Introduction
Myoclonic epilepsy with ragged-red fibers (MERRF) is
a neurological disorder that is characterized by muscle twitches,
weakness and progressive stiffness that affects numerous muscles of
the body. Patients with MERRF can additionally exhibit recurrent
seizures, difficulty coordinating movements, peripheral neuropathy
and the slow deterioration of intellectual function (1). MERRF is a maternally inherited
mitochondrial encephalomypathy caused by a mtDNA mutation. The most
common mutation is the m.8344A>G mutation in the mtDNA gene,
MT-TK, which encodes mitochondrial transfer (t)RNA lysine. The
mutation causes poor aminoacylation of the mutant tRNA and
premature termination of translation at lysine codons (2), which subsequently decreases the
activity of respiratory chain complexes I and IV, as well as the
respiration rate and the mitochondrial membrane potential (3). Ragged red fibers consist of a large
collection of abnormal-appearing mitochondria (4).
To date, the principal treatment for MERRF is
symptomatic. However, standard traditional antiepileptic drug
therapy appears to be effective, and coenzyme Q10 is often used in
an attempt to improve mitochondrial function (5,6).
MERRF is a very rare condition in the Chinese population, as is the
8344A>G mutation (3.9% of all pathogenic mtDNA mutations)
(7). The present study reports the
case of a 25-year-old male with mitochondrial encephalomyopathy,
who presented with myoclonic epilepsy.
Case report
A 25-year-old male presented with paroxysmal left
upper limb tics and weakness that had been ongoing for two years.
The involuntary limb tics exhibited a sudden onset and lasted for
seconds, but were not accompanied by consciousness disturbance. The
patient had approximately 10 attacks per day, which were
accompanied by limb weakness. A magnetic resonance imaging (MRI)
scan was performed initially and was found to be normal. The
patient had received irregular diazepam administration from the
onset of the disease; however, the symptoms became increasingly
more serious. The patient was prescribed 600 mg per day valproate
sodium on admission to hospital to control the seizures, but
experienced one or two attacks per month subsequent to the
administration of valproate sodium. The past medical history of the
patient was unremarkable. On examination, the patient was alert and
his pupils adjusted to light. Neurological examination revealed
intact cranial nerves, but decreased deep tendon reflexes and a
decreased sensation of touch, pain and vibration. The gait of the
patient was broad and he was unable to walk in a straight line.
Full strength was observed in all the muscle groups. The results of
the Romberg, heel-knee-shin and finger-to-nose tests were normal.
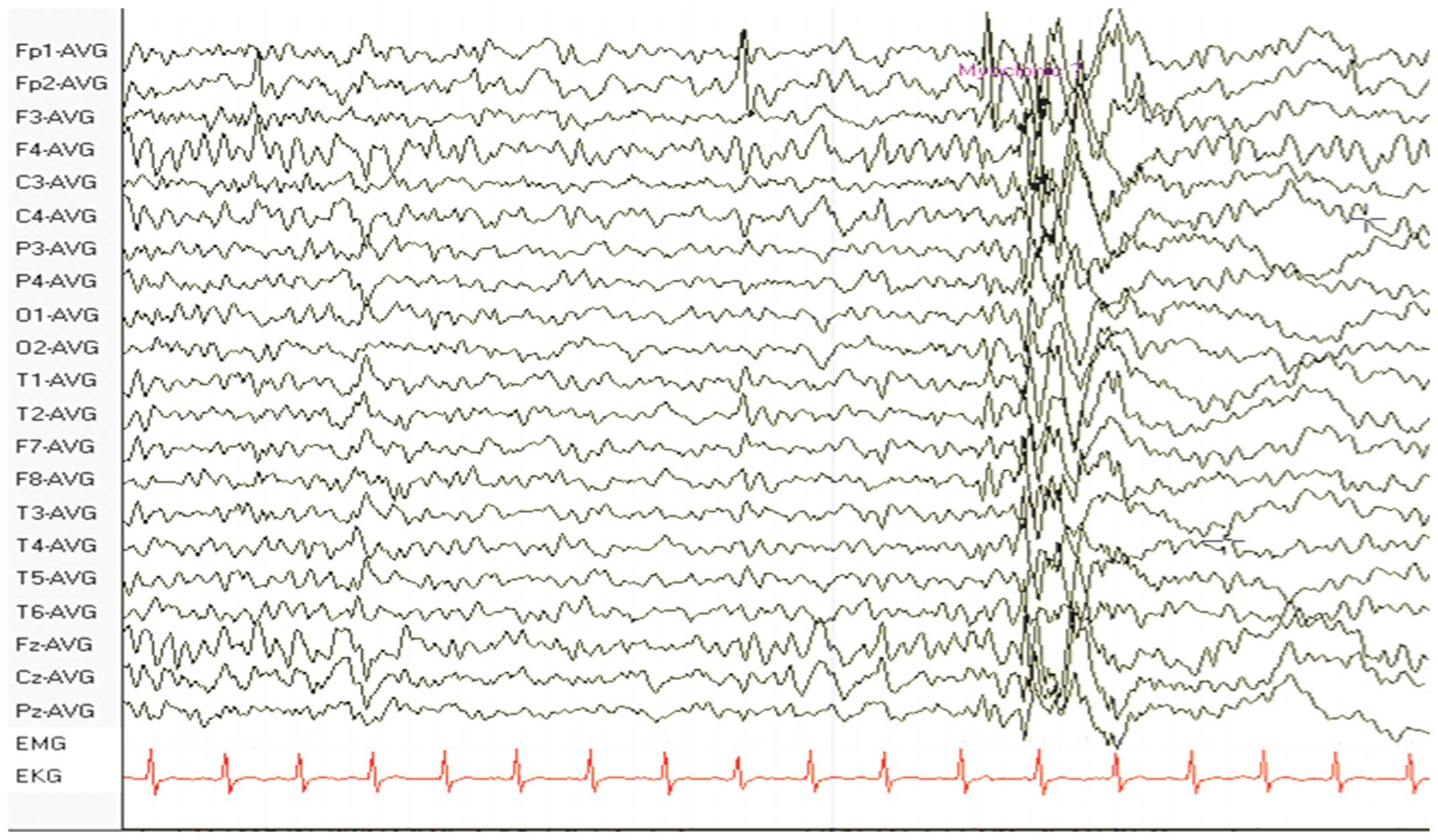
An electroencephalogram (EEG) revealed diffuse spikes and slow
waves, predominantly in the frontal and temporal lobes (Fig. 1). A further MRI scan was performed
and revealed increased signal density on T2-weighted imaging and
decreased signal density on T1-weighted imaging in the right
temporal occipital cortical lesions. Local cortical atrophy was
also observed in the left temporal-occipital cortex. In addition,
the lactic acid concentration (5.2 mmol/l) had markedly increased.
The results of the carotid ultrasound and electromyography were
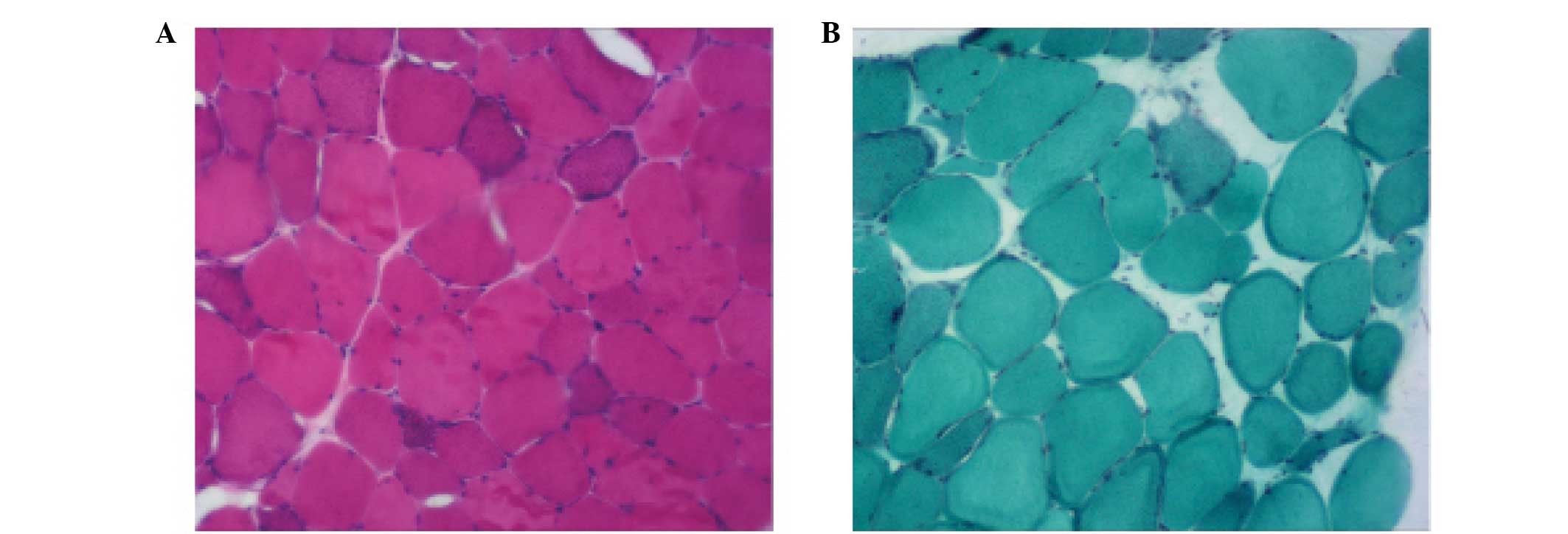
normal. A biopsy of the biceps muscle demonstrated a variation in
fiber size and the presence of ragged-red fibers (Fig. 2). In addition to the prescribed 600
mg per day valproate sodium, the patient was administered 10 mg per
day coenzyme Q10 for approximately 2 years. Two years later his
symptoms relieved and an EEG showed less spikes and slow waves than
it had previously shown.
Discussion
Mitochondrial encephalomyopathies are a group of
disorders characterized by impaired oxidative metabolism, which
result in the impairment of skeletal muscles and the central
nervous system. The clinical presentation of a mitochondrial
encephalomyopathy is highly variable and diagnosis is often
difficult, requiring an extensive series of clinical studies and
muscle biopsies or DNA testing.
The present case report described a young male with
involuntary tics and a diagnosis of MERRF based on EEG findings
(8). Notably, the patient also
presented with a number of associated symptoms, including weakness
and a broad gait while walking. In a previous study (9), abnormal brain MRI observations were
reported in patients with mitochondrial encephalomyopathy. The most
frequent abnormalities in patients with mitochondrial
encephalomyopathy are widespread white matter hyperintensity and
supratentorial cortical and cerebellar atrophy. In certain cases
(10), brain abnormalities are
absent. In the present case, the first MRI scan was normal. The
patient subsequently developed right temporal-occipital cortical
long T1 and T2 signals in the MRI scans. Local cortical atrophy was
also observed in the left temporal-occipital cortex. This
observation was consistent with brain MRI abnormalities in patients
with mitochondrial encephalomyopathy.
Pathologically, MERRF is characterized by a
variation in fiber size and ragged-red fibers. In the present
study, the muscle biopsy and pathological findings were consistent
with MERRF. Therefore, the present study hypothesizes that imaging
observations and follow-up examinations are important for patients
with myoclonic epilepsy.
Acknowledgements
The authors thank Medjaden Bioscience Ltd. (Hong
Kong, China) for assisting in the preparation of this study.
References
|
1
|
DiMauro S, Hirano M, Kaufmann P, et al:
Clinical features and genetics of myoclonic epilepsy with ragged
red fibers. Adv Neurol. 89:217–229. 2002.PubMed/NCBI
|
|
2
|
Enriquez JA, Chomyn A and Attardi G: MtDNA
mutation in MERRF syndrome causes defective aminoacylation of
tRNA(Lys) and premature translation termination. Nat Genet.
10:47–55. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
James AM, Sheard PW, Wei YH and Murphy MP:
Decreased ATP synthesis is phenotypically expressed during
increased energy demand in fibroblasts containing mitochondrial
tRNA mutations. Eur J Biochem. 259:462–469. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Black JT, Judge D, Demers L and Gordon S:
Ragged-red fibers. A biochemical and morphological study. J Neurol
Sci. 26:479–488. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De la Mata M, Garrido-Maraver J, Cotán D,
et al: Recovery of MERRF fibroblasts and cybrids pathophysiology by
coenzyme Q10. Neurotherapeutics. 9:446–463. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bindoff LA and Engelsen BA: Mitochondrial
diseases and epilepsy. Epilepsia. 53(Suppl 4): 92–97. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mancuso M, Orsucci D, Angelini C, et al:
Phenotypic heterogeneity of the 8344A>G mtDNA ‘MERRF’ mutation.
Neurology. 80:2049–2054. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brackmann F, Abicht A, Ahting U, Schröder
R and Trollmann R: Classical MERRF phenotype associated with
mitochondrial tRNA(Leu) (m.3243A>G) mutation. Eur J Pediatr.
171:859–862. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zsurka G, Becker F, Heinen M, et al:
Mutation in the mitochondrial tRNA(Ile) gene causes progressive
myoclonus epilepsy. Seizure. 22:483–486. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chuang CS, Lo MC, Lee KW and Liu CS:
Magnetic resonance spectroscopy study in basal ganglia of patients
with myoclonic epilepsy with ragged-red fibers. Neurol India.
55:385–387. 2007. View Article : Google Scholar : PubMed/NCBI
|