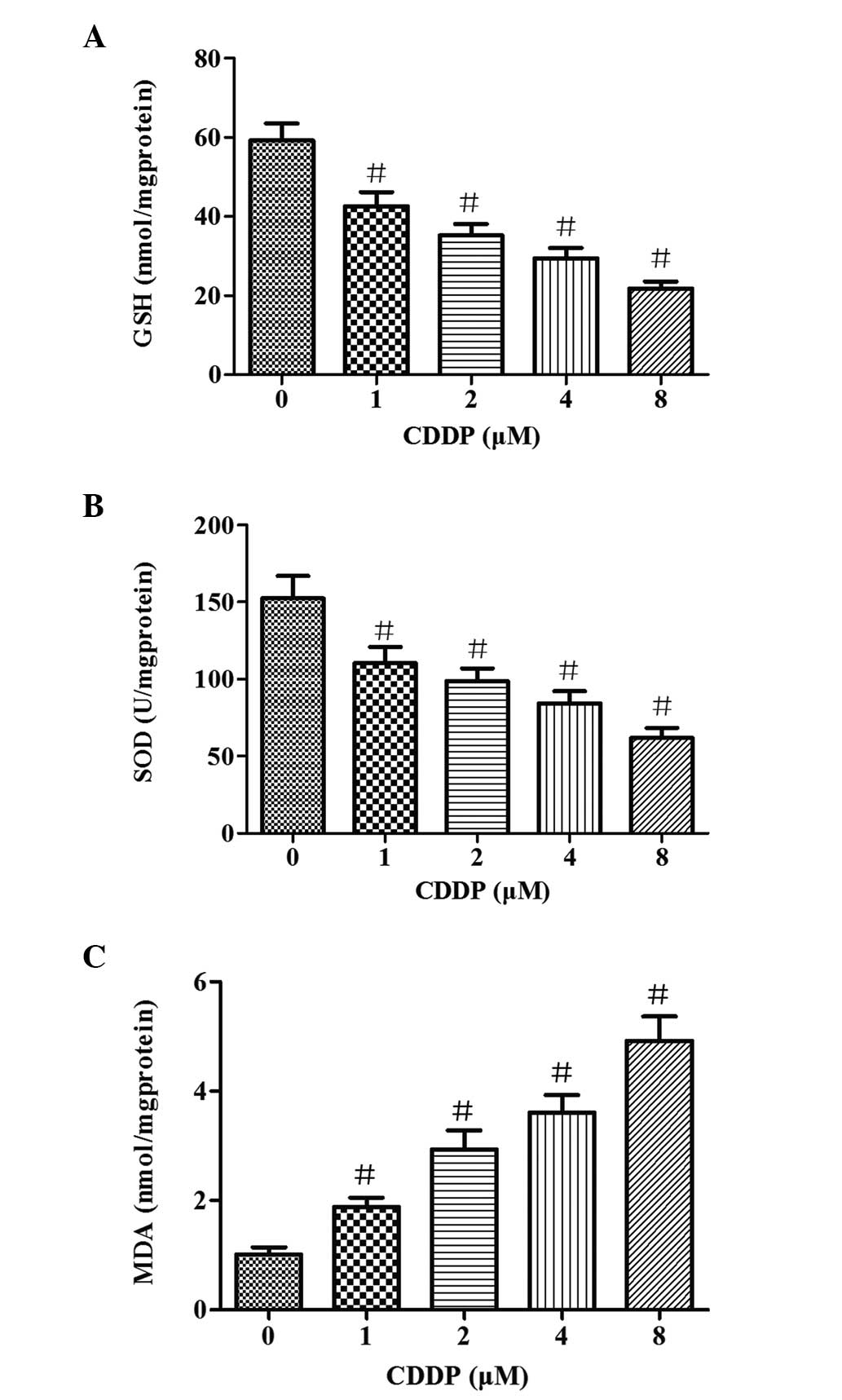 | Figure 3Impact on GSH, MDA and SOD levels in
HepG2 cells of exposure to different concentrations of CDDP (1, 2,
4 and 8 μM) for 48 h. (A) GSH levels, (B) SOD activity and (C) MDA
levels following the exposure of cultured cells to CDDP. Bars
represent the means ± SD of results obtained with three independent
cultures per experimental point. GSH, MDA and SOD levels were
determined as described in Materials and methods.
#P<0.05, vs. the control group, as determined by one
way analysis of variance and Dunnett’s test. GSH, glutathione; MDA,
malondialdehyde; SOD, superoxide dismutase; CDDP, cisplatin. |
NAC protects HepG2 cells against
CDDP-induced growth suppression
Following pretreatment of the HepG2 cells for 3 h
with various concentrations of NAC prior to exposure to CDDP, the
cell viability was determined by an MTT assay. The results are
presented in Fig. 1B. They show
that NAC was able to attenuate the growth suppression of HepG2
cells induced by CDDP. The survival rate of the cells that were
pre-treated with NAC was higher than that of cells treated with
CDDP alone. However, the viability of cells was not affected by a
low concentration of NAC. Following pretreatment of the cells with
50, 100 and 200 μM of NAC, the cellular viability increased by 5.1,
16.5 and 23.2%, respectively, compared with that in the cells that
were not pretreated with NAC.
The apoptotic rate of HepG2 cells, determined by
staining with annexin V-FITC/PI and flow cytometric analysis is
shown in Fig. 2B. The apoptotic
rate was 39.2% in cells treated with 2 μM CDDP for 48 h. Following
pretreatment with different concentrations of NAC (50, 100 and 200
μM), the apoptotic rates reduced to 35.7, 29.8 and 24.6%,
respectively. These results indicate that NAC protected apoptosis
in a concentration-dependent manner.
NAC ameliorates CDDP-induced DNA
damage
To further investigate the mechanism by which NAC
protected the HepG2 cells from growth inhibition, DNA damage was
evaluated by SCGE assay. The results of experiments in which NAC
was applied to protect cells against CDDP-induced DNA damage are
summarized in Fig. 4. Longer DNA
tail lengths, tail moments and olive tail moments were observed in
cells exposed to CDDP compared with the control group. Data for
tail length, tail moment and olive tail moment in the CDDP-treated
cells that were pretreated with 50 μM NAC revealed a slight
reduction of migration compared with that of the cells treated with
CDDP alone, although the difference was not statistically
significant (P>0.05). However, treatment with 100 or 200 μM NAC
prior to exposure to CDDP resulted in a statistically significant
reduction of migration (P<0.05). NAC showed a protective effect
on DNA damage.
Protective effects of NAC against
oxidative damage
The GSH and MDA concentrations and SOD activity in
the cells are presented in Table
II. The results show that CDDP (1 μM) caused significant
reductions of the cellular levels of GSH and SOD compared with
those in the control. Pretreatment of the cells with NAC for 3 h
restored the GSH and SOD levels in a concentration-dependent
manner. Exposure of the cells to CDDP resulted in a significant
increase in the formation of MDA, which was attenuated when the
cultures were supplemented with NAC.
| Table IIResults of pre-treatment with NAC for
3 h prior to incubation with CDDP (2 μM) in HepG2 cells. |
Table II
Results of pre-treatment with NAC for
3 h prior to incubation with CDDP (2 μM) in HepG2 cells.
| NAC concentration
(μM) | GSH (nmol/mg
protein) | SOD (U/mg
protein) | MDA (nmol/mg
protein) |
|---|
| 0 | 34.82±2.74 | 97.92±8.58 | 2.87±0.33 |
| 50 | 39.14±2.91a | 112.46±9.73a | 2.42±0.31a |
| 100 | 42.27±3.17b | 119.32±9.52b | 2.03±0.27b |
| 200 | 46.38±3.29b |
126.75±10.04b | 1.79±0.19b |
Discussion
CDDP, an alkylating agent used for the treatment of
various solid tumors, is a known inducer of DNA damage. Thus,
strategies for minimizing CDDP toxicity are of clinical interest.
In the present study, the aim was to investigate the protective
role of NAC against CDDP-induced injury in cultured HepG2 cells.
The results demonstrated that CDDP inhibited cell growth, in
addition to inducing cell apoptosis and DNA double-strand breaks.
NAC was shown to ameliorate CDDP-induced DNA damage and oxidative
stress in the HepG2 cells.
In the present study, treatment of the cells with 1
μM CDDP resulted in a significant reduction of cell viability. CDDP
exerts cytotoxicity by its ability to form complexes with nucleic
acids when ligand dissociation from CDDP occurs, leaving a reactive
complex that interacts with deoxyribonucleic acid. Subsequently,
CDDP causes inter- and intra-strand crosslinks, probably between
N7 and O6 of the adjacent guanine molecules,
which results in local denaturing of the DNA chain (19,20).
This eventually leads to cellular death via apoptotic or
non-apoptotic pathways. The DNA damage induced by CDDP is revealed
by the SCGE data in the present study. A bigger DNA tail area and a
longer DNA tail length compared with those of the control group
were identified following the exposure of the HepG2 cells to CDDP.
A recent study also reported an increase of comet formation
following the treatment of HepG2 cells with platinum compounds at
similar concentrations to those of CDDP used in the present study
(21). Pretreatment with NAC was
able to reduce the DNA injury that was observed in the CDDP group.
NAC significantly decreased the cytotoxicity of CDDP due to a
protective effect against DNA damage. When the cells were exposed
to NAC, the apoptotic and necrotic cell populations were clearly
reduced. Also, in this experiment, a clear concentration-dependent
effect was observed. Apoptosis of cells is closely associated with
DNA damage. Serpeloni et al have previously shown that
lutein protects against CDDP-induced DNA damage in HepG2 cells and
increases the survival rate of the cells (21).
Mistry et al (22) hypothesized that the detachment of
the chlorines from CDDP, resulting in the platinum becoming
positively charged, would attract the electronegative sulfur moiety
of GSH. GSH-CDDP conjugates have been isolated from cells treated
with CDDP and from the serum of CDDP-treated rats (23). In the present study, it was
observed that NAC reduced CDDP-induced cell apoptosis. NAC, as a
precursor of GSH, is one of the most important low molecular weight
antioxidants in vivo. It has been suggested NAC may block
the CDDP-dependent oxidation of intracellular GSH (24,25).
The increased concentration of GSH is likely to have attenuated the
accumulative DNA damage caused by platinum in the HepG2 cells.
Oxidative stress is involved in the toxicities of
CDDP. A number of studies have demonstrated the contribution of
several ROS in CDDP-induced cell damage (26,27).
For a clearer elucidation of the possible protective
properties of NAC, a number of biochemical components associated
with the redox status of cells were tested. SOD and GSH are
important in the antioxidant defense system, while MDA is regarded
as a major marker of lipid peroxidation in tissue. Lipid
peroxidation is a consequence of impaired activities of antioxidant
enzymes and GSH. GSH, which is a tripeptide, acts as a key ROS
scavenger to protect cells in the liver against oxidative stress,
and its depletion in hepatic cells could endanger the antioxidant
defense system, leading to the accumulation of ROS (28). The present study shows that the GSH
content of the HepG2 cells was reduced following exposure to CDDP.
The results indicate that CDDP may induce lipid peroxidation
products (LPO) by exhausting GSH, leading to the formation of
pro-mutagenic exocyclic DNA adducts. The data in the present study
regarding markers associated with oxidative stress in the
CDDP-treated cells are concordant with the findings of a previous
study (29). NAC exhibits
antioxidative properties through increasing the concentrations of
GSH in cells.
SOD, a scavenger of superoxide, is the most
important protective enzyme that provides the first line of
enzymatic antioxidant defense against oxidative stress in various
organs and tissues (30–32), including the liver. It provides
protective effects against oxygen free radicals as it catalyzes the
removal of the superoxide radical (O2•−), which damages
membranes and biological structures. In the present study, it was
found that CDDP significantly decreased the SOD level in HepG2
cells in a concentration-dependent manner. Exposure to CDDP at 1–8
μM for 48 h resulted in a significant reduction of SOD activity,
whereas NAC pre-treatment partially reversed this effect.
SOD activity and MDA concentration are usually
measured together. MDA is one of the end-products of membrane lipid
peroxidation (33). It has been
postulated that the mechanism of peroxidation involves the
formation of prostaglandin-like endoperoxides from polyunsaturated
fatty acids that have two or more double bonds (34). Therefore, in the present study, the
MDA concentration was determined as another marker of oxidative
damage. The MDA level significantly increased compared with that of
control group when the cells were treated with various
concentrations of CDDP. This result indicates that lipid
peroxidation products were produced as well as DNA damage in the
cells. Pretreatment of the cells with NAC for 3 h prior to exposure
to 2 μM CDDP, resulted in the MDA levels being reduced.
A reduction or loss of antioxidant enzyme activity
in HepG2 cells may result in oxidative stress and induce lipid
peroxidation, DNA damage, and cell injury, and subsequent cell
death. Leonardo et al (35)
demonstrated that PC12 cells treated with CDDP exhibited an
increased generation of ROS, which may be one of the main
mechanisms by which CDDP exerts DNA genotoxicity. In CDDP-induced
renal damage, the increased production of ROS can lead to an
enhancement in the expression of proinflammatory mediators, which
could intensify the cytotoxic effects (36). In a previous study it was found
that the exposure of cells to 2,4,5,2′,4′,5′-hexachlorobiphenyl
(PCB153) decreased SOD activity and the concentration of MDA and
increased cell apoptosis, and that NAC pretreatment had a
protective effect as it significantly reduced the cell apoptosis
(37). In the present study,
increased MDA levels and reduced GSH levels and SOD activity were
observed in association with CDDP-induced cytotoxicity. The
imbalance of the intracellular antioxidant/oxidant system
exacerbated HepG2 cell apoptosis. NAC treatment was able to
ameliorate the oxidative stress and cytotoxicity.
In summary, the results of the present study suggest
that CDDP induces pronounced oxidative stress in HepG2 cells that
is associated with DNA damage, and that supplementation with the
antioxidant NAC may protect against these adverse effects caused by
the platinum compound. These findings suggest that NAC may be a
useful supplement in chemotherapy involving platinum compounds.
Acknowledgements
This study was supported by funding from the Key
Science and Technology Program of Hangzhou (grant no.
20130733Q30).
References
|
1
|
Acharya M and Lau-Cam CA: Comparison of
the protective actions of N-acetylcysteine, hypotaurine and taurine
against acetaminophen-induced hepatotoxicity in the rat. J Biomed
Sci. 17(Suppl 1): S352010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Balansky R, Ganchev G, Iltcheva M, Steele
VE and De Flora SD: Prevention of cigarette smoke-induced lung
tumors in mice by budesonide, phenethyl isothiocyanate, and
N-acetylcysteine. Int J Cancer. 126:1047–1054. 2010.PubMed/NCBI
|
|
3
|
Baumgardner JN, Shankar K, Hennings L,
Albano E, Badger TM and Ronis MJ: N-acetylcysteine attenuates
progression of liver pathology in a rat model of nonalcoholic
steatohepatitis. J Nutr. 138:1872–1879. 2008.PubMed/NCBI
|
|
4
|
Arfsten DP, Johnson EW, Wilfong ER, Jung
AE and Bobb AJ: Distribution of radio-labeled N-acetyl-l-cysteine
in Sprague-Dawley rats and its effect on glutathione metabolism
following single and repeat dosing by oral gavage. Cutan Ocul
Toxicol. 26:113–134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Atkuri KR, Mantovani JJ and Herzenberg LA
and Herzenberg LA: N-acetylcysteine - a safe antidote for
cysteine/glutathione deficiency. Curr Opin Pharmacol. 7:355–359.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zwingmann C and Bilodeau M: Metabolic
insights into the hepatoprotective role of N-acetylcysteine in
mouse liver. Hepatology. 43:454–463. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Raftos JE, Whillier S, Chapman BE and
Kuchel PW: Kinetics of uptake and deacetylation of N-acetylcysteine
by human erythrocytes. Int J Biochem Cell Biol. 39:1698–1706. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mansour HH, Hafez HF, Fahmy NM and Hanafi
N: Protective effect of N-acetylcysteine against radiation induced
DNA damage and hepatic toxicity in rats. Biochem Pharmacol.
75:773–780. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Slim R, Toborek M, Robertson LW, Lehmler
HJ and Hennig B: Cellular glutathione status modulates
polychlorinated biphenyl-induced stress response and apoptosis in
vascular endothelial cells. Toxicol Appl Pharmacol. 166:36–42.
2000. View Article : Google Scholar
|
|
10
|
Eastman A: Activation of programmed cell
death by anticancer agents: cisplatin as a model system. Cancer
Cells. 2:275–280. 1990.PubMed/NCBI
|
|
11
|
van Moorsel CJ, Pinedo HM, Smid K, et al:
Schedule-dependent pharmacodynamic effects of gemcitabine and
cisplatin in mice bearing Lewis lung murine non-small cell lung
tumours. Eur J Cancer. 36:2420–2429. 2000.PubMed/NCBI
|
|
12
|
Kuhad A, Tirkey N, Pilkhwal S and Chopra
K: Renoprotective effect of Spirulina fusiformis on
cisplatin-induced oxidative stress and renal dysfunction in rats.
Renal Failure. 28:247–254. 2006.
|
|
13
|
Cloven NG, Re A, Mchale MT, et al:
Evaluation of D-methionine as a cytoprotectant in cisplatin
reatment of an animal model for ovarian cancer. Anticancer Res.
20:4205–4209. 2000.PubMed/NCBI
|
|
14
|
Dehne N, Lautermann J, Petrat F, et al:
Cisplatin ototoxicity: involvement of iron and enhanced formation
of superoxide anion radicals. Toxicol Appl Pharmacol. 174:27–34.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thomas Dickey D, Muldoon LL, Kraemer DF,
et al: Protection against cisplatin ototoxicity by N-acetylcysteine
in a rat model. Hear Res. 193:25–30. 2004.PubMed/NCBI
|
|
16
|
Wang FG and Xi JJ: The influence of
acetylcysteine on cytokines of alcoholic liver injury in rat. Chin
J Public Health. 25:336–337. 2009.(In Chinese).
|
|
17
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singh NP, McCoy MT, Tice RR and Schneider
EL: A simple technique for quantitation of low levels of DNA damage
in individual cells. Exp Cell Res. 175:184–191. 1988. View Article : Google Scholar
|
|
19
|
Zwelling LA, Anderson T and Kohn KW:
DNA-protein and DNA interstrand cross-linking by cis- and
trans-platinum (II) diamminedichloride in L1210 mouse leukemia
cells and relation to cytotoxicity. Cancer Res. 39:365–369.
1979.PubMed/NCBI
|
|
20
|
Blommaert FA, van Dijk-Knijnenburg HC,
Dijt FJ, et al: Formation of DNA adducts by the anticancer drug
carboplatin: different nucleotide sequence preferences in vitro and
in cells. Biochemistry. 34:8474–8480. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Serpeloni JM, Barcelos GR, Friedmann
Angeli JP, et al: Dietary carotenoid lutein protects against DNA
damage and alterations of the redox status induced by cisplatin in
human derived HepG2 cells. Toxicol In Vitro. 26:288–294. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mistry P, Lee C and McBrien DC:
Intracellular metabolites of cisplatin in the rat kidney. Cancer
Chemother Pharmacol. 24:73–79. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen G, Hutter KJ and Zeller WJ: Positive
correlation between cellular glutathione and acquired cisplatin
resistance in human ovarian cancer cells. Cell Biol Toxicol.
11:273–281. 1995. View Article : Google Scholar
|
|
24
|
Godbout JP, Pesavento J, Hartman ME,
Manson SR and Freund GG: Methylglyoxal enhances cisplatin-induced
cytotoxicity by activating protein kinase Cdelta. J Biol Chem.
277:2554–2561. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu YJ, Muldoon LL and Neuwelt EA: The
chemoprotective agent N-acetylcysteine blocks cisplatin-induced
apoptosis through caspase signaling pathway. J Pharmacol Exp Ther.
312:424–431. 2005.PubMed/NCBI
|
|
26
|
Chirino YI, Trujillo J, Sánchez-González
DJ, et al: Selective iNOS inhibition reduces renal damage induced
by cisplatin. Toxicol Lett. 176:48–57. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Antunes LM, Darin JD and Bianchi MLP:
Effects of the antioxidants curcumin or selenium on
cisplatin-induced nephrotoxicity and lipid peroxidation in rats.
Pharmacol Res. 43:145–150. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cetin R, Devrim E, Kiliçoğlu B, Avci A, et
al: Cisplatin impairs antioxidant system and causes oxidation in
rat kidney tissues: possible protective roles of natural
antioxidant foods. J Appl Toxicol. 26:42–46. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shan XQ, Aw TY and Jones DP:
Glutathione-dependent protection against oxidative injury.
Pharmacol Ther. 47:61–71. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Powers SK, Quindry JC and Kavazis AN:
Exercise-induced cardioprotection against myocardial
ischemia-reperfusion injury. Free Radic Biol Med. 44:193–201. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nishino Y, Takemura S, Minamiyama Y, et
al: Targeting superoxide dismutase to renal proximal tubule cells
attenuates vancomycin-induced nephrotoxicity in rats. Free Radic
Res. 37:373–379. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oktem F, Arslan MK, Ozguner F, Candir O,
Yilmaz HR, et al: In vivo evidences suggesting the role of
oxidative stress in pathogenesis of vancomycin-induced
nephrotoxicity: protection by erdosteine. Toxicology. 215:227–233.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Termini J: Hydroperoxide-induced DNA
damage and mutations. Mutat Res. 450:107–124. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lykkesfeldt J: Malondialdehyde as
biomarker of oxidative damage to lipids caused by smoking. Clin
Chim Acta. 380:50–58. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mendonça LM, dos Santos GC, dos Santos RA,
et al: Evaluation of curcumin and cisplatin-induced DNA damage in
PC12 cells by the alkaline comet assay. Hum Exp Toxicol.
29:635–643. 2010.PubMed/NCBI
|
|
36
|
Francescato HD, Costa RS, Scavone C and
Coimbra TM: Parthenolide reduces cisplatin-induced renal damage.
Toxicology. 230:64–75. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gao M, Wu NX, Song Y, Jin LZ, Lou JL and
Tao H: PCB153-induced oxidative stress and cell apoptosis on
cultured rat Sertoli cells. Toxicol Res. 2:173–179. 2013.
View Article : Google Scholar
|