Introduction
Colorectal cancer (CRC) is a malignant tumor that is
a major threat to human life due to its high morbidity and
mortality rates. The incidence of CRC ranks third among other
cancer types, while the mortality rate of CRC ranks fourth
worldwide (1). Similar to the
majority of cancer types, therapeutic regimens for CRC include
traditional therapy, such as aggressive surgery, chemotherapy and
radiotherapy. However, surgery itself damages the health of a
patient and reduces their resistance to other diseases. Thus, novel
therapeutic interventions, particularly biological agents,
molecular targeted therapy and gene therapy, have been studied for
their ability to treat aspects of CRC that traditional therapy is
unable to overcome, including tumor apoptosis-resistance and
recurrence (2).
Gene therapy offers a promising strategy for
patients who are resistant to traditional therapies, due to its
advantage of selectively correcting or eradicating defective
tissues and targeting defects in malignant cells (3). The most important issues concerning
gene therapy for cancer treatment include the efficiency of
transfection and the reliability of expression. Adenovirus vectors
have attracted increasing attention in human cancer gene therapy
due to their preferential replication in tumor cells (4). An increasing number of clinical
trials on oncolytic adenoviruses have been conducted in the last
two decades (5). However,
non-replicating adenoviruses have little effect in eradicating
tumor cells. To overcome such a limitation, the replication
competents of adenoviruses that replicate specifically in tumor
cells and release virus progeny to further infect and destroy
neighboring cancer cells have been developed (6).
Apoptin, a protein derived from chicken anemia
virus, has received significant attention as a selective killer of
cancer cells (7). Gene expression
differences between normal and tumor cells may account for the
sensitivity of tumor cells to apoptin. Apoptin is located
predominantly in the nucleus of tumor cells, whereas in normal
cells, the protein is expressed in the cytoplasm (8). Apoptin expression induces apoptosis
in human tumor and transformed cells; however, there has been shown
to be little or no cytotoxic effect in a number of normal human
cell lines derived from different tissues (9). The combination of apoptin expression
with adenovirus-based delivery was selected for cancer therapy due
to both molecules having diverse, multiple, yet partly overlapping
targets in the cell. Previous studies have indicated that
interference with survivin expression facilitates apoptin function.
Human telomerase reverse transcriptase (hTERT) is a catalytic
subunit of human telomerase that is expressed at a substantially
higher level in tumor cells than in normal cells. Telomerase
activity influences hTERT expression (10). Thus, hTERT may be a good tumor
marker due to the high telomerase activity in ~90% of cancer cells
(11). In addition, the hTERT
promoter has been used for the tumor-specific expression of
transgenes.
In a previous study, an oncolytic adenovirus was
combined with the hTERT promoter and the apoptin gene, which
functioned as a cancer cell selective apoptosis-inducing gene
(12). The oncolytic adenovirus
has the ability to inhibit tumor-specific growth and tumor-specific
replication (12,13). The present study aimed to determine
whether the recombinant Ad-Apoptin-hTERT-E1a vector was able to
target CRC cells and induce apoptosis selectively in vitro
and in vivo.
Materials and methods
Cell lines, animals and viruses
A human CRC cell line (SW1116), mouse CRC cell line
(CT26; syngeneic to C57BL/6 mice) and human gastric epithelium cell
line (GES) were obtained from the Type Culture Collection of the
Chinese Academy of Sciences (Shanghai, China). The cells were
cultured in Dulbecco’s modified Eagle’s medium (Invitrogen Life
Technologies, Beijing, China), which was supplemented with 10%
heat-inactivated fetal bovine serum (Hyclone Biochemical Product
Co. Ltd., Beijing, China), 100 U/ml penicillin and 100 μg/ml
streptomycin. The cell lines were passaged for no more than six
months following receipt and were subcultured every 48–72 h. A
total of 50 female BALB/c mice (age, 6–8 weeks) were purchased from
the Experimental Animal Center of the Academy of Military Medical
Sciences (Beijing, China) and housed in a pathogen-free facility
for all the experiments, following institutional guidelines. The
construction and characterization of the dual cancer-specific
oncolytic adenovirus, Ad-Apoptin-hTERT-E1a, and the control viruses
(Ad-mock, Ad-apoptin and Ad-hTERT-E1a) used in the study have been
described previously (12).
Cell viability assay
Cell viability was determined using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT;
Sigma-Aldrich, St. Louis, MO, USA) assay, as described previously
(13). Briefly, the cells were
seeded in a 96-well plate at a density of 5×103/ml, and
incubated at 37°C overnight in a humidified environment of 95% air
and 5% CO2. Cells were infected with disparate
concentrations [1, 10 and 100 multiplicity of infection (MOI)] of
the recombinant adenoviruses for 12 h. Next, 20 μl MTT (5 mg/ml)
was added to each well and incubation was continued at 37°C for 4
h. The culture medium was aspirated and 150 μl dimethylsulfoxide
was added to dissolve the insoluble purple formazan product into a
colored solution; absorbance was subsequently measured at 490 nm.
Thereafter, the absorbance of each well was determined using an
automated plate reader (Spectramax 190; Molecular Devices,
Sunnyville, CA, USA). The percentage of viable cells was calculated
using the background-corrected absorbance as follows: 100 ×
[(control cells - experimental cells)/control cells]. Cell
viability was measured every 12 h over a four-day period. Untreated
cells were used as controls.
Acridine orange and ethidium bromide
(AO/EB) staining
Morphological observations of apoptosis were
detected by AO/EB staining using a fluorescence microscope (VANOX
BX51; Olympus Corporation, Tokyo, Japan). Briefly, the cells were
seeded in six-well plates at a density of 1×106 cells
per culture well, and cultured for 24 h at 37°C with 5%
CO2. The cells were infected with the various
recombinant adenoviruses (100 MOI) and incubated for 48 h. The
treated cells were harvested and washed three times in
phosphate-buffered saline (PBS). A 250-μl aliquot was added to a
microcentrifuge tube and stained with 4 μl AO-EB (Sigma, St. Louis,
MO, USA). Subsequently, 20-μl samples were placed on a microscopic
slide and images were collected utilizing the fluorescence
microscope. The images were obtained and analyzed using an
Image-Pro Plus (version 5.0.2) software program (Media Cybernetics,
Inc., Rockville, MD, USA). Untreated cells were used as
controls.
Annexin V/propidium iodide (PI) staining
analysis
Annexin V-fluorescein isothiocyanate (FITC)/PI (BD
Biosciences, Franklin Lakes, NJ, USA) staining was performed to
detect the apoptotic cells by assaying translocated
phosphatidylserine (14). In
brief, the cells were incubated in six-well plates at a density of
1×106 cells per culture well for 24 h. The cells were
cultured for 48 h following infection with the various recombinant
adenoviruses (100 MOI). The cells were harvested, washed once with
PBS and resuspended in binding buffer. The cells were subsequently
stained with FITC-labeled annexin V (Annexin V-FITC Apoptosis
Detection kit; BioVision, Inc., Mountain View, CA, USA), according
to the manufacturer’s instructions, with simultaneous dye exclusion
of PI. The samples were analyzed by flow cytometry (FACScan; BD
Biosciences). Untreated cells were used as controls.
Measurement of the mitochondrial membrane
potential (MMP)
The laser dye, rhodamine 123 (Rho123;
Sigma-Aldrich), was used to detect the MMP. Briefly, cells
(1×106) were cultured for 48 h following infection with
the various recombinant adenoviruses (100 MOI). The treated cells
were trypsinized and centrifuged at 1,000 × g at 4°C for 5 min.
Rho123 (10 μl) was added to the samples, which were subsequently
incubated at 37°C for 30 min. Thereafter, the samples were washed
with PBS three times. The MMP was quantified by flow cytometry
(FACScan), and untreated cells were used as controls.
Reactive oxygen species (ROS) assay
To quantify the intracellular level of ROS,
2′,7′-dichlorfluorescein-diacetate (DCFH-DA; Sigma) was used.
Briefly, cells (1×106) were cultured for 48 h following
infection with the various recombinant adenoviruses (100 MOI). The
treated cells were trypsinized and centrifuged at 1,000 × g at 4°C
for 5 min. ROS levels were determined by treating the cells with 10
μmol/l DCFH-DA at 37°C for 30 min. Data were quantified by flow
cytometric analysis (FACScan). Untreated cells were used as
controls.
Caspase analysis
Caspase Activity Assay kits (Beyotime Institute of
Biotechnology, Haimen, China) were used to detect the activity
levels of caspase-3, 6 and 7 in the treated SW1116 and GES cells.
In brief, the cells were infected with the recombinant adenoviruses
at 100 MOI for 48 h, trypsinized and washed once with PBS. The
cells (1×106) were resuspended in lysis buffer and the
total proteins were extracted. The activity levels of caspase-3, 6
and 7 were then analyzed according to the manufacturer’s
instructions. The untreated HepG-2 or L02 cells were used as
controls.
Cell fractionation and cytochrome c
analysis
Cytoplasmic and mitochondrial fractions were
separated, and the cytochrome c levels were detected by
western blotting. Briefly, the cells (1×106) were
infected with the recombinant adenoviruses at 100 MOI for 48 h. The
treated cells were then resuspended in lysis buffer [10 mM Tris-HCl
(pH 7.8), 1% Nonidet P-40, 10 mM β-mercaptoethanol, 0.5 mM
phenylmethylsulfonyl fluoride, 1 mg/ml aprotinin and 1 mg/ml
leupeptin] and sheared by passing through a 22-gauge needle. The
nuclear fraction was removed by centrifugation at 600 × g for 5
min, and the ‘low-speed’ supernatant was centrifuged at 10,000 × g
for 30 min to obtain the mitochondrial fraction (pellet) and the
cytosolic fraction (supernatant). The mitochondrial fraction was
further lysed in buffer [10 mM Tris (pH 7.4), 150 mM NaCl, 1%
Triton X-100 and 5 mM EDTA (pH 8.0)]. The proteins of the extracted
samples were separated by SDS-PAGE and transferred onto Hybond-C
membranes (GE Healthcare, Pittsburgh, PA, USA). The blots were
incubated with rabbit anti-cyto c polyclonal antibody (1:1,000;
#4272; Cell Signaling Technology, Inc., Danvers, MA, USA) for 2 h,
followed by incubation for 2 h with a horseradish peroxidase
labeled goat anti-rabbit IgG secondary antibody (1:1,000; #7074;
Cell Signaling Technology, Inc.) labeled with horseradish
peroxidase. Signals were visualized using an enhanced
chemiluminescence western blotting substrate kit (Pierce
Biotechnology, Inc., Rockford, IL, USA).
Animal experiments
Two methods were used to induce tumors in the mice.
In the first model, 1×106 CT26 cells were implanted
subcutaneously into the right flank of the BALB/c mice. The
tumor-burdened mice were randomly assigned into five groups (five
mice per group) following one week of tumor growth. The mice in the
first model were treated with the various recombinant adenoviruses,
via intratumoral injection at a dose of 1×109
plaque-forming units, in 50 μl saline. The control group received
50 μl saline alone. The injections were administered every two days
for the first week (days 6, 8 and 10 following implantation) and
once per week for two further weeks (days 17 and 21 following
implantation) (15). Tumor size
was assessed by caliper measurements of two perpendicular diameters
of the implant twice a week. Tumor volume (in cm3) was
estimated using the following formula: 1/2a × b2, where
‘a’ is the long diameter and ‘b’ is the short diameter (in cm). The
tumor doubling time (TDT) and the tumor growth delay (TGD) were
evaluated at the end of the experiment (16). In the second model, CT26
(1×106) cells were injected into the mice via the tail
vein to represent a pulmonary metastasis model. The tumor-burdened
mice were randomly assigned into five groups (five mice per group)
following one week of tumor growth. The mice were treated
intravenously according to the injection protocol of the first
model. The animal experiments were conducted in the animal facility
of the Institute of Military Veterinary Medicine at the Academy of
Military Medical Sciences (Changchun, China), in accordance with
governmental and institutional guidelines.
Statistical analysis
The statistical significance of differences was
assessed using one-way analysis of variance, where P<0.05 was
considered to indicate a statistically significant difference.
Log-rank tests were performed for survival analysis, and data from
all the animals were represented in Kaplan-Meier survival plots.
All statistical tests were performed using GraphPad Prism 5.0
software (GraphPad, San Diego, CA, USA).
Results
Recombinant adenoviruses inhibit the
growth of CRC cells
An MTT assay was used to measure the viability of
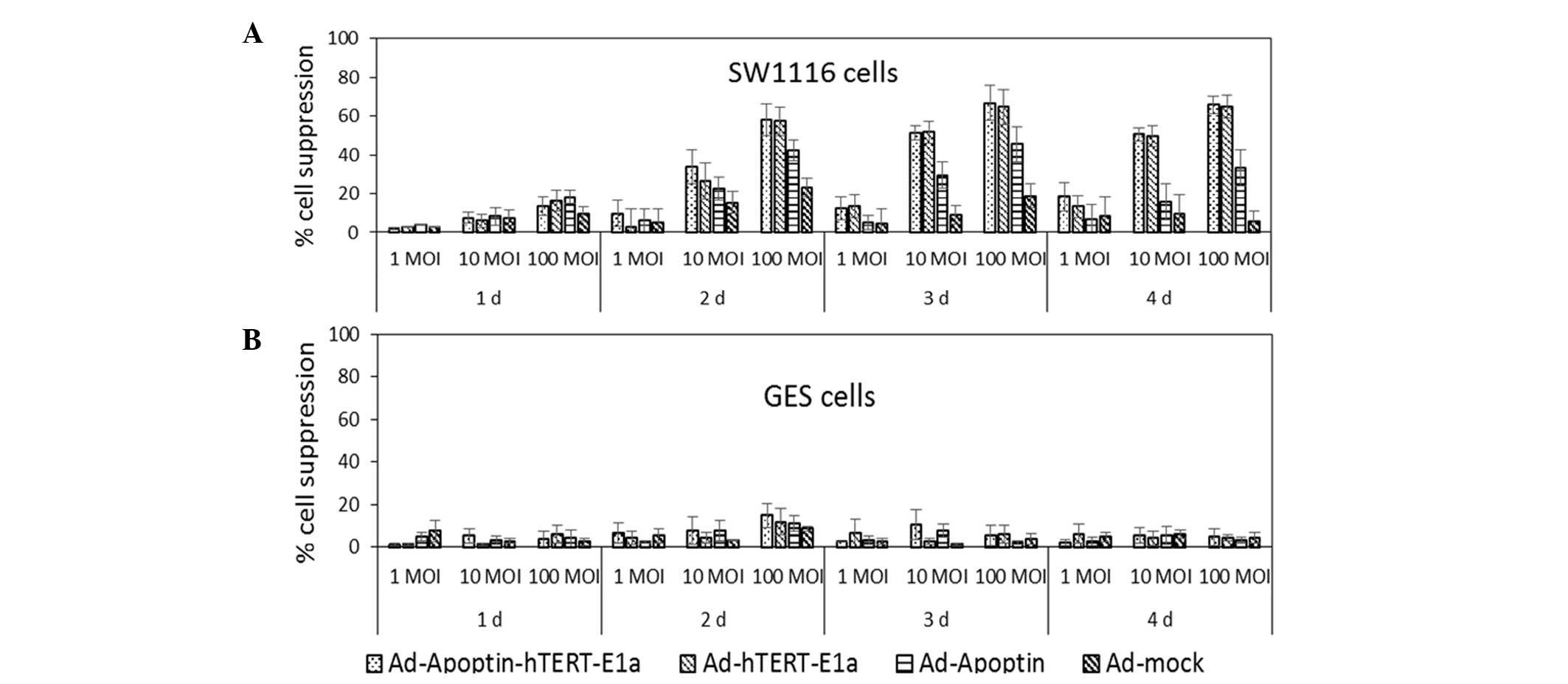
the cells infected with the various adenoviruses (17). As shown in Fig. 1A, in the early stages of infection,
the adenovirus did not cause significant inhibition of the CRC
cells. However, as the infection time extended,
Ad-Apoptin-hTERT-E1a, Ad-hTERT-E1a and Ad-Apoptin were shown to
inhibit SW1116 tumor cells, with the level of inhibition increasing
with an increased infection dose. In the SW116 cells infected with
Ad-mock, after two days, the infective doses of 1, 10 and 100 MOI
induced cell growth inhibition of 0–5, 15–20 and 20–25%,
respectively. The Ad-mock treated group demonstrated a gradual
attenuation of inhibition. Although cells infected with Ad-Apoptin
induced a higher level of inhibition compared with the
Ad-mock-treated group, the level of inhibition decreased after
three days due to the absence of the replication gene. By contrast,
cells treated with the replication-competent adenoviruses
(Ad-Apoptin-hTERT-E1a and Ad-hTERT-E1a) showed significant
suppression of cell growth, which correlated with the infection
doses. When infected with 1, 10 and 100 MOI Ad-Apoptin-hTERT-E1a or
Ad-hTERT-E1a, after two days, the cell growth was inhibited by
5–10, 30–35 and 55–60%, respectively. Inhibition in the
Ad-Apoptin-hTERT-E1a and Ad-hTERT-E1a groups peaked at a MOI of
100, with the suppression rates between 70 and 75% after three
days. The inhibition induced by Ad-Apoptin-hTERT-E1a was slightly
higher compared with that of Ad-hTERT-E1a. In addition, the cell
growth suppression observed following infection with
Ad-Apoptin-hTERT-E1a or Ad-hTERT-E1a at 10 MOI was similar to that
following infection with 100 MOI Ad-Apoptin. Inhibition by the
combined replication-competent adenoviruses decreased a little
after four days. However, in the normal GES cells (Fig. 1B), regardless of the recombinant
adenovirus infection time and infection dose, cells treated with
the recombinant adenoviruses did not show a marked inhibitory
effect. Thus, the adenoviruses with the dual cancer-specific genes
were more effective compared with the normal
replication-incompetent adenoviruses in inhibiting cancer cell
growth. The interaction of infection time and MOI was complex and
synergistic, and cell viability revealed a non-rigorous dependent
association with the two factors. Therefore, the in vitro
experiments were performed 48 h following infection at a MOI of
100.
Recombinant adenoviruses induce the
apoptosis of CRC cells
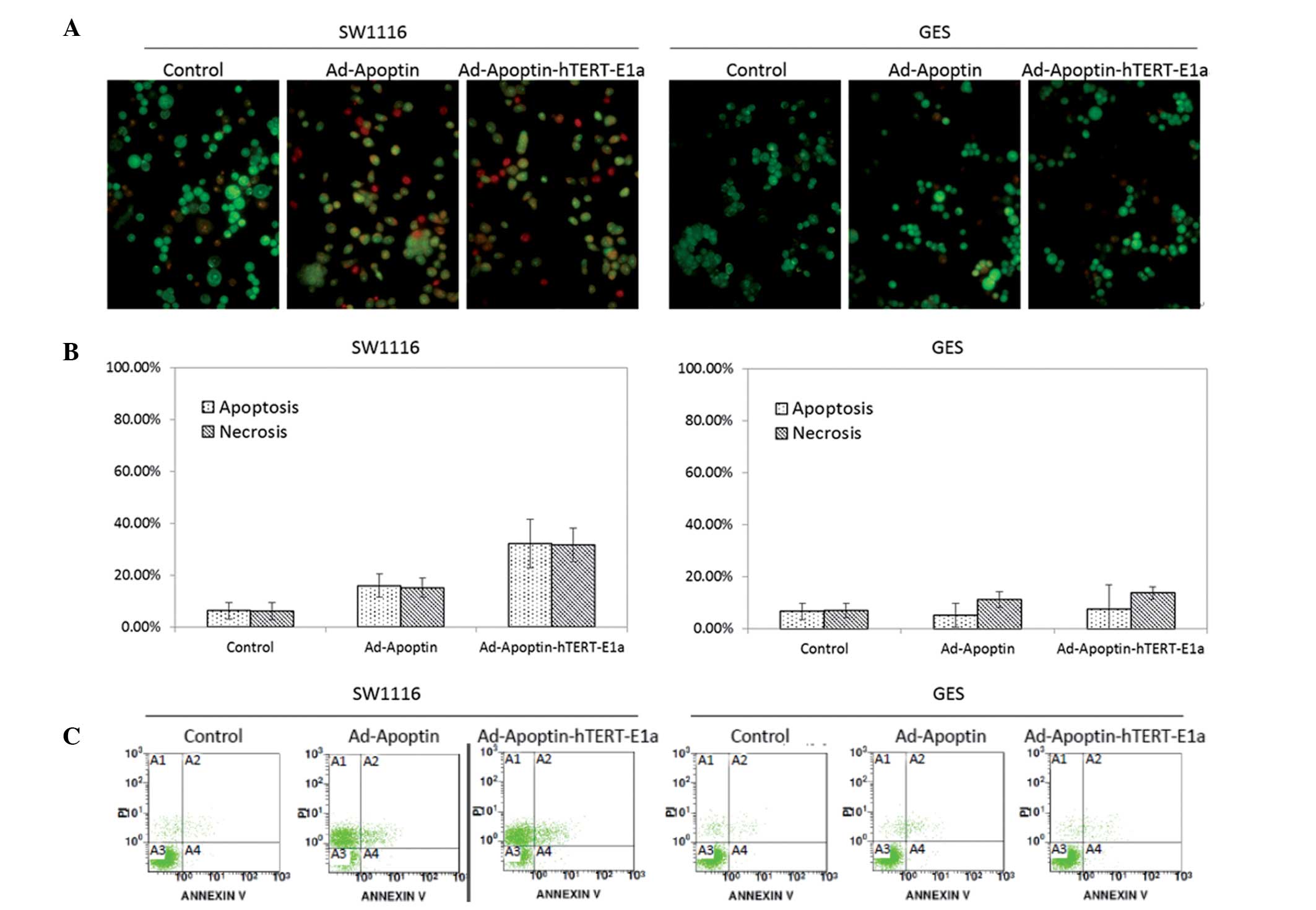
AO dye is unable to permeate the intact cell
membrane, which stains live cells with bright green fluorescence,
while EB can only enter the membrane of damaged cells and stains
the nuclei orange (18). Images of
AO/EB staining and the results of Image-Pro Plus analysis are shown
in Fig. 2A and B. Fig. 2A compares the morphological changes
for SW1116 and GES cells treated with the recombinant adenoviruses.
Live cells are shown with a normal green nucleus; early apoptotic
cells have a bright green nucleus with condensed or fragmented
chromatin; late apoptotic cells exhibit condensed and fragmented
orange chromatin; and cells that have died from direct necrosis
have structurally normal orange nuclei. As shown in Fig. 2B, the proportion of necrotic and
apoptotic cell populations in the control and treated SW1116 or GES
cells was significantly different. In the SW1116 cells, infection
with Ad-Apoptin-hTERT-E1a resulted in apoptosis (32.2%) and
necrosis (31.5%). However, the proportion of cells undergoing
apoptosis or necrosis in the Ad-Apoptin-hTERT-E1a-treated GES cells
was similar to the uninfected control GES cells. The percentage of
apoptotic cells following recombinant adenovirus treatment was
quantified by flow cytometry. As shown in Fig. 2C, in contrast to the GES cells,
infection with Ad-Apoptin-hTERT-E1a resulted in the apoptosis of
SW1116 cells. These results indicated that Ad-Apoptin-hTERT-E1a
specifically induced apoptosis in CRC cells.
Recombinant adenoviruses induce apoptosis
via the mitochondrial pathway
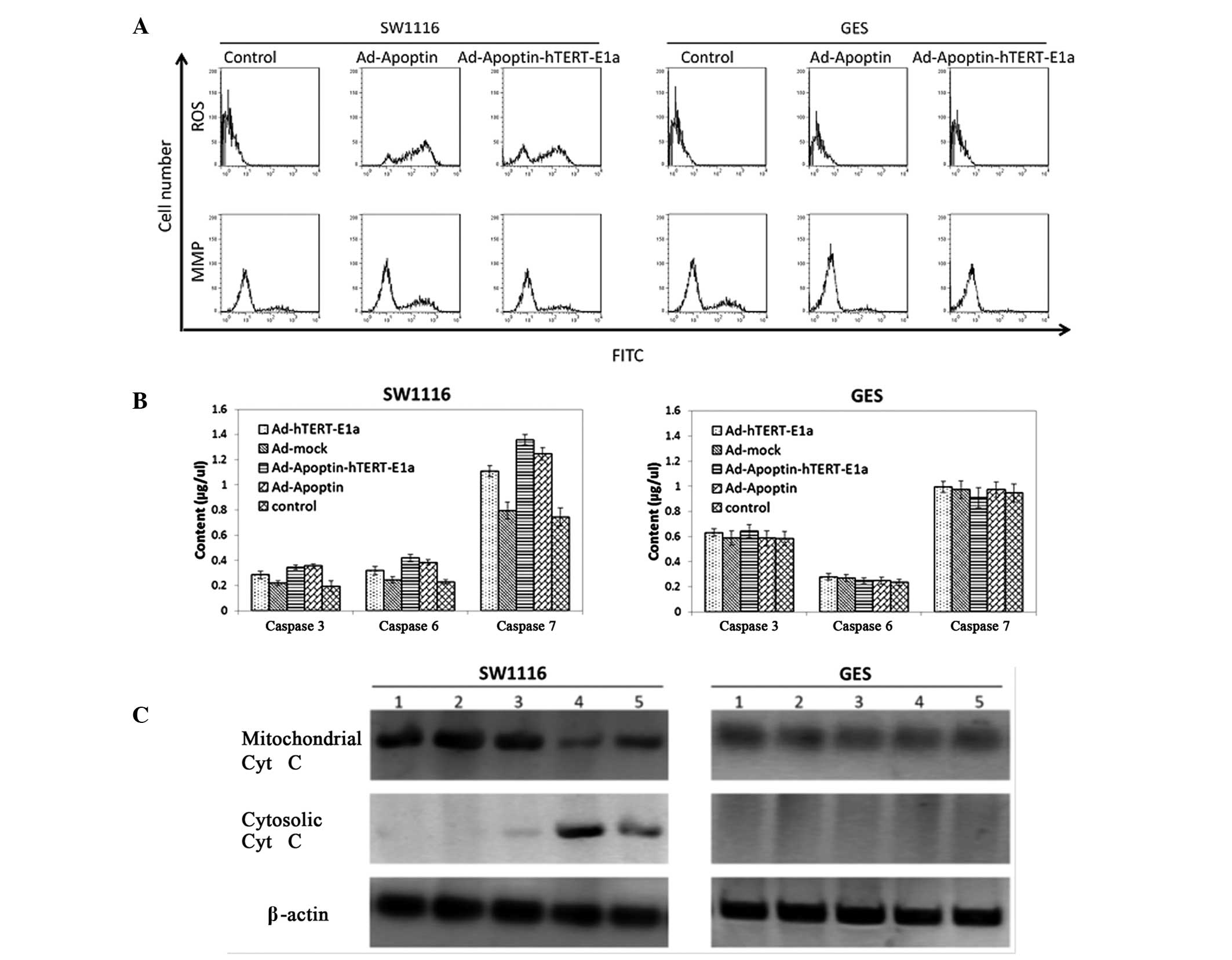
The effects of the recombinant adenoviruses on the
MMP and ROS levels in SW1116 cells were determined. As shown in
Fig. 3A,
Ad-Apoptin-hTERT-E1a-infected SW1116 cells showed a significant
increase in ROS levels compared with the untreated control group.
In the GES cells, infection with Ad-Apoptin-hTERT-E1a resulted in a
slight increase in the level of ROS compared with the control
group. The results of MMP analysis were similar to that of the ROS
detection. SW1116 cells infected with Ad-Apoptin-hTERT-E1a showed a
significant decrease in the MMP, while in GES cells,
Ad-Apoptin-hTERT-E1a treatment did not result in a MMP decrease, as
compared with the control group (Fig.
3A). In addition, the activity levels of caspases were
determined (Fig. 3B). Infection of
SW1116 cells with Ad-Apoptin-hTERT-E1a caused a marked increase in
the activity levels of caspses-3, 6 and 7. By contrast, caspase
activity was not detected in the GES cells infected with the
recombinant adenoviruses. Furthermore, significant quantities of
cytochrome c were detected in the cytosol of the
Ad-Apoptin-hTERT-E1a-infected SW1116 cells (Fig. 3C). The levels of cytochrome
c in the cells treated with Ad-Apoptin-hTERT-E1a were more
evident than in the control groups. However, Ad-Apoptin-hTERT-E1a
exhibited no significant effects on cytochrome c release in
the GES cells (Fig. 3C). These
results indicated that the specific apoptosis of SW1116 cells
triggered by Ad-Apoptin-hTERT-E1a was associated with the release
of mitochondrial cytochrome c, a decrease in the MMP and an
increase in the levels of ROS.
Ad-Apoptin-hTERT-E1a suppresses
subcutaneous primary tumor growth
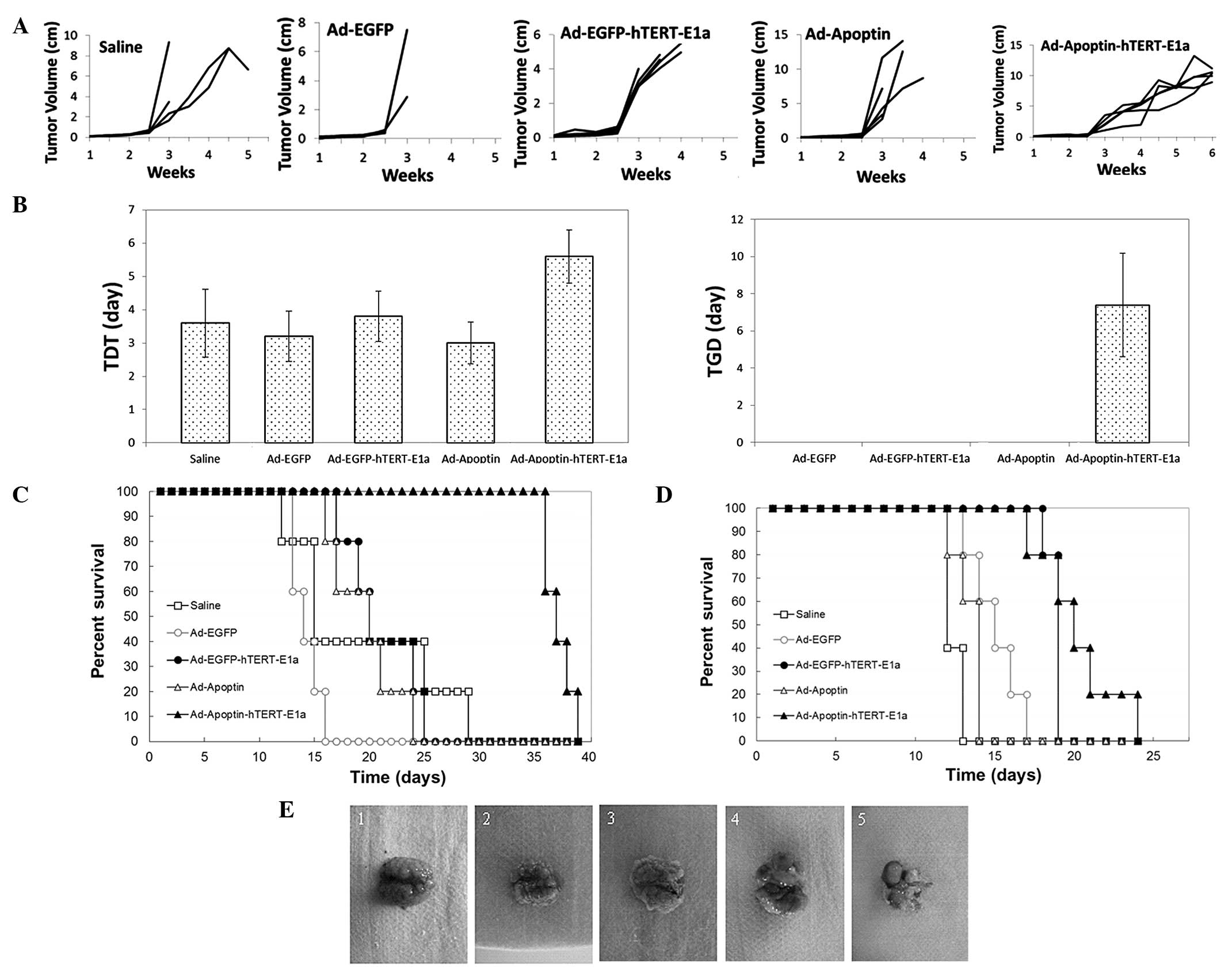
The antitumor potential of Ad-Apoptin-hTERT-E1a was
examined in a mouse CT26 tumor model. The growth kinetics of the
tumors following treatment are shown in Fig. 4A. Compared with the saline control
and the Ad-enhanced green fluorescent protein (EGFP) group, the
growth of the tumors in the recombinant adenovirus groups was
suppressed. However, following three injections, the tumors
infected with Ad-EGFP, Ad-EGFP-hTERT-E1a and Ad-Apoptin resumed
their usual growth. By contrast, the majority of
Ad-Apoptin-hTERT-E1a-infected tumors grew slowly. Furthermore, the
intratumoral injection of Ad-Apoptin-hTERT-E1a significantly
increased the TDT and TGD. When compared with the saline control
groups, Ad-Apoptin-hTERT-E1a significantly increased the TDT from
3.6 to 5.6 days (Fig. 4B;
P<0.05). In addition, treatment with Ad-Apoptin-hTERT-E1a
delayed tumor growth by 7.4 days, whereas the other recombinant
adenoviruses exhibited no tumor delaying effects (Fig. 4B; P<0.05). The ability of the
recombinant adenoviruses to prolong the mean survival times of the
tumor-bearing mice was also investigated. As shown in Fig. 4C, the mice treated with
Ad-Apoptin-hTERT-E1a had the longest survival times. The mean
survival times were 19.2±3.3 days for the saline-treated mice,
14.2±0.6 days for the Ad-EGFP-infected mice, 21±1.5 days for the
Ad-EGFP-hTERT-E1a-infected mice, 19.6±1.4 days for the
Ad-Apoptin-infected mice and 37.2±0.6 days for the
Ad-Apoptin-hTERT-E1a-infected mice. The results indicated that
intratumoral injections of Ad-Apoptin-hTERT-E1a conferred
significant survival benefits in vivo.
Systemic delivery of Ad-Apoptin-hTERT-E1a
reduces the number of metastatic lung nodules
Survival analysis revealed that Ad-Apoptin-hTERT-E1a
treatment significantly increased the survival times of the mice in
the lung metastasis model group when compared with treatment using
the other recombinant adenoviruses or with saline (Fig. 4D). The mean survival times were
12.4±0.2 days for the saline-treated mice, 15±0.7 days for the
Ad-EGFP-infected mice, 18.8±0.2 days for the
Ad-EGFP-hTERT-E1a-infected mice, 13.4±0.4 days for the
Ad-Apoptin-infected mice and 20.2±1.2 days for the
Ad-Apoptin-hTERT-E1a-infected mice. As shown by the representative
metastatic nodules in Fig. 4E,
Ad-Apoptin-hTERT-E1a significantly decreased the tumor burden of
the mice. The lungs of the mice infected with Ad-Apoptin-hTERT-E1a
exhibited minimal metastatic nodules, whereas the lungs from the
control or treated groups exhibited severe metastasis. Therefore,
systemic delivery of Ad-Apoptin-hTERT-E1a was shown to
significantly reduce the tumor burden and provide survival benefits
in a lung metastatic cancer model.
Discussion
Cancer is a serious threat to public health. The
pathogenesis of cancer is that normal cells are transformed into a
malignant cells, in which apoptosis is reduced (19). Thus, promoting the apoptosis of
cancer cells plays a vital role in oncotherapy. Oncolytic
adenoviruses are promising tools in cancer therapeutics due to
their ease of manipulation and multiple, distinct anticancer
mechanisms, including direct lysis, apoptosis induction, expression
of toxic proteins, autophagy and the inhibition of protein
synthesis, as well as the induction of antitumoral immunity
(20,21). Assessing therapeutic genes to
insert into the viral genome has been a major focus in cancer
virotherapy, and the types of transgenes considered for this
purpose have included tumor suppressor, proapoptotic,
antiangiogenic, ‘suicide’ and immunomodulatory genes (13). Apoptin is the VP3 protein of
chicken infectious anemia virus. The protein is p53-independent,
Bcl-2-insensitive and apoptotic (22). Apoptin resides in the cytoplasm of
normal cells, whereas it localizes in the nucleus of cancer cells
(23). The nuclear translocation
of apoptin largely depends on phosphorylation (14). Apoptin has the specific ability to
kill human cancer cells and transform cells without interfering
with normal cell proliferation (15). Apoptin induces various types of
human cancer cell lines to undergo apoptosis via classical
apoptotic pathways (16).
In cancer gene therapy, the specificity of the
promoter that controls the expression of an exogenous gene to
target cells determines the treatment validity (17). Telomeres are repeated DNA sequences
that provide protection for chromosomes (18). Telomerase, in which hTERT functions
as the catalytic protein, adds telomere repeats to chromosomes
(19). Telomerase activity is
closely associated with hTERT expression (24). The activity of the hTERT promoter
has been associated with cancer and has been detected in a number
of invasive cancer types; however, the promoter is repressed in
normal somatic tissues or benign tumors (25). The hTERT promoter has been used to
drive the expression of a number of genes for cancer therapeutics
(26,27). In a previous study, a
tumor-specific apoptosis-inducing gene (apoptin) and a
cancer-specific promoter (hTERTp) were inserted into the RAPAd
adenoviral vector to obtain a novel, dual-specific antitumor
oncolytic adenovirus, Ad-Apoptin-hTERT-E1a, as well as the control
recombinant adenoviruses, Ad-Apoptin, Ad-EGFP and Ad-EGFP-hTERT-E1a
(13). In the present study, the
antitumor effects of these novel oncolytic viruses were evaluated
in CRC cells in vitro and in vivo. In order to avoid
the influence of EGFP gene expressed by Ad-EGFP on the fluorescence
experiment, the Ad-EGFP virus was not used in the in vitro
studies. While, to facilitate the in vivo imaging studies
(data not shown), Ad-EGFP was used in the animal experiments.
As shown in Fig. 1,
the cell viability showed a non-rigorous dependence on the
infection time and MOI. With increasing infection times and
increasing infective doses, the inhibitory effects on SW1116 cells
treated with Ad-Apoptin-hTERT-E1a became more evident than in cells
infected with the other recombinant adenoviruses. By contrast,
Ad-Apoptin-hTERT-E1a had a limited inhibitory effect on GES cells.
The AO/EB staining assay was used to analyze cell death and
quantify the relative proportions of live, apoptotic and necrotic
recombinant adenovirus-infected cells (Fig. 2A and B). The results indicated that
Ad-Apoptin-hTERT-E1a significantly induced apoptosis and necrosis
in SW116 cells, but had no effect on the normal GES cells. In
addition, annexin V assays indicated that Ad-Apoptin-hTERT-E1a was
able to suppress the growth of SW1116 cells through the induction
of apoptosis (Fig. 2C), while the
normal GES cells showed little sensitivity to the recombinant
adenovirus. Furthermore, Ad-Apoptin-hTERT-E1a caused an apparent
increase in the levels of ROS, significantly reduced the MMP and
activated caspases in the SW1116 cells (Fig. 3A and B). The release of cytochrome
c was also detected in Ad-Apoptin-hTERT-E1a-infected SW1116
cells (Fig. 3C). By contrast, the
effects of Ad-Apoptin-hTERT-E1a on GES cells were minimal.
Therefore, the results confirmed the previous observations that
Ad-Apoptin-hTERT-E1a has the potential to specifically kill CRC
cells by inducing the apoptosis pathway.
The in vivo antitumor activities of the
recombinant adenoviruses were also evaluated in a CRC mouse model,
which further confirmed the efficacies observed in vitro. As
shown in Fig. 4A–C,
Ad-Apoptin-hTERT-E1a exhibited significant antitumor effects
compared with the other recombinant adenoviruses in the primary
tumor model. Injection of Ad-Apoptin-hTERT-E1a directly into the
tumors resulted in a complete response to treatment and the longest
mean survival time, which were the best outcomes compared with the
results from the other recombinant adenovirus-treated groups. In
the in vivo experiments, the antitumoral effects of
Ad-Apoptin-hTERT-E1a were also observed on metastatic tumors. The
data indicated that Ad-Apoptin-hTERT-E1a inhibited the formation of
metastatic tumors successfully (Fig.
4D and E). Furthermore, no toxic effects were observed
following the injection of Ad-Apoptin-hTERT-E1a.
In conclusion, the effects of Ad-Apoptin-hTERT-E1a
on the CRC SW1116 cell line were investigated in vitro. The
results demonstrated that Ad-Apoptin-hTERT-E1a specifically
replicated in human SW1116 tumor cells and restricted the growth of
these cells selectively, while showing no adverse effects on GES
cells. In addition, the results obtained from the in vivo
tumor model indicated that Ad-Apoptin-hTERT-E1a not only inhibited
primary transplanted tumors, but also played a key role in
suppressing the metastasis of tumors. These results highlight the
need for further evaluation of Ad-Apoptin-hTERT-E1a as a novel
class of drugs for the clinical treatment of CRC.
Acknowledgements
The study was supported in part by grants from the
National Science and Technology Major Projects for ‘Major New Drugs
Innovation and Development’ (no. 2010ZX09401-305-14), the National
Natural Science Foundation of China (nos. 81072210 and 81101140)
and the Key Technologies R&D Program of Jilin Province (nos.
10ZDGG007, 20130206041NY, 201015166 and 201101066).
References
|
1
|
Li ST and Chi P: Evolution of the
management of colorectal cancer using integrative medicine. Chin J
Integr Med. 17:73–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Waldner MJ and Neurath MF: The molecular
therapy of colorectal cancer. Mol Aspects Med. 31:171–178. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barnes MN and Pustilnik TB: Current
strategies in gene therapy for ovarian cancer. Curr Opin Obstet
Gynecol. 13:47–51. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shirakawa T: The current status of
adenovirus-based cancer gene therapy. Mol Cells. 25:462–466.
2008.PubMed/NCBI
|
|
5
|
Kang E and Yun CO: Current advances in
adenovirus nanocomplexes: more specificity and less immunogenicity.
BMB Rep. 43:781–788. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pesonen S, Kangasniemi L and Hemminki A:
Oncolytic adenoviruses for the treatment of human cancer: focus on
translational and clinical data. Mol Pharm. 8:12–28. 2011.
View Article : Google Scholar
|
|
7
|
Panigrahi S, Klonisch T and Los M: The art
of killing: double stroke with apoptin and survivin as a novel
approach in cancer therapy. Cancer Biol Ther. 7:1061–1062. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hashimoto Y, Tazawa H, Teraishi F, et al:
The hTERT promoter enhances the antitumor activity of an oncolytic
adenovirus under a hypoxic microenvironment. PLoS One.
7:e392922012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kovalenko OA, Kaplunov J, Herbig U, et al:
Expression of (NES-)hTERT in cancer cells delays cell cycle
progression and increases sensitivity to genotoxic stress. PLoS
One. 5:e108122010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chan G, Kamarudin MN, Wong DZ, et al:
Mitigation of H(2)O(2)-induced mitochondrial-mediated apoptosis in
NG108-15 cells by novel mesuagenin C from Mesua kunstleri (King)
Kosterm. Evid Based Complement Alternat Med. 2012:1565212012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Byczkowska A, Kunikowska A and Kaźmierczak
A: Determination of ACC-induced cell-programmed death in roots of
Vicia faba ssp minor seedlings by acridine orange and ethidium
bromide staining. Protoplasma. 250:121–128. 2013. View Article : Google Scholar :
|
|
12
|
Li X1, Liu Y, Wen Z, et al: Potent
anti-tumor effects of a dual specific oncolytic adenovirus
expressing apoptin in vitro and in vivo. Mol Cancer. 9:102010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang M, Wang J, Li C, et al: Potent
growth-inhibitory effect of a dual cancer-specific oncolytic
adenovirus expressing apoptin on prostate carcinoma. Int J Oncol.
42:1052–1060. 2013.PubMed/NCBI
|
|
14
|
Los M, Panigrahi S, Rashedi I, et al:
Apoptin, a tumor-selective killer. Biochim Biophys Acta.
1793:1335–1342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Backendorf C, Visser AE, de Boer AG, et
al: Apoptin: therapeutic potential of an early sensor of
carcinogenic transformation. Annu Rev Pharmacol Toxicol.
48:143–169. 2008. View Article : Google Scholar
|
|
16
|
Pavet V, Portal MM, Moulin JC, Herbrecht R
and Gronemeyer H: Towards novel paradigms for cancer therapy.
Oncogene. 30:1–20. 2011. View Article : Google Scholar
|
|
17
|
Ye F, Zhong B, Dan G, et al: Therapeutic
anti-tumor effect of exogenous apoptin driven by human survivin
gene promoter in a lentiviral construct. Arch Med Sci. 9:561–568.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
O’Sullivan RJ and Karlseder J: Telomeres:
protecting chromosomes against genome instability. Nat Rev Mol Cell
Biol. 11:171–181. 2010.
|
|
19
|
Antoniou KM, Margaritopoulos GA, Proklou
A, et al: Investigation of telomerase/telomeres system in bone
marrow mesenchymal stem cells derived from IPF and RA-UIP. J
Inflamm (Lond). 9:272012. View Article : Google Scholar
|
|
20
|
Vähä-Koskela MJ, Heikkilä JE and Hinkkanen
AE: Oncolytic viruses in cancer therapy. Cancer Lett. 254:178–216.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nettelbeck DM: Cellular genetic tools to
control oncolytic adenoviruses for virotherapy of cancer. J Mol Med
(Berl). 86:363–377. 2008. View Article : Google Scholar
|
|
22
|
Wu Y, Zhang X, Wang X, et al: Apoptin
enhances the oncolytic properties of Newcastle disease virus.
Intervirology. 55:276–286. 2012. View Article : Google Scholar
|
|
23
|
Jiang J, Cole D, Westwood N, et al:
Crucial roles for protein kinase C isoforms in tumor-specific
killing by apoptin. Cancer Res. 70:7242–7252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kang X, Chen W, Kim RH, Kang MK and Park
NH: Regulation of the hTERT promoter activity by MSH2, the hnRNPs K
and D, and GRHL2 in human oral squamous cell carcinoma cells.
Oncogene. 28:565–574. 2009. View Article : Google Scholar
|
|
25
|
Xie X, Hsu JL, Choi MG, et al: A novel
hTERT promoter-driven E1A therapeutic for ovarian cancer. Mol
Cancer Ther. 8:2375–2382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tan J, Li W and Wang P: Telomerase reverse
transcriptase promoter-driven expression of iodine pump genes for
targeted radioiodine therapy of malignant glioma cells. Chin J
Cancer. 30:574–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu L, Wu W, Zhu G, et al: Therapeutic
efficacy of an hTERT promoter-driven oncolytic adenovirus that
expresses apoptin in gastric carcinoma. Int J Mol Med. 30:747–754.
2012.PubMed/NCBI
|