Introduction
Glaucoma is a neurodegenerative disease in which
progressive retinal ganglion cells (RGCs) loss and irreversible
visual field defects occur. Elevated intraocular pressure (IOP) is
considered to be a major causative factor (1). However, in clinical practice, even
when the IOP has been well controlled, deterioration of the visual
function of a patient may continue to proceed in certain cases.
Currently, there are no effective treatments available for the
prevention of glaucoma.
Retinal ischemia/reperfusion (RIR) injury is a
common clinical condition that is the main cause of loss of vision
in humans (2). RIR occurs in a
variety of ocular pathologies, including acute glaucoma, diabetic
retinopathy and retinal vascular occlusion (2,3). An
animal model of RIR injury, which mimics the clinical situation of
acute glaucoma, is frequently used to study RGC loss or dysfunction
following ischemic injury (4).
However, the mechanism of RGC death in glaucoma is not fully
understood. Despite the efforts that have been made to understand
the pathological processes responsible for the insults to the
retina, it is currently not possible to prevent this
complication.
Autophagy is an evolutionarily conserved process
that leads to the degradation of long-lived proteins and recycling
of cellular constituents (5).
According to its mechanism and function, autophagy can be divided
into three pathways in all eukaryotic cells: macroautophagy,
microautophagy and chaperone-mediated autophagy (CMA) (6). Basal autophagy is crucial,
particularly in neurons. It occurs in various organs and assists in
maintaining homeostatic function during protein and organelle
turnover (6,7). Under various pathophysiological forms
of stress, autophagy can be induced to meet the needs of
developmentally related structural remodeling, and to clear toxic
or misfolded proteins, superfluous or damaged organelles and
invading microorganisms (8,9).
During autophagy, proteins and organelles are degraded and form
double- or multiple-membrane autophagic vesicles, the
autophagosomes, which subsequently fuse with the lysosomal
compartment to form autolysosomes (10). In some cells, the excessive
accumulation of autophagosomes and autophagolysosomes leads to cell
death, which is known as autophagic cell death or type II
programmed cell death (PCD) (7).
Autophagic death is activated in the pathophysiology in a variety
of disorders, particularly in neuronal cell death associated with
chronic neurodegenerative disorders and acute ischemia/hypoxia
neuronal injury (11). Despite
recent studies which have shown that autophagy is activated in RGCs
following optic nerve transection and glaucoma, autophagic
processes in RGC death remain poorly understood (12,13).
Although apoptosis is sometimes equated with PCD, it
has become evident that several non-apoptotic forms of PCD,
including autophagic cell death and cytoplasmic cell death exist
(14). A type of PCD, dubbed
paraptosis, that often exists in parallel with apoptosis was
originally identified in 2000 (15). Paraptosis is characterized by
extensive cytoplasmic vacuolization without nuclear fragmentation
that begins with progressive swelling of the endoplasmic reticulum
(ER) and/or mitochondria. Apoptotic morphology and DNA
fragmentation are absent. Paraptosis typically does not respond to
caspase inhibitors (z-VAD-fmk, boc-aspartate fluoromethylketone,
p35 and XIAP) or Bcl-XL; nor does it involve activation of
poly(ADP-ribose) polymerase (PARP) cleavage and chromatin
condensation (16). Paraptosis can
be triggered by the TNF receptor family member TAJ/TROY, the
apoptotic protein Bax, and human insulin-like growth factor I
receptor (17). However, due in
part to the lack of specific markers, paraptosis has not been
thoroughly investigated and may thus have been underestimated.
Paraptosis can take place in certain pathological conditions, such
as excitotoxicity, ischemia and neurodegeneration. In
neurodegenerative diseases, such as Huntington’s disease and
amyotrophic lateral sclerosis, not all neuronal cell death appears
to occur via apoptosis (18,19).
Paraptosis has begun to garner attention as a significant
contributor to ischemia-associated damage (12). There are, however, few reports on
retinal insult through mechanisms involving paraptosis (20).
The aim of this study was to investigate the
involvement of various cell death mechanisms in a model of RIR
injury induced by acute IOP increase. Changes of the features of
autophagy and paraptosis were investigated at various time points
following RIR injury.
Materials and methods
Experimental animals
A total of 48 adult male Sprague-Dawley rats
weighing 220–250 g were provided by the Experimental Animal Center
of the Medical School of Xi’an Jiaotong University (Xi’an, China).
They were housed in individually ventilated cages containing wood
shavings and maintained in a temperature-controlled environment
with free access to food and water and a 12-h light-dark cycle. All
experiments were performed in accordance with the Statement on the
Use of Animals of the Association for Research in Vision and
Ophthalmology, and were approved by the ethics committee of Xi’an
Jiaotong University (Xi’an, China).
Induction of ischemia and
reperfusion
Rats were deeply anesthetized by intraperitoneal
injection of 10% chloral hydrate (40 mg/kg). Topical analgesia was
achieved using 0.4% oxybuprocaine hydrochloride eye drops (Santen,
Osaka, Japan). Pupils were dilated with 1% tropicamide eye drops
(Santen). A 30-gauge infusion needle, connected to a pressure
device, was inserted into the anterior chamber of the right eye.
The IOP was elevated to 110 mmHg for 60 min. The cannulating needle
was removed after 60 min of retinal ischemic and the IOP was
normalized. For each animal, the left eye served as a non-ischemic
control. At 6, 12, 24 h, 3 days and 7 days after ischemia, rats
were sacrificed by an intraperitoneal overdose injection of chloral
hydrate. The right eyes were rapidly enucleated and cut through the
pars plana, anterior segment and the vitreous was removed. The
harvested retina was immersed in liquid nitrogen prior to analysis.
Of the 8 rats sacrificed following ischemia, the control and model
eyes of three rats were used for analysis by transmission electron
microscopy (TEM) and the eyes from the other five rats were used
for cryosections.
Immunohistochemical staining
For histological examination, the fixed retinas were
cut in 12-μm sections using a cryostat microtome. After
preincubating with 10% goat serum (Boster Biological Technology
Co., Ltd., Wuhan, China) for 30 min, sections were incubated
overnight at 4°C with a primary antibody against
microtubule-associated protein 1 light chain 3 (polyclonal;
anti-rat LC3; rabbit IgG, 1:500; MBL International Corporation, Des
Plaines, IL, USA), followed by incubation with fluorescently
labeled secondary antibody [fluorescein isothiocyanate (FITC)
anti-mouse IgG, 1:200; Beijing CoWin Biosciences Co., Ltd.,
Beijing, China] for 2 h at room temperature. The sections were
counterstained for 10 min with 0.1 mg/ml DAPI (Roche Applied
Science, Mannheim, Germany) and rinsed with phosphate-buffered
saline. Sections were observed under a fluorescence microscope
(Olympus BX51; Olympus Corporation, Tokyo, Japan). DAPI-positive
cell were counted at ×400 magnification and images were captured in
6 different areas per retinal with ~442 μm retinal length per area.
DAPI-positive cells were counted using Image Pro Plus 6.0 software
(Media Cybernetics, Inc. Rockville, MD, USA) and 4–6 samples per
group were used for this analysis.
TEM
Tissue samples were obtained from the three pairs of
control and model eyes and 10 fields of each eye were examined by
TEM (H-7650; Hitachi, Tokyo, Japan). Specimens were post-fixed with
4% glutaraldehyde in 0.1 mmol/l phosphate buffer (pH 7.4) for 2–4 h
and then with 1% osmium tetroxide in 0.1 mmol/l phosphate buffer
for 2 h. After dehydrating in applied acetone, the retinal sections
were embedded in pure acetone medium. Ultrathin sections (80 nm)
were sliced and stained with uranyl acetate and lead citrate (Ted
Pella, Inc., Redding, CA, USA). Photographs were taken with a
digital CCD camera (Olympus-SIS Veleta, Münster, Germany). Image
Pro Plus 6.0 (Media Cybernetics, Inc.) and Photoshop (Adobe, San
Jose, CA, USA) software were used to further quantify the TEM
images.
Statistical analysis
Statistical analysis was performed using SPSS
statistical software version 13.0 (SPSS, Inc., Chicago, IL, USA).
The results are expressed as mean ± standard deviation (SD).
One-way analysis of variance (ANOVA) with subsequent post hoc tests
was used to compare differences among groups. A P-value <0.05
was considered to indicate a statistically significant
difference.
Results
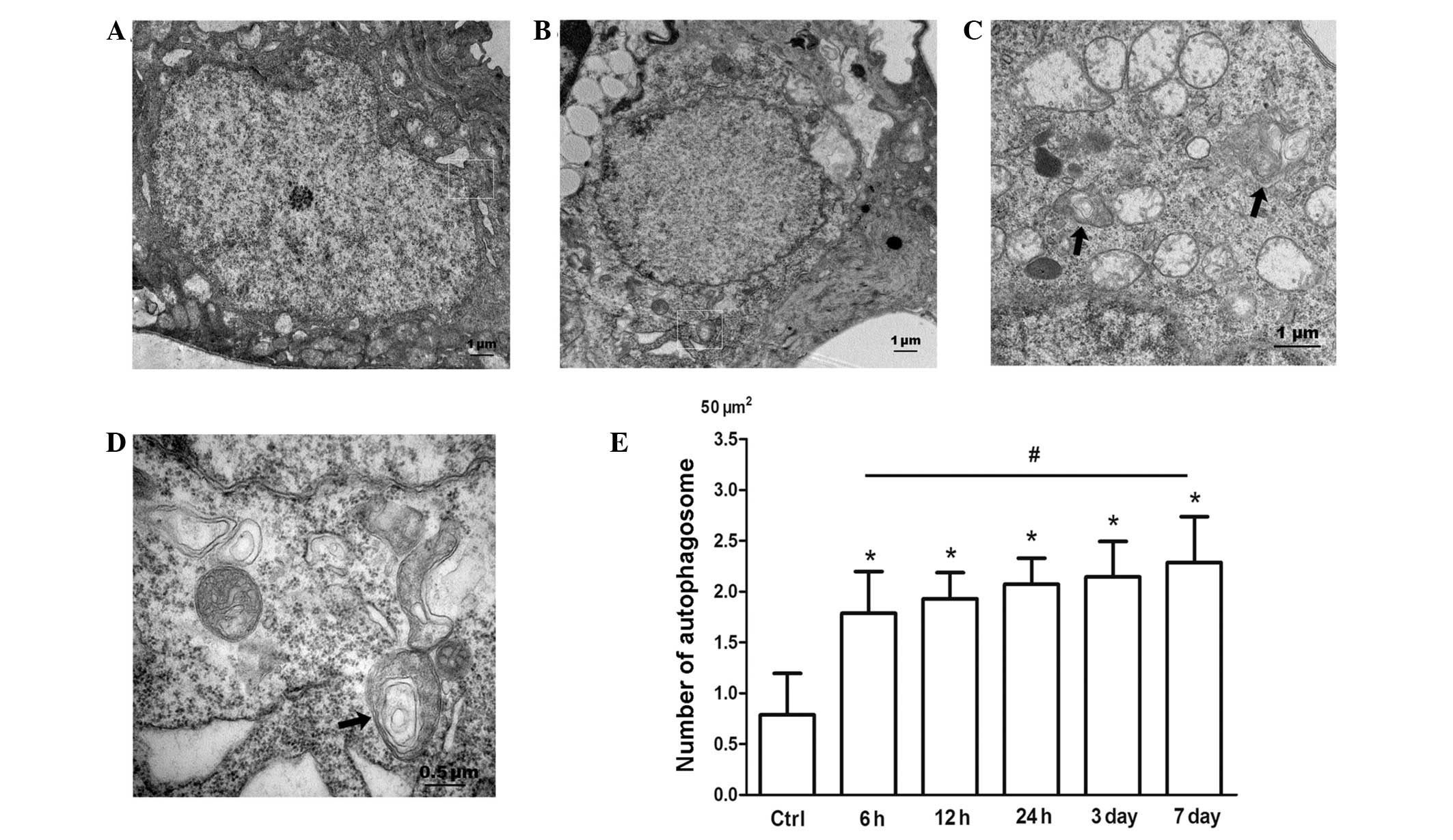
Ultrastructural features of autophagy
following RIR injury
Morphometric analysis by TEM cell imaging clearly
confirmed whether autophagic activity was altered in RGCs following
RIR at different time-points (6, 12, 24 h, 3 days and 7 days). The
TEM manifestation of double-membrane vesicles surrounding
cytoplasmic structures remains the gold standard for identifying
autophagosomes (8). Double- and
multiple-membrane vacuoles surrounding electron-dense and compacted
amorphous contents or whorls of membranous material are properties
of autophagosomes (8). Prior to
transient ischemia, autophagic events were occasionally observed in
the cytoplasm of RGCs in the ganglion cell layer (GCL; Fig. 1A). However, autophagosomes were
readily detectable in the cytoplasm of RGCs in the GCL 6 h after
RIR injury (Fig. 1C) and were
sustained throughout the experimental period. The average number of
autophagic vacuoles in the cytoplasm of RGCs was ~0.79/50
μm2 prior to IOP elevation. However, RIR insult markedly
increased the number of autophagic vacuoles in RGCs in the GCL
(Fig. 1B–D). The average number of
autophagosomes was a maximum at 7 days after reperfusion at
~2.29/50 μm2 (Fig. 1E).
Autophagic activities were exhibited as an autophagosome enclosing
a damaged mitochondrion, and an autophagic vacuole surrounding
partially degraded membranous material. These observations indicate
that stimulated autophagic flux in the cytoplasm of RGCs in the GCL
is involved in the pathogenesis of RIR injury.
Immunolocalization of LC3 following IOP
elevation
Using immunofluorescence against LC3, a specific
form of autophagic protein, the activation of autophagy in RGCs
following RIR was analyzed by comparison with the control group.
Relatively weak LC3 (green) staining was present in the GCL and
inner plexiform layer (IPL). The negative control for
immunofluorescence, in which the LC3 primary antibody was omitted,
showed no detectable staining. Cell nuclei were counterstained with
DAPI. LC3 immunoreactivity, which was observed as clusters of
small, intensely stained granules, significantly increased in the
GCL and IPL 6 h after the RIR insult. After 6 h of RIR injury, LC3
expression was enhanced in the cytoplasm of RGCs in the GCL, and
sustained throughout the experimental period. LC3 immunoreactivity
decreased in the IPL 3 days after reperfusion. DAPI-positive cells
in the GCL were counted under ×400 magnification. The average
number of DAPI-positive cells in the GCL was 26.2±1.92 cells in the
control, and 7 days after RIR insult, it was decreased to 16.4±1.67
cells. The reduction in the number of DAPI-positive cells indicated
that RIR injury led to extensive loss of neurons in the GCL. The
thickness of the IPL and inner nuclear layer (INL) at 7 days after
RIR injury also markedly decreased, reflecting the destruction of
inner retinal elements. These results indicate that IOP elevation
induced the activation of autophagy. In addition, the activation of
autophagy apparently increased in the cytoplasm of RGCs 7 days
after RIR insult, which coincided with the period of RGC death.
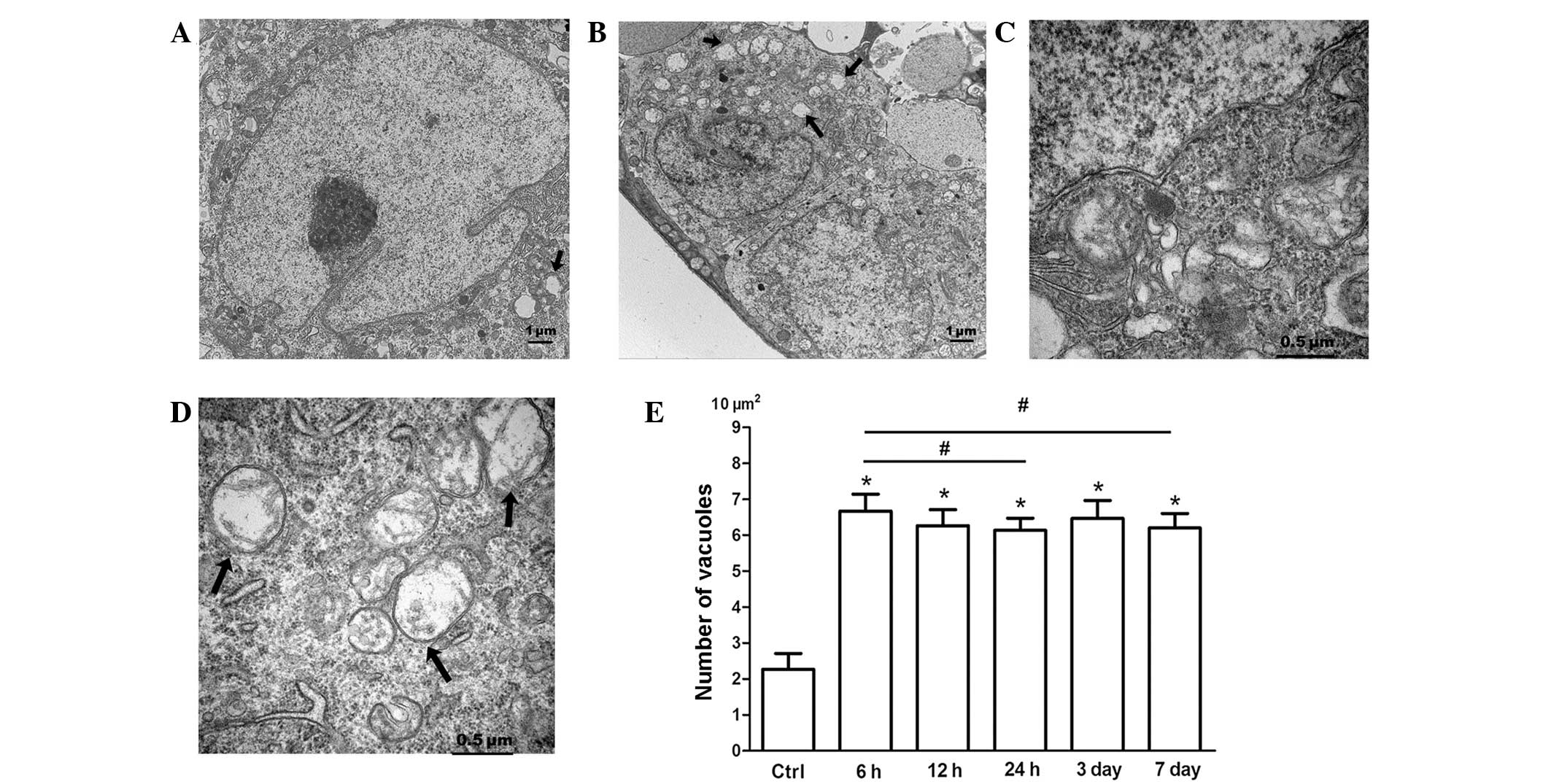
Ultra-structural features of
paraptosis-like cell death induced by RIR injury
Following RIR injury, extensive cytoplasmic
vacuolization in RGCs was significant at different end-points (6 h,
12 h, 24 h, 3 days and 7 days). In the control group, intracellular
vacuoles surrounding the cell nucleus were occasionally observed
(Fig. 2A). Cytoplasmic
vacuolization, in which the vacuoles appear clear with no
cytoplasmic material in the vacuoles, is a typical feature of
paraptosis (15). The present TEM
study verified that cytoplasmic vacuolation was evident at 6 h
after RIR injury; however, it was accompanied by the simultaneous
occurrence of a necrotic-like morphology, autophagy and apoptosis,
and therefore showed specific characteristics. When the reperfusion
time was increased, RGCs with cytoplasmic vacuolization were more
pronounced and extensive cytoplasmic vacuolation occurred at 6 h
(Fig. 2B). As illustrated in
Fig. 2D, RIR injury caused
progressive swelling of the ER and/or mitochondria of RGCs, and the
cristae of the mitochondria appeared diffuse. As shown in Fig. 2E, RIR injury significantly
increased the number of cytoplasmic vacuoles among the RGCs.
Cytoplasmic vesicles were occasionally observed in the RGCs
(Fig. 3A and C). The average
number of cytoplasmic vacuoles in the RGCs was ~2.27/10
mm2 prior to IOP elevation. However, following RIR
insult, the number of RGCs displaying cytoplasmic vesicles in the
GCL markedly increased. The peak average number of cytoplasmic
vesicles appeared at 6 h after IOP elevation, and was ~6.7/10
mm2 (Fig. 3E).
Increased numbers of cytosolic vesicles remained in the RGCs in the
GCL until 7 days after RIR injury. These observations correspond to
the pattern of paraptosis in which paraptotic cells are
characterized by physical enlargement of the mitochondria and ER
(15). Therefore, RIR injury
induced RGC death in part via paraptosis. The appearance of swollen
organelles may suggest disruption of intracellular homoeostasis due
to RIR injury.
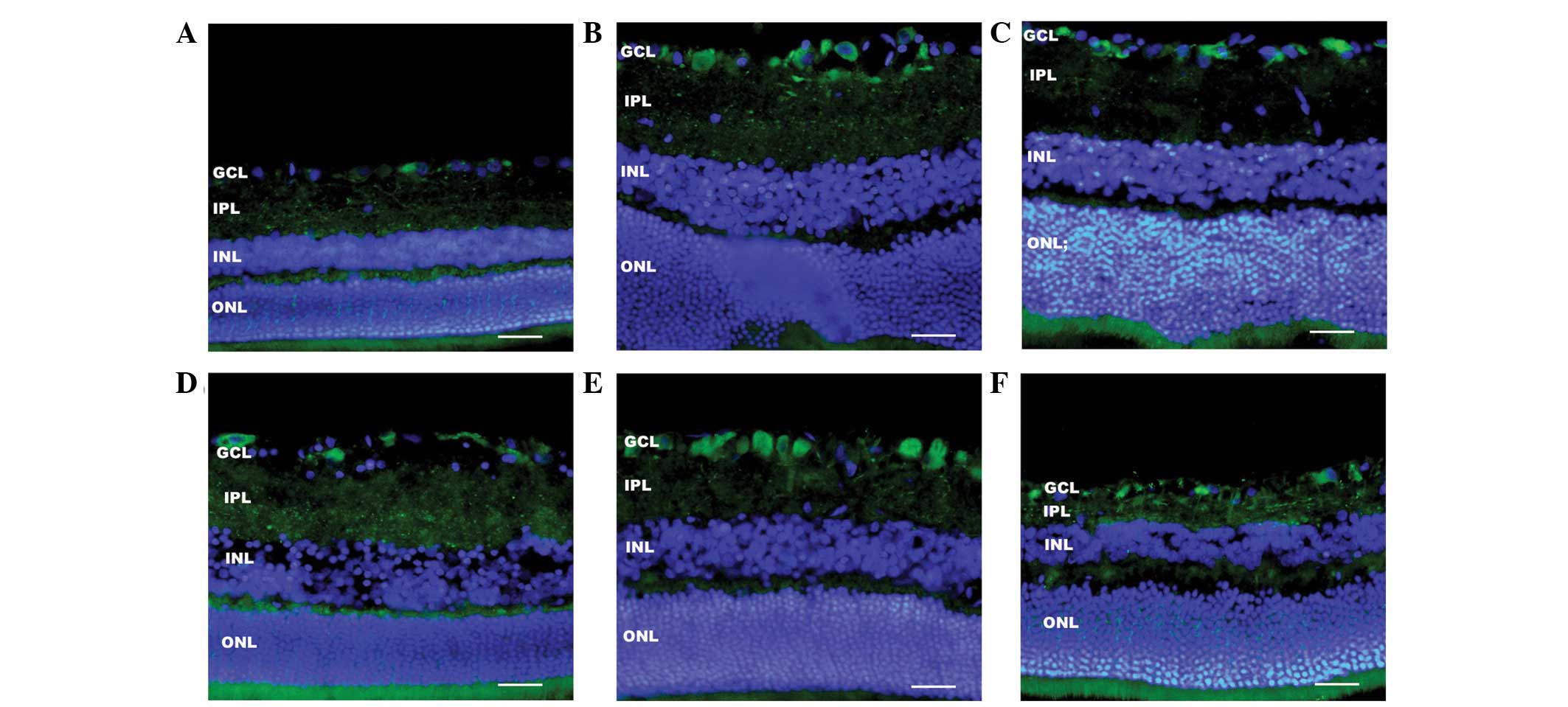 | Figure 2Immunofluorescence showed the time
course of LC3 (green) expression in all retina layers after RIR.
(A) Control shows relatively weak staining of LC3 in the IPL and
GCL. After RIR insult, LC3 immunoreactivity gradually increases in
the IPL, and more intense immunoreactivity is observed in the GCL
at (B) 6 h, (C) 12 h and (D) 24 h. (E) Three days after RIR, LC3
immunoreactivity decreases markedly in the IPL. (F) LC3 expression
is enhanced in the cytoplasm of RGCs in the GCL until 7 days after
RIR. Cell nuclei were counterstained with DAPI. The thickness of
IPL and INL at 7 days after RIR injury also markedly decreased.
Three eyes were used in each experimental period. Scale bars, 50
mm. RIR, retinal ischemia/reperfusion; IPL, inner plexiform layer;
GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer
nuclear layer. |
Discussion
Results from the present study indicate that the
activation of autophagy and paraptosis is associated with retinal
damage caused by RIR insult in an acute hypertensive glaucoma
model. In comparison to the control group, there was evidence that
autophagy markedly increased in the retina following RIR injury, as
demonstrated by changes in the immunohistological staining and
changes observed by TEM. Following RIR injury, the accumulation of
cytoplasmic vacuoles was observed in the RGCs, suggesting that
paraptosis, a non-apoptotic form of PCD, was associated with RIR
injury. The occurrence of paraptosis is further supported by the
observation of mitochondrial and ER swelling.
Glaucoma is a leading cause of blindness globally.
It is a neurodegenerative disease of the optic nerve that can
ultimately lead to irreversible damage to visual function (1,2). A
transient increase in the IOP threshold can trigger a cascade of
damage to the RGCs. Even when pathological IOP is lowered to the
normal range, neural damage in the retina may continue to proceed.
A number of studies demonstrate that all kinds of PCD, including
apoptosis and autophagy, play important roles in the glaucomatous
retina of glaucoma patients and mammalian models (12,14,20).
Methods aimed at improving the understanding of molecular
mechanisms within each PCD and blocking the PCD may be helpful for
neuronal survival and preservation of visual function. The use of
animal models of RIR injury induced by the transient elevation of
IOP, mimicking clinical situations of acute angle-closure glaucoma,
may increase the knowledge of the mechanisms underlying the retinal
neuronal death of glaucoma and thus may provide new insights into
the disease therapy.
The present study showed that in the animal model,
the involvement of autophagy in the neurodegenerative processes
accompanying ischemia in the GCL was sustained following acute IOP
elevation. Immunostaining analysis demonstrated that the expression
of LC3 in the GCL increased 6 h after IOP elevation. This preceded
significant RGC loss, and was maintained throughout the
experimental period. However, following a period of significant
RGCs loss at ~7 days after IOP induction, LC3 immunoreactivity in
the cytoplasm of RGCs in the GCL markedly increased. However, an
increase of LC3 expression does not appear to be specific for
autophagy. In addition to its important role in autophagy, LC3 has
been reported to be involved in non-autophagic cytoplasmic
vacuolization (21). Moreover,
previous studies have indicated a significant reduction in retinal
thickness at 7 days after RIR (12,13).
Consistent with these studies, the ocular hypertension model in the
present study also exhibited a marked reduction in retinal
thickness at 7 days after RIR injury, reflecting the destruction of
the inner retinal elements. Ultrastructural features of double- or
multiple-membrane acidic autophagic vesicles were observed by TEM
in the RGCs until 7 days after IOP elevation. This increase of
autophagy in the glaucomatous retina may act to recycle damaged
material and lead to cell death. Depending on the cellular milieu,
autophagy, as a lysosome-mediated self-degradation process of
eukaryotic cells, can either promote survival or act as an
alternative mechanism of PCD for neurons (6,22).
Autophagy, as a defense mechanism for the removal of toxic
multimeric complexes and aggregated proteins in neurodegenerative
diseases, may also occur for self-digestion and self-clearance and
the suppression of basal autophagy may cause neurodegeneration
(6,8). By contrast, autophagy can promote
cell death through excessive cellular autolysis, and by degrading
fundamental cellular constituents. Autophagic markers such as LC3
participate in various steps of cerebral focal ischemia following
the insult (23).
Paraptosis is a recently defined type of PCD and
little is known about its mechanism. There is a continuing effort
to identify paraptosis-specific changes, and only in the last few
years has the first proteomic analysis of this type of
non-apoptotic PCD been described (16). In the present study, evaluation by
TEM confirmed that RIR insult induced irregular cytoplasmic
vacuolization in RGCs. Further characterization of the cytoplasmic
vacuolization indicated that it was associated with ER and/or
mitochondria dilation along with preservation of nuclear chromatin.
These results indicate that the cytoplasmic vacuolization induced
by RIR insult might be associated with paraptosis, and is
suggestive of paraptotic cell death. Swelling of the ER and
mitochondria has been demonstrated to be associated with a
disruption of intracellular homoeostasis (24). Furthermore, in a previous study it
was found that autophagy inhibitors did not prevent but, on the
contrary, enhanced the formation of cytoplasmic vacuolization
(25). Paraptosis occurs during
cell differentiation as the nervous system develops, as well as in
many neurodegenerative diseases (16). The association between paraptosis
and neurodegeneration has been demonstrated by several studies
highlighting the fact that neurons are vulnerable to stress-related
signals involved in the paraptosis process (15,26).
Further studies of paraptosis are required to elucidate the
mechanism of PCD and may facilitate the identification of future
therapeutic approaches for ischemia. Therapies for the induction of
non-apoptotic forms of PCD such as paraptosis might suppress the
multi-drug resistant phenotype often concerned with resistance to
apoptosis (27). Thus, methods of
targeting PCD may be beneficial as alternative to apoptosis-based
therapeutics.
The results of the present study indicated that
paraptosis and autophagy occurred simultaneously in the
RIR-insulted RGCs. All three types of PCD, namely paraptosis,
autophagy and apoptosis, can be induced by RIR insult in RGCs. In a
previous study, Kim and Park found that acute IOP elevation was
associated with a variety of changes in cell death and cell
survival pathways in RGCs (28).
Although paraptosis and autophagy were induced in RIR-insulted
RGCs, they may not have contributed to cell death; they may have
served as a mechanism of cell survival. The preliminary
observations in this study focus on the high incidence of autophagy
and paraptosis in the GCL where RGCs die by this nonapoptotic
pathway. The roles of individual types of cell death and the
coexistence of multiple cell death types (paraptosis, autophagy and
apoptosis) have been reported in recent studies concerning ischemic
injury, neurodegeneration and viral infection (25,27,29,30).
For the identification of the type of cell death, and even for the
quantification of certain processes, EM remains one of the most
accurate methods; for example, paraptosis was first discovered by
TEM (15). In addition, EM is able
to demonstrate the basic characteristics of cell death and
unexpected associations of subcellular features in the same cell,
such as apoptotic, paraptotic and autoschizic changes, which may
suggest a kind of cell reprogramming. Ultrastructural features of
RGCs that die in the retina are useful in making a clear
morphological comparison between different activated PCD programs
in a variety of clinical statuses, not only for diagnosis and
prognosis, but also to establish appropriate therapeutic
interventions. The identification of biochemical markers of PCD and
other methodological improvements should increase our understanding
of the multiple roles of cell death programs in neuronal fate.
In summary, the results of the present study confirm
previous reports that apoptosis produces retinal cell death after
RIR injury, and also demonstrate that dysregulated autophagy and
paraptosis may participate in the death of RGCs under transiently
elevated IOP. An emerging consensus is that autophagy and
paraptosis have dual roles, acting as a prosurvival mechanism or as
a process of progressive deterioration leading to cell death.
Numerous microenvironmental factors may induce and/or activate one
or more biochemical pathways of autophagy and paraptosis. These
molecular events can be visualized by TEM at the cellular level,
and the type of death can be characterized morphologically.
Therefore, these observations enhance our understanding of the
mechanism of non-apoptotic cell death in the retina following RIR
injury. Targeting autophagy and paraptosis, either by inhibition or
by enhancement, could represent a potential approach for new and
supporting therapeutic interventions for diseases of the nervous
system, in retinal ischemia as well as for other neurodegenerative
diseases.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (NSFC; no. 30772373) and the Science
and Technology Development Project of Shaanxi Province [no.
2012K16-11(04)]. The authors thank Mingxia Chen (Room of Electron
Microscopy, Medical School, Xi’an Jiaotong University) for
excellent technical assistance in these studies.
References
|
1
|
Quigley HA: Glaucoma. Lancet.
377:1367–1377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Osborne NN, Casson RJ, Wood JP, Chidlow G,
Graham M and Melena J: Retinal ischemia: mechanisms of damage and
potential therapeutic strategies. Prog Retin Eye Res. 23:91–147.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kaur C, Foulds WS and Ling EA:
Hypoxia-ischemia and retinal ganglion cell damage. Clin Ophthalmol.
2:879–889. 2008. View Article : Google Scholar
|
|
4
|
Sun MH, Pang JH, Chen SL, et al: Retinal
protection from acute glaucoma-induced ischemia-reperfusion injury
through pharmacologic induction of heme oxygenase-1. Invest
Ophthalmol Vis Sci. 51:4798–4808. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hotchkiss RS, Strasser A, McDunn JE and
Swanson PE: Cell death. N Engl J Med. 361:1570–1583. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hara T, Nakamura K, Matsui M, et al:
Suppression of basal autophagy in neural cells causes
neurodegenerative disease in mice. Nature. 441:885–889. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fimia GM, Stoykova A, Romagnoli A, et al:
Ambra1 regulates autophagy and development of the nervous system.
Nature. 447:1121–1125. 2007.PubMed/NCBI
|
|
10
|
Nijholt DA, de Graaf TR, van Haastert ES,
et al: Endoplasmic reticulum stress activates autophagy but not the
proteasome in neuronal cells: implications for Alzheimer’s disease.
Cell Death Differ. 18:1071–1081. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wong E and Cuervo AM: Autophagy gone awry
in neurodegenerative diseases. Nat Neurosci. 13:805–811. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Piras A, Gianetto D, Conte D, Bosone A and
Vercelli A: Activation of autophagy in a rat model of retinal
ischemia following high intraocular pressure. PLoS One.
6:e225142011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park HY, Kim JH and Park CK: Activation of
autophagy induces retinal ganglion cell death in a chronic
hypertensive glaucoma model. Cell Death Dis. 3:e2902012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sperandio S, Poksay KS, Schilling B,
Crippen D, Gibson BW and Bredesen DE: Identification of new
modulators and protein alterations in non-apoptotic programmed cell
death. J Cell Biochem. 111:1401–1412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sperandio S, de Belle I and Bredesen DE:
An alternative, nonapoptotic form of programmed cell death. Proc
Natl Acad Sci USA. 97:14376–14381. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Broker LE, Kruyt FA and Giaccone G: Cell
death independent of caspases: a review. Clin Cancer Res.
11:3155–3162. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Y, Li X, Wang L, Ding P, Zhang Y, Han
W and Ma D: An alternative form of paraptosis-like cell death,
triggered by TAJ/TROY and enhanced by PDCD5 overexpression. J Cell
Sci. 117:1525–1532. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Turmaine M, Raza A, Mahal A, Mangiarini L,
Bates GP and Davies SW: Nonapoptotic neurodegeneration in a
transgenic mouse model of Huntington’s disease. Proc Natl Acad Sci
USA. 97:8093–8097. 2000. View Article : Google Scholar
|
|
19
|
Fombonne J, Padron L, Enjalbert A, Krantic
S and Torriglia A: A novel paraptosis pathway involving
LEI/L-DNaseII for EGF-induced cell death in somato-lactotrope
pituitary cells. Apoptosis. 11:367–375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Valamanesh F, Torriglia A, Savoldelli M,
Gandolphe C, Jeanny JC, BenEzra D and Behar-Cohen F:
Glucocorticoids induce retinal toxicity through mechanisms mainly
associated with paraptosis. Mol Vis. 13:1746–1757. 2007.PubMed/NCBI
|
|
21
|
Karl R, Singha PK, Venkatachalam MA and
Saikumar P: A novel role for MAP1 LC3 in nonautophagic cytoplasmic
vacuolation death of cancer cells. Oncogene. 28:2556–2568. 2009.
View Article : Google Scholar
|
|
22
|
Xie BS, Zhao HC, Yao SK, et al: Autophagy
inhibition enhances etoposide-induced cell death in human hepatoma
G2 cells. Int J Mol Med. 27:599–606. 2011.PubMed/NCBI
|
|
23
|
Rami A, Langhagen A and Steiger S: Focal
cerebral ischemia induces upregulation of Beclin 1 and
autophagy-like cell death. Neurobiol Dis. 29:132–141. 2008.
View Article : Google Scholar
|
|
24
|
Hoa N, Myers M, Douglass T, et al:
Molecular mechanisms of paraptosis induction: implications for a
non-genetically modified tumor vaccine. PLoS One. 4:e46312009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang WB, Feng LX, Yue QX, et al:
Paraptosis accompanied by autophagy and apoptosis was induced by
celastrol, a natural compound with influence on proteasome, ER
stress and Hsp90. J Cell Physiol. 227:2196–2206. 2012. View Article : Google Scholar
|
|
26
|
Pehar M, O’Riordan KJ, Burns-Cusato M, et
al: Altered longevity-assurance activity of p53:p44 in the mouse
causes memory loss, neurodegeneration and premature death. Aging
Cell. 9:174–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tardito S, Isella C, Medico E, et al: The
thioxotriazole copper(II) complex A0 induces endoplasmic reticulum
stress and paraptotic death in human cancer cells. J Biol Chem.
284:24306–24319. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim HS and Park CK: Retinal ganglion cell
death is delayed by activation of retinal intrinsic cell survival
program. Brain Res. 1057:17–28. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Danaila L, Popescu I, Pais V, et al:
Apoptosis, paraptosis, necrosis, and cell regeneration in
posttraumatic cerebral arteries. Chirurgia (Bucur). 108:319–324.
2013.
|
|
30
|
Pais V, Danaila L and Pais E:
Ultrastructural patterns of the activated cell death programs in
the human brain. Ultrastruct Pathol. 37:110–120. 2013. View Article : Google Scholar : PubMed/NCBI
|