Introduction
Alzheimer’s disease (AD) is a neurodegenerative
disorder of the human brain and is associated with loss of memory
and cognitive abilities (1). AD is
characterized by amyloid plaques, neurofibrillary tangles and
neuronal loss (2). In AD brains,
there is also an abundance of two abnormal structures: Senile
plaques composed of β-amyloid peptide (Aβ), that are deposited
outside neuronal bodies, and neurofibrillary tangles, which are
aggregates of hyperphosphorylated tau proteins that bind to
microtubules within the neurons (3). Synaptic dysfunction in AD may be
caused by accumulation of aggregated amyloid peptides (3). Aβ, a 40–42 amino acid peptide
fragment of the Aβ precursor, has been shown to be a key
pathological feature in the formation of senile plaques (4). Aβ peptides induce cell death,
decrease survival rate, and increase inflammation, oxidative stress
and neurotoxicity in in vitro models used to study AD
(5,6).
Brain-derived neurotrophic factor (BDNF) is an
important neurotrophin that has been extensively studied and that
may play a role in the pathology of AD. BDNF is involved in the
structural and functional plasticity of the brain (7), protects neurons in the brain against
insults (8) and plays a role in
neural development and maintenance of the central and peripheral
neurons (9). Another important
intracellular regulatory protein is glycogen synthase kinase-3β
(GSK3β). This protein is phoshorylated by growth factor-stimulated
signaling pathways (10). GSK3β is
a protein kinase that also has regulatory effects on neuronal
survival and plasticity. Previously, a study indicated that GSK3β
may play a part in AD and that its deregulation may account for a
number of the pathological hallmarks of AD (11).
In the present study, the impact of BDNF on the
toxic effects of the Aβ-induced apoptosis was examined via the
phosphoinositide 3-kinase (PI3K)/Akt signaling pathway in SH-SY5Y
neuroblastoma cells.
Materials and methods
Cell culture
Human SH-SY5Y neuroblastoma cells were maintained in
Dulbecco’s modified Eagles’s medium (DMEM) and F-12 (Gibco-BRL,
Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum
(FBS; HyClone, Logan, UT, USA) in a humidified atmosphere of 5%
CO2 and 95% air at 37°C. The media was replaced every
two days. Prior to the experiments, the SH-SY5Y cells were plated
in 96-well plates at a density of ~1.5×104 cells per
well (for MTT) and in six-well plates at 8×106 cells per
well (all other assays). For the experiments, the cells were
incubated with agents for 24 h at 37°C. For a single experiment,
each treatment was performed in triplicate.
Reagents
Aβ25–35 and scrambled peptides were
purchased from Bachem (Weil am Rhein, Germany). BDNF and LY294002
were purchased from Sigma-Aldrich (St Louis, MO, USA). Antibodies
against phosphorylated-(p-)Akt (product no. 9271), Akt (product no.
9272), p-GSK3β (product no. 9336) and GSK3β (product no. 9315) were
from Cell Signaling Technology, Inc. (Danvers, MA, USA). The
antibody against BNDF (product no. 546) was from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA) and the antibody against
Actin (product no. 3280) was from Abcam (Cambridge, MA, USA).
Cell proliferation and MTT assay
A cell survival analysis was performed according to
the MTT (Cell Titer 96 Aqueous Cell Proliferation Assay kit;
Promega, Madison, WI, USA) assay method. Briefly, the cells were
cultured with Aβ (0–20 μM) or BDNF (0–30 ng/ml), and 10 μl of 4
mg/ml MTT solution was added to each well of the 96-well plate. The
cells were subsequently incubated for 4 h in the dark. The
absorbance was measured in a microplate reader at 490 nm, and the
results were expressed as a percentage of the control.
Evaluation of apoptotic cells by
Annexin-V-FITC
Apoptosis was induced by incubating cells with
culture medium containing Aβ (20 μM), BDNF (10 ng/ml) and LY294002
(20 μM). The cells were stained with Annexin-V-FITC according to
the manufacturer’s instructions (Molecular Probes, Eugene, OR,
USA). Approximately 1×105 cells were harvested and
washed with phosphate-buffered saline. The cells were resuspended
in 100 μl of Annexin-V binding buffer (10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid, 140 mM NaCl
and 2.5 mM CaCl2; pH 7.4), incubated with 5 μl of Annexin-V-FITC
for 15 min at room temperature and counterstained with propidium
iodide (final concentration, 1 μg/ml). Following the incubation
period, the cells were diluted with 190 μl of Annexin-V binding
buffer. The cells were analyzed by flow cytometry using a
Becton-Dickinson FACScan flow cytometer with Cell Quest software
(Becton-Dickinson, Mountain View, CA, USA).
Western blotting
The cells were washed in fresh phosphate-buffered
saline, homogenized in lysis buffer and centrifuged. Protein
concentration was determined by the Bradford assay. The purified
proteins were separated by polyacrylamide gel electrophoresis
(SDS-PAGE) and the resolved proteins were transferred to a
nitrocellulose membrane. Each membrane was incubated overnight with
a 1:1000 dilution of the primary antibody at 4°C. The membranes
were treated with a 1:1000 dilution of peroxidase-conjugated
secondary anti-rabbit or anti-mouse antibodies for 2 h. The
proteins were detected using the enhanced chemiluminescence western
blotting method (GE Healthcare, Piscataway, NJ, USA). Densitometric
quantification of the bands was performed using ImageJ software
version 1.29× (National Institutes of Health, Bethesda, MD, USA)
(12).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the cells following the
Promega Total RNA Isolation System manual. RT-qPCR was performed on
an ABI Prism 7500 Sequence Detection System (Applied Biosystems,
Inc., Foster City, CA, USA), following the manufacturer’s
instructions, with SYBR Green (Toyobo Corp., Osaka, Japan) used as
a double-stranded DNA specific fluorescent dye. The specific
primers were as follows: GSK3β forward,
5′-ATCCTTATCCCTCCTCACGC-3′; and reverse,
5′-GTTATTGGTCTGTCCACGGTCT-3′; Akt forward,
5′-AGGCATCCCTTCCTTACAGC-3′; and reverse,
5′-CAGCCCGAAGTCCGTTATCT-3′; and β-actin forward,
F-5′-CGTTGACATCCGTAAAGACCTC-3′; and reverse,
5′-TAGGAGCCAGGGCAGTAATCT-3′.
Statistical analysis
Data were obtained from three separate cultures and
expressed as the mean ± standard error of the mean. Statistical
comparison was determined by an analysis of variance test with the
Student’s t-test as the post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
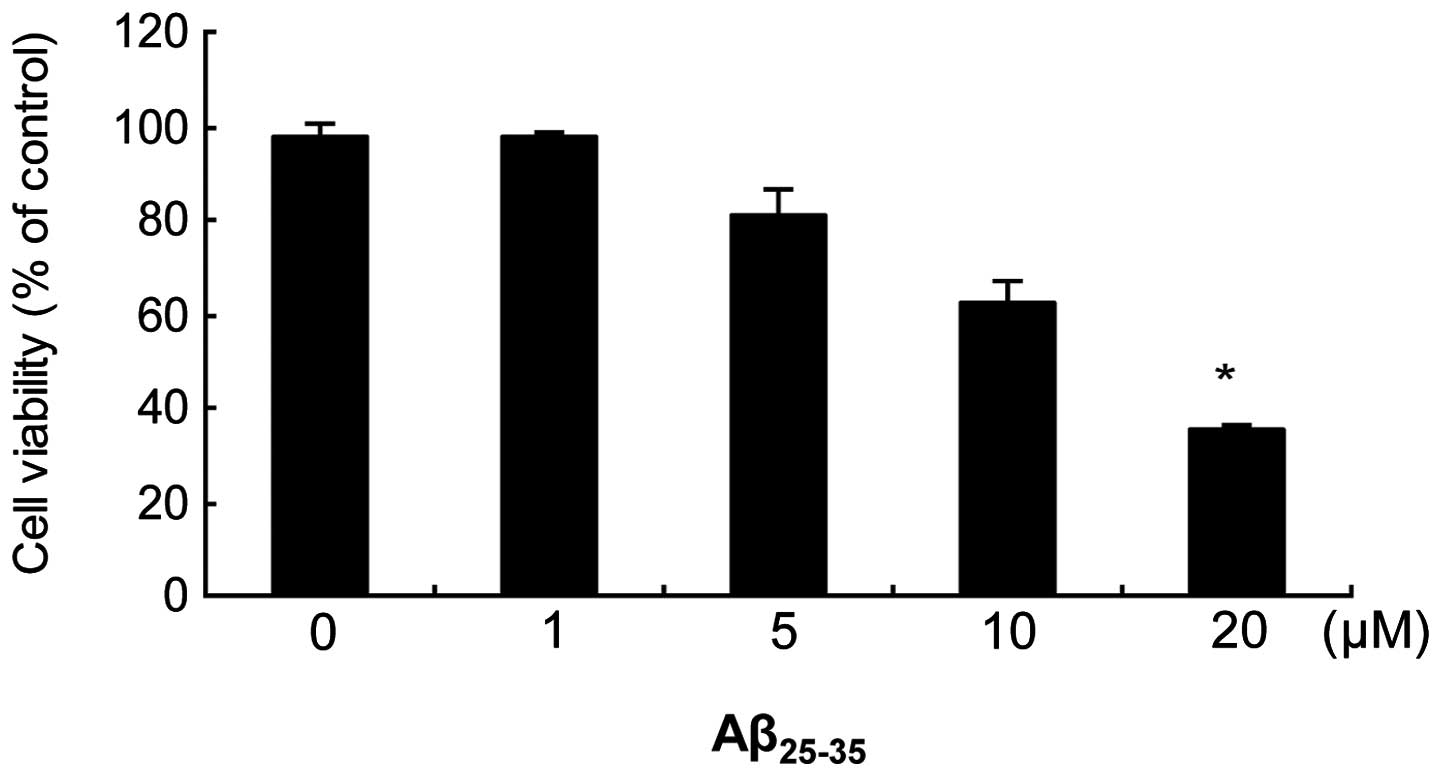
Effect of increasing concentrations of
Aβ25–35 on cell viability
The MTT assay was used to determine
Aβ25–35-induced toxicity (Fig. 1). A 24 h exposure to
Aβ25–35 induced a toxic, dose-dependent effect on
SH-SY5Y cells, with a maximal effect of ~40% noted at a
concentration of 20 μM. Thus, 20 μM was used as the concentration
for Aβ25–35 in all further experiments. The scrambled
peptide did not affect cell survival in comparison with the
vehicle-treated control group: 100±3% at 20 μM in the
Aβ25–35 group versus 98±2% in the control group.
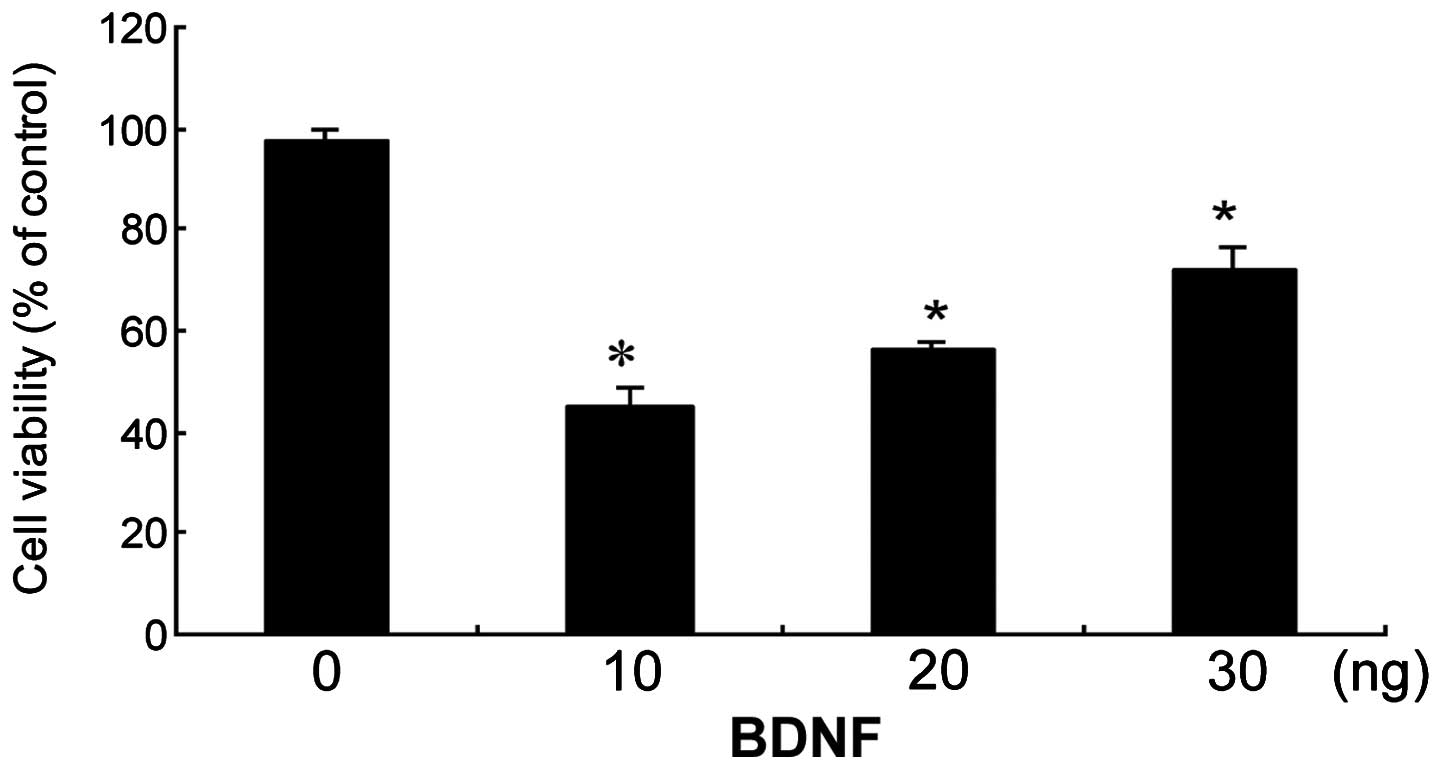
Effect of increasing BDNF concentrations
on Aβ25–35-treated cells
The ability of BDNF to protect SH-SY5Y cells against
Aβ25–35-induced toxicity using the MTT assay was
examined (Fig. 2). BDNF was able
to protect SH-SY5Y cells from 20 μM Aβ25–35-induced
toxicity. This protective effect of BDNF was dose-dependent, and
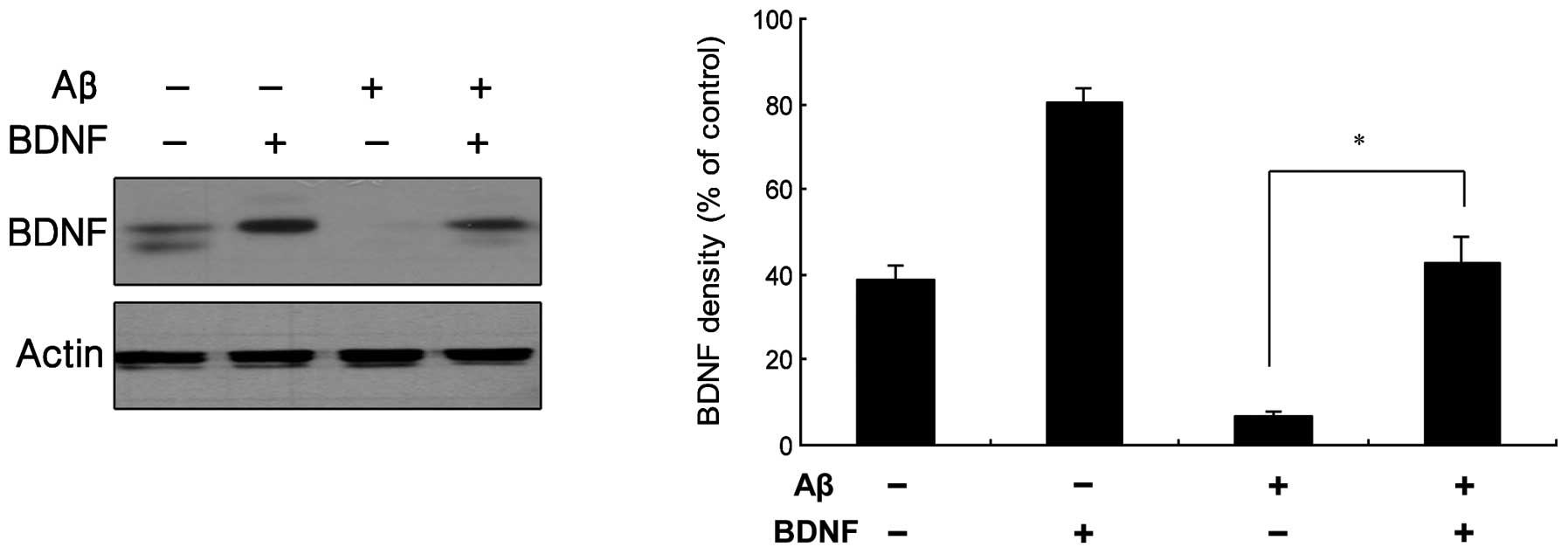
the effect was significant ≥10 ng/ml. In Fig. 3, the results from a western blot
analysis are shown, indicating that BDNF levels increased following
exposure of the cells to treatment with a combination of 20 μM
Aβ25–35 and 10 ng/ml BDNF.
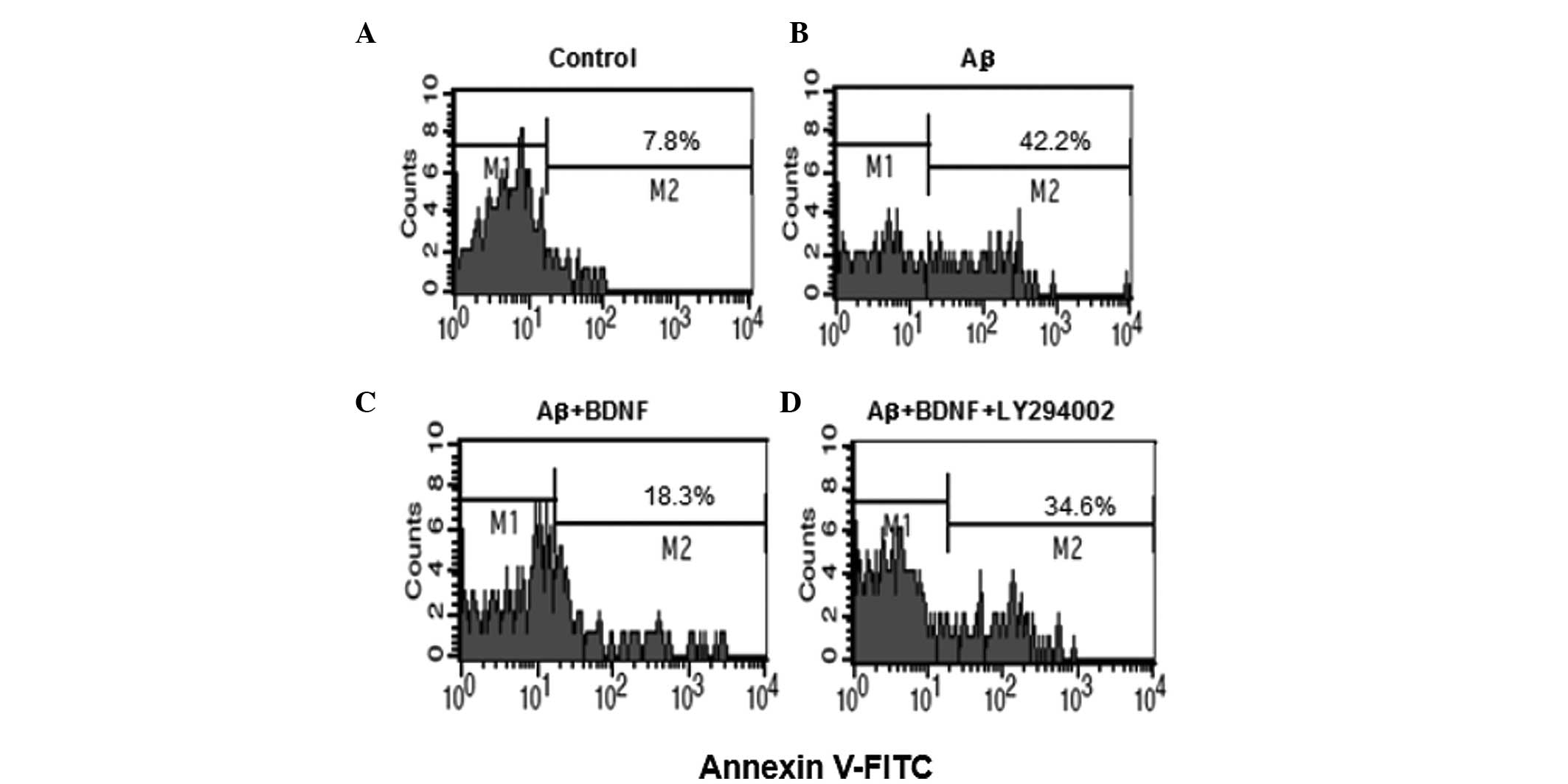
BDNF reduces the apoptosis in SH-SY5Y
cells
To examine whether β25–35-induced cell
death is apoptotic-like, the flow cytometry assay was performed
(Fig. 4). In the control,
apoptotic cells comprised 7.8% of the total number of cells.
Following exposure to 20 μM Aβ25–35 for 24 h, the number
of apoptotic cells increased to 42.2%. This increase was prevented
by addition of BDNF (10 ng/ml). To further investigate, LY294002
(20 μM), the inhibitor of Akt, was used. The apoptosis of cells
increased to 34.6% following the use of LY294002 (20 μM).
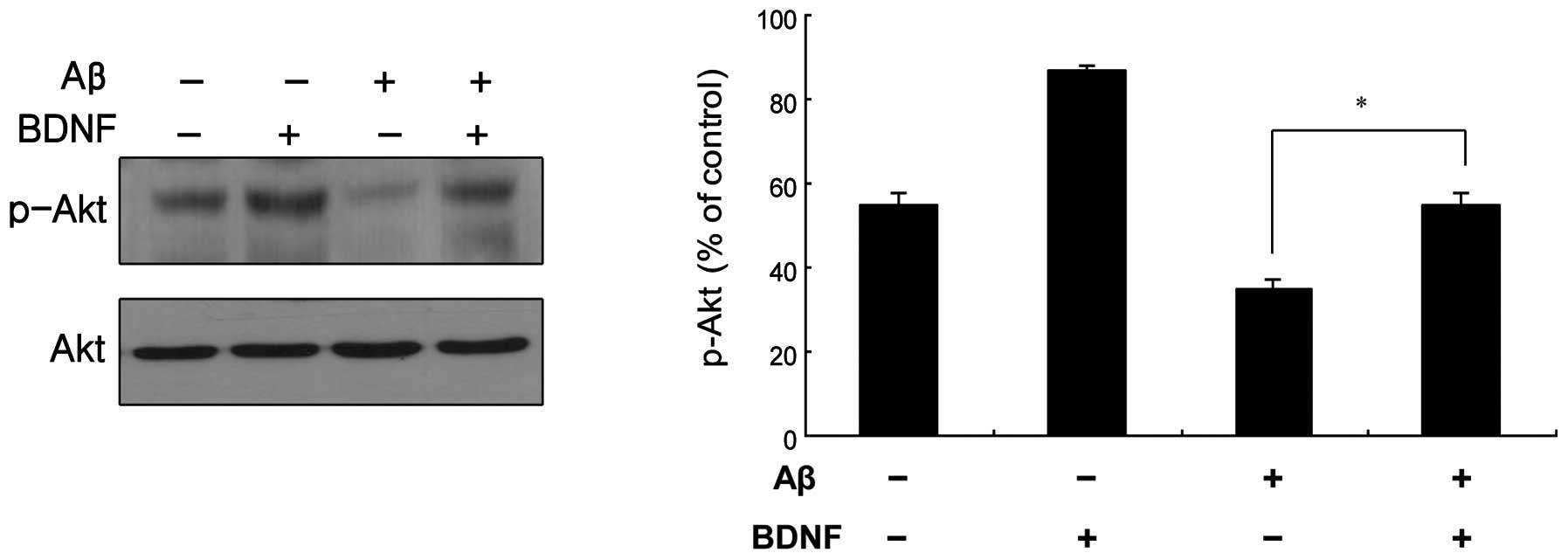
BDNF-mediated signal transduction in
SH-SY5Y cells
Akt is a known pro-survival kinase that is activated
by phosphorylation via PI3K (13).
The BDNF-mediated phosphorylation of Akt was measured, as shown in
Fig. 5. The treatment of cells
with a combination of Aβ25–35 (20 μM) and BDNF (10
ng/ml) induced a significant increase in the level of p-Akt
compared to that in the cells treated only with Aβ25–35
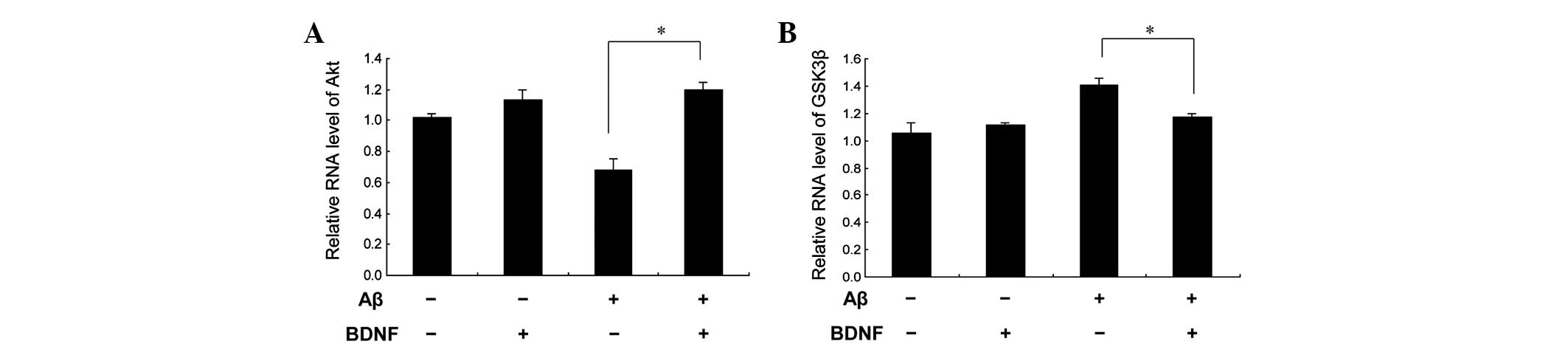
(20 μM). This was supported by the measurement of the Akt
mRNA levels, as shown in Fig.
6A.
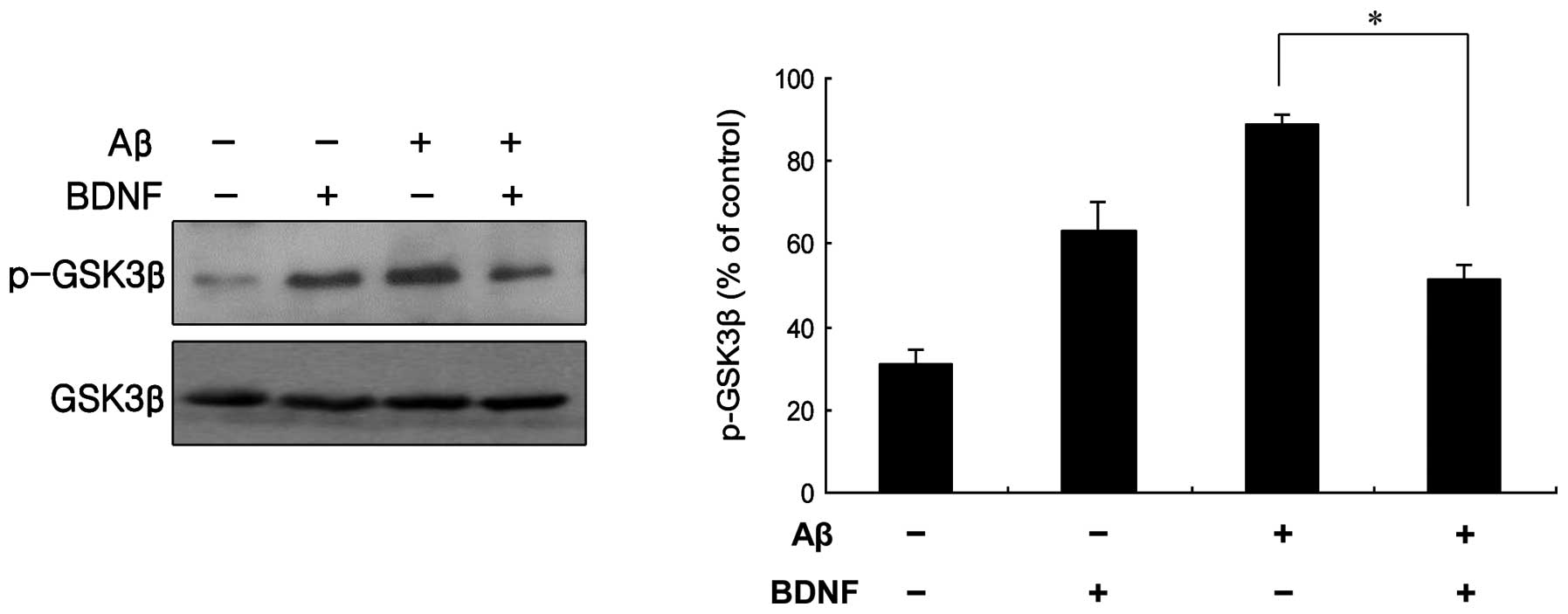
GSK3β modulation of BDNF-mediated
signaling
GSK3β is known to be inhibited by serine
phosphorylation mediated by Akt, which in turn is induced by growth
factor signaling (11). Treatment
of cells with a combination of Aβ25–35 (20 μM) and BDNF
(10 ng/ml) induced a significant decrease in the level of p-GSK3β,
compared with cells treated only with 20 μM Aβ25–35
(Fig. 7). The mRNA levels obtained
for GSK3β supported this finding (Fig. 6B). These data indicate that GSK3β
contributed to the neuroprotective effect of BDNF against the
neurotoxicity of Aβ25–35.
Discussion
Aβ is a 40–42 amino acid peptide fragment derived by
proteolysis from the integral membrane protein known as Aβ
precursor protein (14). Aβ, the
central constituent of senile plaques in AD, is known to exert
toxic effects on neurons (15).
Aβ25–35 is the shorter toxic fragment corresponding to
amino acids 25–35, which encompasses the β sheet of the full
protein (16). Therefore,
Aβ25–35 has been used to assess toxicity in the AD in
vitro models (10). However,
the mechanisms by which the Aβ peptide exerts its neurotoxic effect
are not yet understood.
BDNF levels have been observed to decrease in the
parietal cortex and hippocampus of patients with AD (17). BDNF serum concentrations have been
reported to correlate with the severity of dementia (18). Furthermore, in AD brains, neurons
containing neurofibrillary tangles do not contain
BDNF-immunoreactive material, whereas neurons that stain intensely
for BDNF are devoid of tangles (19).
In the present study, the protective effect of BDNF
against toxicity induced by Aβ25–35 in SH-SY5Y cells was
assessed. The results indicated that exposure of SH-SY5Y cells to
Aβ25–35 caused a decrease in viability and the cell
apoptosis rate increased, while the expression of p-Akt decreased
and p-GSK3β increased. In addition, p-Akt and p-GSK3β may be
associated with the Aβ25–35-induced cell apoptosis.
The PI3K/Akt pathway is important for cell survival.
PI3K enhances neuroprotection by regulating the level of
phosphorylation and activation of Akt. Akt activity can be
modulated by phosphorylation of either Thr308 or Ser473 (20). Phosphorylation of Thr308 has been
reported to be stimulated by dopamine receptor activation, while
phosphorylation of Ser473 is regulated by the activation of the
N-methyl-D-aspartate receptor (21–23).
Therefore, phosphorylation and activation of Akt may underlie the
observed protective effect of BDNF.
GSK3β is a substrate of Akt, which phosphorylates
and inhibits GSK3β. Furthermore, GSK3β is subject to inhibitory
regulation by growth factors that activate the PI3K/Akt pathway
(24). Beaulieu et al
(25) reported that activation of
the PI3K/Akt pathway can increase the phosphorylation of GSK3β,
thereby inhibiting the activity of the latter; thus, Akt may be a
regulator of GSK3β. The present study indicated that the
neuroprotective effect of BDNF against Aβ25–35 toxicity
was mediated by the inhibitory effect of Akt on GSK3β. In
conclusion, the data showed that administration of BDNF exerts
neuroprotective actions against the toxic effect of the
Aβ25–35-induced apoptosis in SH-SY5Y cells, which
involved PI3K/Akt activation and GSK3β phosphorylation. The
mechanism and signaling pathways underlying neuronal degeneration
induced by the Aβ peptide remain to be further elucidated.
Acknowledgements
This work was funded by a grant to Jin Hee Kim in
2013 from Korea University.
References
|
1
|
Mesulam MM: Neuroplasticity failure in
Alzheimer’s disease: bridging the gap between plaques and tangles.
Neuron. 24:521–529. 1999.
|
|
2
|
Dickson DW: Neuropathology of Alzheimer’s
disease and other dementias. Clin Geriatr Med. 17:209–228.
2001.
|
|
3
|
Song B, Davis K, Liu XS, Lee HG, Smith M
and Liu X: Inhibition of Polo-like kinase 1 reduces
beta-amyloid-induced neuronal cell death in Alzheimer’s disease.
Aging (Albany NY). 3:846–851. 2011.PubMed/NCBI
|
|
4
|
Selkoe DJ: Alzheimer’s disease is a
synaptic failure. Science. 298:789–791. 2002.
|
|
5
|
Yankner BA: Mechanisms of neuronal
degeneration in Alzheimer’s disease. Neuron. 16:921–932. 1996.
|
|
6
|
Götz J and Ittner LM: Animal models of
Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci.
9:532–544. 2008.
|
|
7
|
Poo MM: Neurotrophins as synaptic
modulators. Nat Rev Neurosci. 2:24–32. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lindvall O, Kokaia Z, Bengzon J, Elmér E
and Kokaia M: Neurotrophins and brain insults. Trends Neurosci.
17:490–496. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lewin GR and Barde YA: Physiology of the
neurotrophins. Annu Rev Neurosci. 19:289–317. 1996. View Article : Google Scholar
|
|
10
|
Cross DA, Alessi DR, Cohen P, Andjelkovich
M and Hemmings BA: Inhibition of glycogen synthase kinase-3 by
insulin mediated by protein kinase B. Nature. 378:785–789. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun L, Guo C, Liu D and Zhao Y, Zhang Y,
Song Z, Han H, Chen D and Zhao Y: Protective effects of bone
morphogenetic protein 7 against amyloid-beta induced neurotoxicity
in PC12 cells. Neuroscience. 184:151–163. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abràmoff MD, Magalhães PJ and Ram SJ:
Image processing with ImageJ. Biophotonics International. 11:36–42.
2004.
|
|
13
|
Zhao R, Zhang Z, Song Y, Wang D, Qi J and
Wen S: Implication of phosphatidylinositol-3 kinase/Akt/glycogen
synthase kinase-3β pathway in ginsenoside Rb1’s attenuation of
beta-amyloid-induced neurotoxicity and tau phosphorylation. J
Ethnopharmacol. 133:1109–1116. 2011.PubMed/NCBI
|
|
14
|
Amtul Z, Uhrig M and Beyreuther K:
Additive effects of fatty acid mixtures on the levels and ratio of
amyloid β40/42 peptides differ from the effects of individual fatty
acids. J Neurosci Res. 89:1795–1801. 2011.PubMed/NCBI
|
|
15
|
Hongpaisan J, Sun MK and Alkon DL: PKC ɛ
activation prevents synaptic loss, Aβ elevation, and cognitive
deficits in Alzheimer’s disease transgenic mice. J Neurosci.
31:630–643. 2011.
|
|
16
|
Kaminsky YG, Marlatt MW, Smith MA and
Kosenko EA: Subcellular and metabolic examination of amyloid-beta
peptides in Alzheimer disease pathogenesis: evidence for
Abeta(25–35). Exp Neurol. 221:26–37. 2010.PubMed/NCBI
|
|
17
|
Peng S, Wuu J, Mufson EJ and Fahnestock M:
Precursor form of brain-derived neurotrophic factor and mature
brain-derived neurotrophic factor are decreased in the pre-clinical
stages of Alzheimer’s disease. J Neurochem. 93:1412–1421.
2005.PubMed/NCBI
|
|
18
|
Laske C, Stransky E, Leyhe T, Eschweiler
GW, Maetzler W, Wittorf A, Soekadar S, Richartz E, Koehler N,
Bartels M, Buchkremer G and Schott K: BDNF serum and CSF
concentrations in Alzheimer’s disease, normal pressure
hydrocephalus and healthy controls. J Psychiatr Res. 41:387–394.
2007.
|
|
19
|
Murer MG, Boissiere F, Yan Q, Hunot S,
Villares J, Faucheux B, Agid Y, Hirsch E and Raisman-Vozari R: An
immunohistochemical study of the distribution of brain-derived
neurotrophic factor in the adult human brain, with particular
reference to Alzheimer’s disease. Neuroscience. 88:1015–1032.
1999.PubMed/NCBI
|
|
20
|
Koide H, Asai T, Furuya K, Tsuzuku T, Kato
H, Dewa T, Nango M, Maeda N and Oku N: Inhibition of Akt (ser473)
phosphorylation and rapamycin-resistant cell growth by knockdown of
mammalian target of rapamycin with small interfering RNA in
vascular endothelial growth factor receptor-1-targeting vector.
Biol Pharm Bull. 34:602–608. 2011. View Article : Google Scholar
|
|
21
|
Mannoury la Cour C, Salles MJ, Pasteau V
and Millan MJ: Signaling pathways leading to phosphorylation of Akt
and GSK-3β by activation of cloned human and rat cerebral
D2 and D3 receptors. Mol Pharmacol.
79:91–105. 2011.
|
|
22
|
Sánchez-Blázquez P, Rodríguez-Muñoz M and
Garzón J: Mu-opioid receptors transiently activate the Akt-nNOS
pathway to produce sustained potentiation of PKC-mediated
NMDAR-CaMKII signaling. PLoS One. 5:e112782010.PubMed/NCBI
|
|
23
|
Soriano FX, Papadia S, Hofmann F,
Hardingham NR, Bading H and Hardingham GE: Preconditioning doses of
NMDA promote neuroprotection by enhancing neuronal excitability. J
Neurosci. 26:4509–4518. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grimes CA and Jope RS: The multifaceted
roles of glycogen synthase kinase 3beta in cellular signaling. Prog
Neurobiol. 65:391–426. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Beaulieu JM, Gainetdinov RR and Caron MG:
Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev
Pharmacol Toxicol. 49:327–347. 2009. View Article : Google Scholar : PubMed/NCBI
|