Introduction
Papillary thyroid cancer (PTC) is a common thyroid
malignancy originating from the thyroid follicular cells. It
accounts for ~85% of all thyroid cancer cases with an estimated
66,200 new cases in the United States in 2013 (1,2). PTC is
more prevalent in females and although it may occur at any age, the
peak incidence of PTC occurs between 30 and 50 years (3,4). Despite
being considered a low grade malignancy, PTC poses a substantial
threat to global health due to the marked rise in the incidence
rate of PTC, with an estimated 2.9-fold increase from 2.7 to 7.7
cases per 100,000 individuals in the United States over the past
three decades (5,6). Although the exact etiology of PTC
remains to be fully elucidated, related studies have proposed an
association between the risk of developing PTC and exposure to
radiation (4,7). Furthermore, with the rapid development
of molecular biology, genetic factors have been demonstrated to
play a critical role in increasing susceptibility to PTC (8). Therefore, in recent years, an
increasing number of studies have attempted to investigate the
exact cellular and molecular mechanisms that lead to the
development of PTC. Epigenetic inactivation of tumor suppressor
genes, including Ras association domain family 1 isoform A
(RASSF1A) and p16, has been suggested to be implicated in PTC
development (9,10).
The p16 protein, also known as multiple tumor
suppressor 1 or cyclin-dependent kinase inhibitor 2A, is a cell
cycle regulator protein belonging to a class of cyclin-dependent
kinase (CDK) inhibitory proteins that control two critical
cell-cycle control pathways, namely the p16-Rb and p14-p53 pathways
(11). It plays an important role in
cell cycle regulation by decelerating cell progression from the G1
to the S phase, specifically binding to CDK4, and preventing the
phosphorylation and subsequent inactivation of the retinoblastoma
protein (pRb) (12). Furthermore,
p16 is a tumor suppressor protein that regulates cellular
proliferation and growth by acting as a CDK4 inhibitor, and is
implicated in the prevention of numerous types of cancer, including
breast and pancreatic cancers (13,14). The
human p16 gene is located on chromosome 9p21, and consists of 3
exons and 2 introns (15). There is
evidence to suggest that inactivation and the aberrant methylation
status of p16 is implicated in the pathogenesis and development of
PTC (9). It has been widely accepted
that methylation is a major etiology for p16 inactivation in PTC
(9). Furthermore, the aberrant
methylation of the p16 gene may give rise to the lack of p16
protein expression and affect its function, which may in turn
provide a selective growth advantage for tumor cells, thereby
contributing to an increased risk of PTC (16).
Ras association domain family 1 isoform A (RASSF1A)
is a microtubule-binding protein that regulates the activation of
RAS effector pathways and has been suggested to contribute to
maintaining genomic stability, stabilizing microtubules, as well as
regulating cell cycle arrest and mitotic progression (17–19). The
RASSF1A gene is located on the small arm of the human chromosome
3p21.3 within an area of common heterozygous and homozygous
deletions, which occur frequently in a variety of human tumors
(20,21). Clinically, reduced expression or
inactivation of RASSF1A has been associated with the pathogenesis
of various human cancers, including hepatocellular, gastric and
bladder carcinomas (22,23). It should be noted that the
inactivation of RASSF1A has been found to be associated with the
methylation of its CpG-island promoter region (24). Furthermore, RASSF1A CpG-island
methylation may lead to transcriptional silencing and thus inhibit
its function of tumor suppression, resulting in the development of
PTC (9,25).
Previous studies have indicated that promoter
methylation of the p16 and RASSF1A genes may play an important role
in the pathogenesis of PTC (9,25).
However, other studies have obtained contradictory results
regarding the association of aberrant promoter methylation of the
p16 and RASSF1A genes with PTC development (26,27).
Therefore, the present study attempted to perform a meta-analysis
of all eligible studies to provide insights into the role of p16
and RASSF1A promoter methylation on the pathogenesis of PTC.
Materials and methods
Literature search and selection
criteria
A range of electronic databases were searched
without language restrictions, as follows: Medline (1966–2013), the
Cochrane Library database (Issue 12, 2013), Embase (1980–2013),
CINAHL (1982–2013), Web of Science (1945–2013) and the Chinese
Biomedical Database (CBM) (1982–2013). The following keywords and
Medical Subject Headings (MeSH) were used, in conjunction with a
highly sensitive search strategy: ‘methylation’, ‘DNA methylation’,
‘demethylation’, ‘hypermethylation’ or ‘promoter methylation’; and
‘thyroid cancer, papillary’, ‘papillary thyroid carcinoma’, ‘PTC’,
‘papillary thyroid cancer’ or ‘papillary thyroid neoplasms’; and
‘cyclin-dependent kinase inhibitor p16’, ‘CDKN2 protein’, ‘p16INK4A
protein’, ‘RASSF1 protein, human’ or ‘RASSF1’. A manual search was
also conducted to identify other potential studies based on the
references listed in the individual studies.
The following criteria were used to determine the
eligibility of the included studies: i) the study design was a
clinical cohort study that focused on the association between
promoter methylation of p16 and RASSF1A and the pathogenesis of
PTC; ii) all patients met the diagnostic criteria for PTC; and iii)
the study provided sufficient information about the frequency of
methylation. Studies that did not meet the inclusion criteria were
excluded. The most recently published studies or those with the
largest sample size were included when the authors had published
several studies using the same subjects.
Data extraction and methodological
assessment
Data were systematically extracted by two authors
from each included study using a standardized form. The form used
for data extraction documented the most relevant items including:
study language, publication year, first author's surname,
geographical location, design of the study, sample size, the source
of the subjects, protein expression levels, source of samples and
protein detection method.
Methodological quality was evaluated separately by
two observers using the Newcastle-Ottawa Scale (NOS) criteria
(28). The NOS criteria included
three scores: i) subject selection, 0–4; ii) comparability of
subjects, 0–2; and iii) clinical outcome, 0–3. NOS scores ranged
from 0–9 and a score ≥7 indicated a study of good quality.
Statistical analysis
Meta-analysis was performed using Stata statistical
software (version 12.0; Stata Corporation, College Station, TX,
USA). The odds ratios (ORs), ratio differences (RDs) and 95%
confidence intervals (95% CIs) were calculated as estimates of the
associations. The Z-test was used to estimate the statistical
significance of pooled ORs. Heterogeneity amongst the studies was
estimated by the Cochran's Q and I2 tests (29). If the Q-test revealed a value of
P<0.05 or the I2 test exhibited a value >50%,
which indicates significant heterogeneity, a random-effects model
was conducted, or else a fixed-effects model was used. The reasons
for the heterogeneity were also explored using meta-regression and
subgroup analyses. In order to evaluate the effect of single
studies on the overall estimate, a sensitivity analysis was
performed. Funnel plots and Egger's linear regression test were
applied to investigate publication bias (30).
Results
Study selection and characteristics of
included studies
Initially, the highly sensitive search strategy
identified 133 studies, of which three were duplicates. The titles
and abstracts of all studies were reviewed and 66 studies were
excluded. The full texts were subsequently reviewed and a further
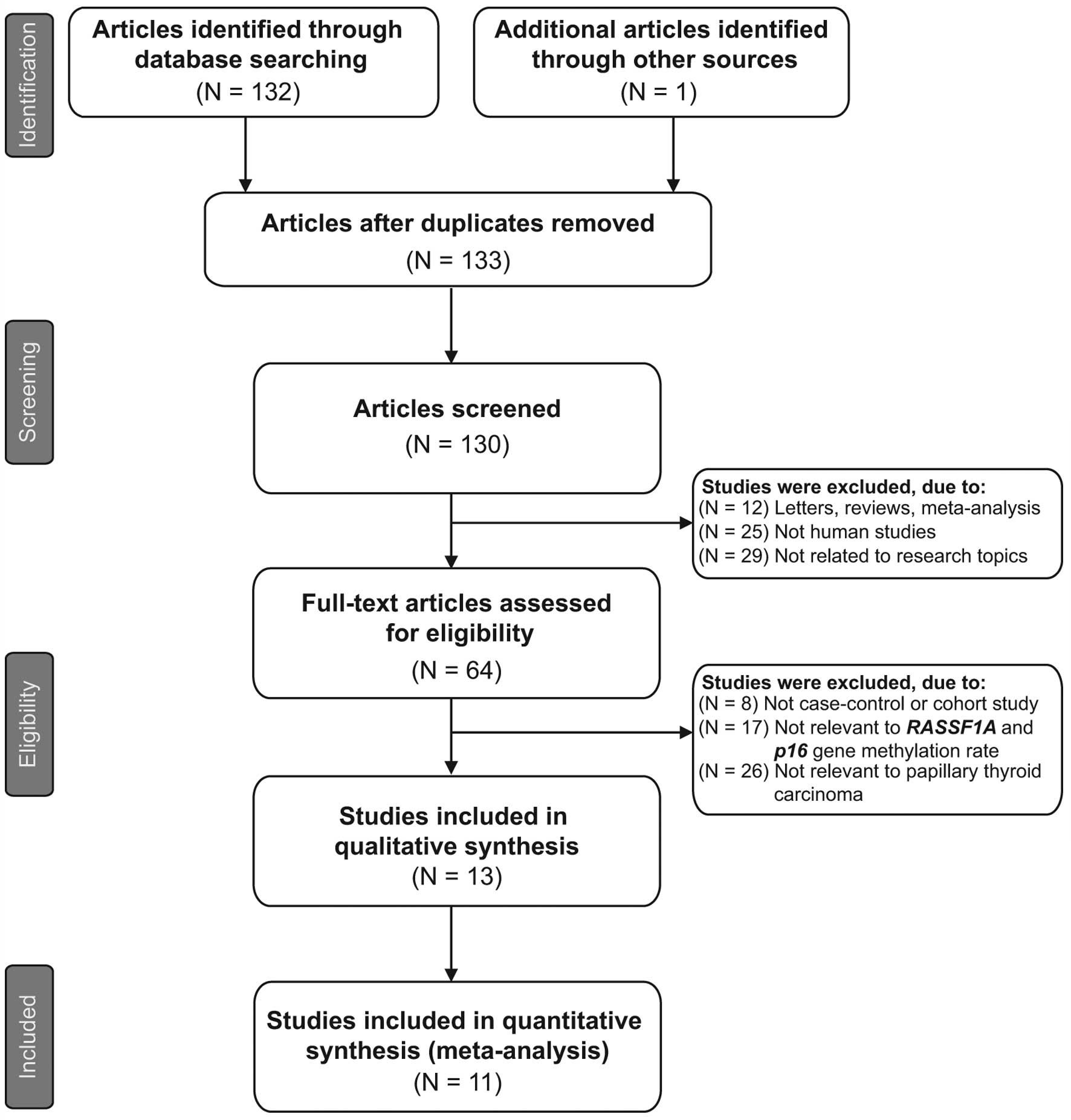
51 studies were excluded. Fig. 1
shows the selection process used to identify eligible studies. A
total of 11 clinical cohort studies with 734 patients with PTC met
the inclusion criteria for qualitative data analysis (9,16,26,27,31–37).
Publication years of the eligible studies ranged from 2002–2013.
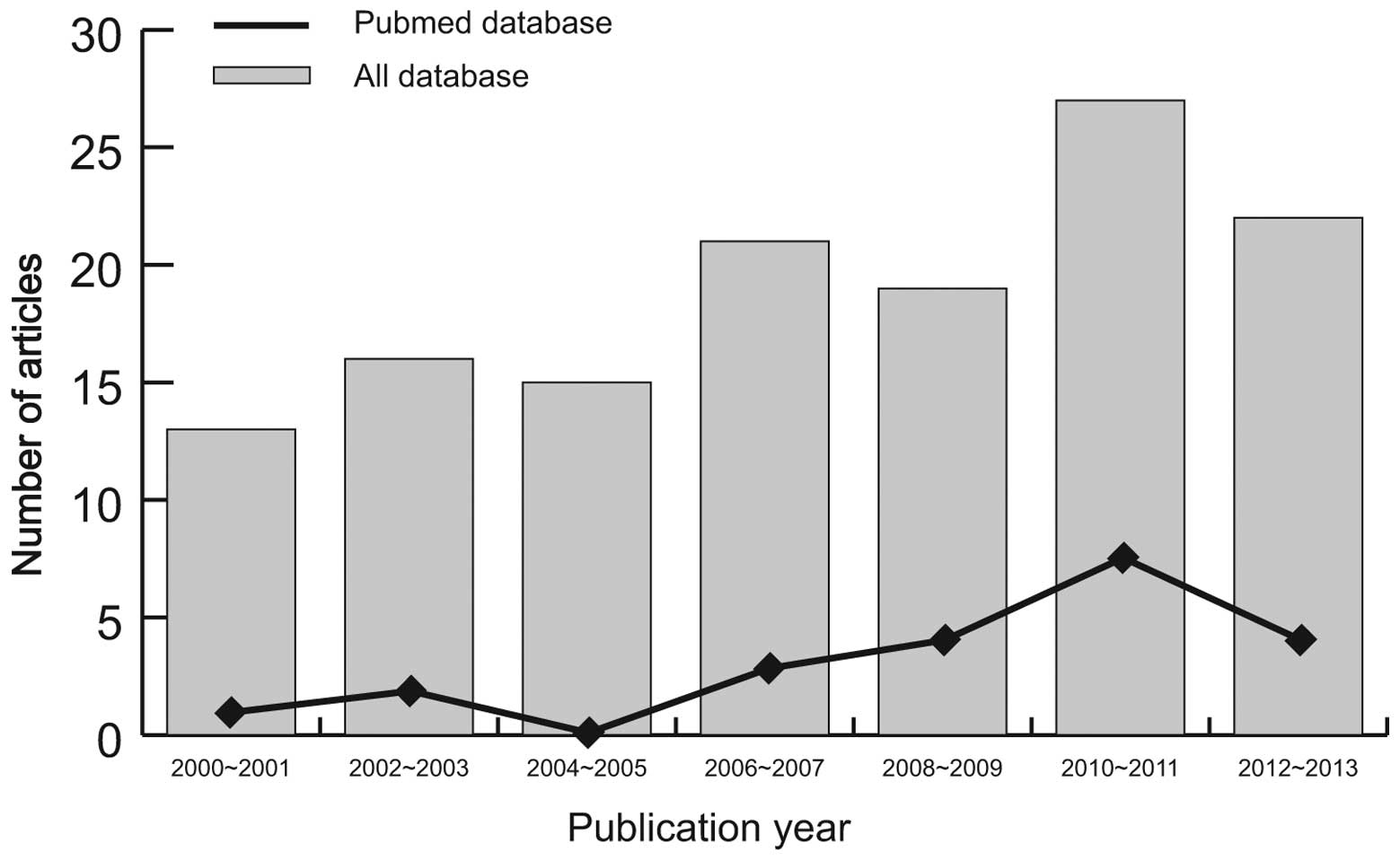
The distribution of the numbers of topic-related studies in
electronic databases during the last decade is shown in Fig. 2. Overall, 10 studies were conducted
among Asian populations and one study among the Caucasian
population. The methylation-specific polymerase chain reaction
(PCR; MSP) method was used in 10 studies and one study used the
combined bisulfite restriction analysis (COBRA) method. The study
characteristics and methodological quality are summarized in
Table I.
 | Table I.Baseline characteristics and
methodological quality of all included studies. |
Table I.
Baseline characteristics and
methodological quality of all included studies.
| Authors | Year | Country | Language | Ethnicity | No. of cases | Gender (M/F) | Age (years) | Gene | Sample | Method | NOS score |
|---|
| Li et al
(32) | 2013 | China | Chinese | Asian | 63 | 14/60 | 41±11.7a, 52±11.8b | p16 | Tissue | nMSP | 8 |
| Wang et al
(16) | 2013 | China | English | Asian | 95 | - | - | p16 | Tissue | MSP | 6 |
| Mohammadi-asl et
al (9) | 2011 | Iran | English | Asian | 50 | - | 56b | p16/RASSF1A | Tissue | COBRA | 7 |
| Wang et al
(36) | 2009 | China | Chinese | Asian | 187 | - | - | p16 | Blood | MSP | 6 |
| Peng et al
(33) | 2006 | China | Chinese | Asian | 34 | - | - | p16 | Tissue | MSP | 5 |
| Huang et al
(26) | 2006 | China | Chinese | Asian | 60 | - | - | p16 | Tissue | MSP | 6 |
| Schagdarsurengin
et al (27) | 2002 | USA | English | Caucasian | 18 | - | - | p16 | Tissue | MSP | 5 |
| Qu and Xue
(34) | 2012 | China | Chinese | Asian | 28 |
6/22 | 43 (28–68) | RASSF1A | Blood | MSP | 6 |
| Dai et al
(31) | 2011 | China | Chinese | Asian | 82 | 15/35 | 40.0±12.0 | RASSF1A | Tissue | MSP | 8 |
| Tang and Su
(35) | 2010 | China | Chinese | Asian | 34 |
5/29 | 41 (20–79) | RASSF1A | Blood | MSP | 6 |
| Wang et al
(37) | 2009 | China | Chinese | Asian | 83 | - | - | RASSF1A | Tissue | MSP | 7 |
Quantitative data synthesis
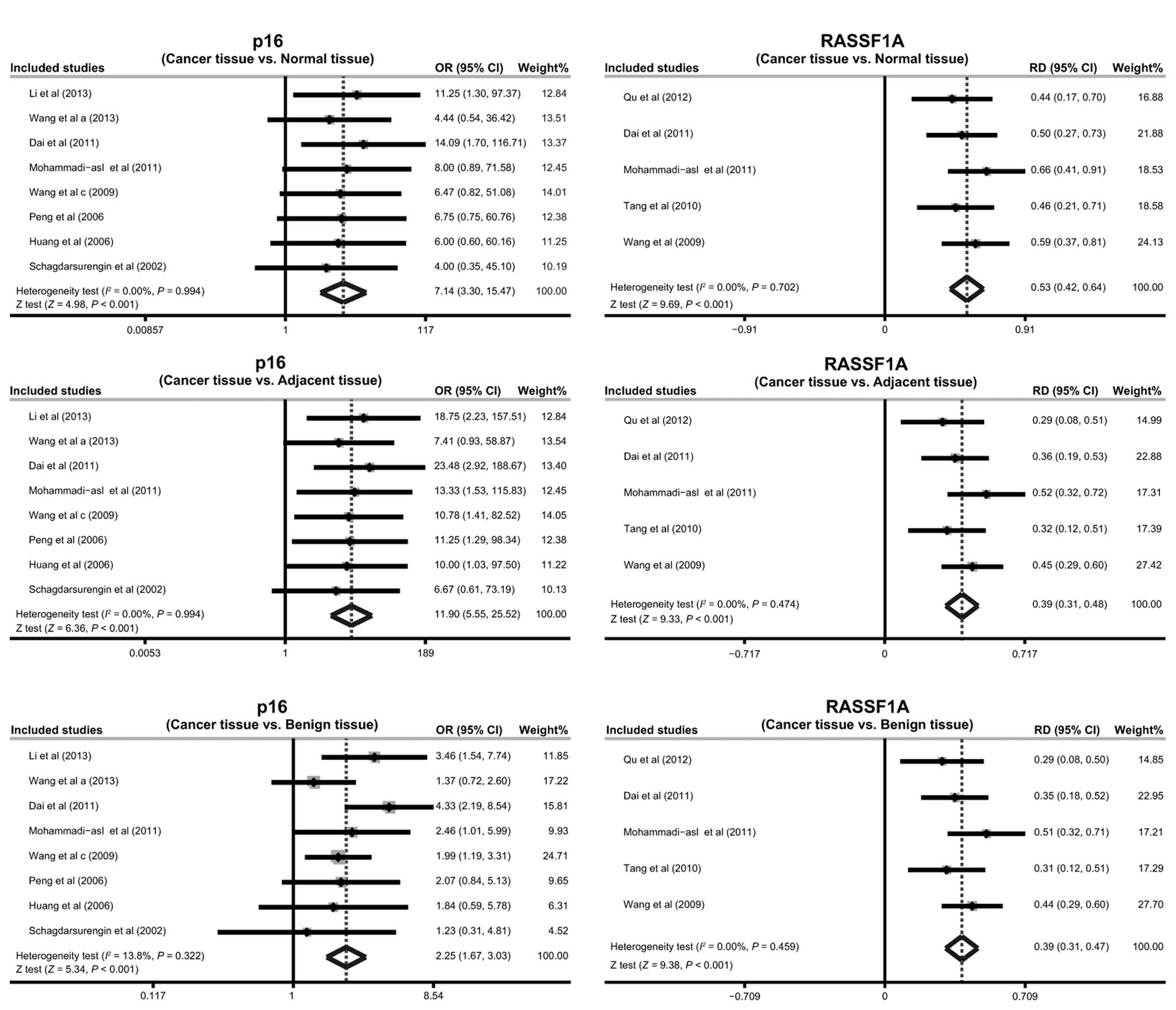
The results of the present meta-analysis indicate
that the frequency of promoter methylation of p16 in the cancer
tissues was significantly higher compared with that in the normal,
adjacent and benign tissues (cancer tissues vs. normal tissues:
OR=7.14; 95% CI, 3.30–15.47; P<0.001; cancer tissues vs.
adjacent tissues: OR=11.90; 95% CI, 5.55–25.52; P<0.001; cancer
tissues vs. benign tissues: OR=2.25, 95% CI: ~1.67–3.03,
P<0.001; respectively; Fig. 3).
The results also suggest that RASSF1A promoter methylation may be
implicated in the pathogenesis of PTC (cancer tissues vs. normal
tissues: RD=0.53; 95% CI, 0.42–0.64; P<0.001; cancer tissues vs.
adjacent tissues: RD=0.39; 95% CI, 0.31–0.48; P<0.001; cancer
tissues vs. benign tissues: RD=0.39; 95% CI, 0.31–0.47; P<0.001;
respectively; Fig. 3).
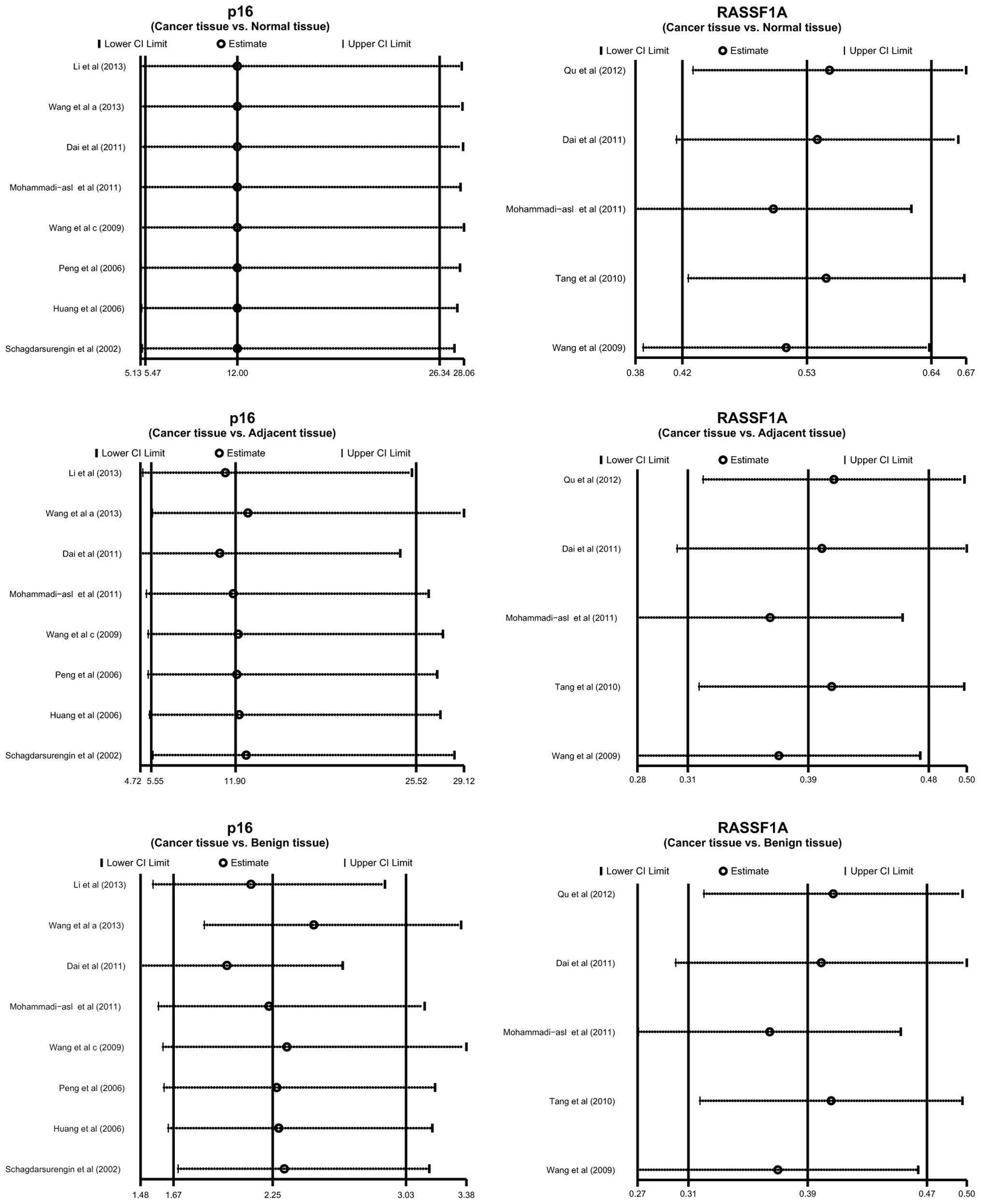
Sensitivity analysis revealed that no single study
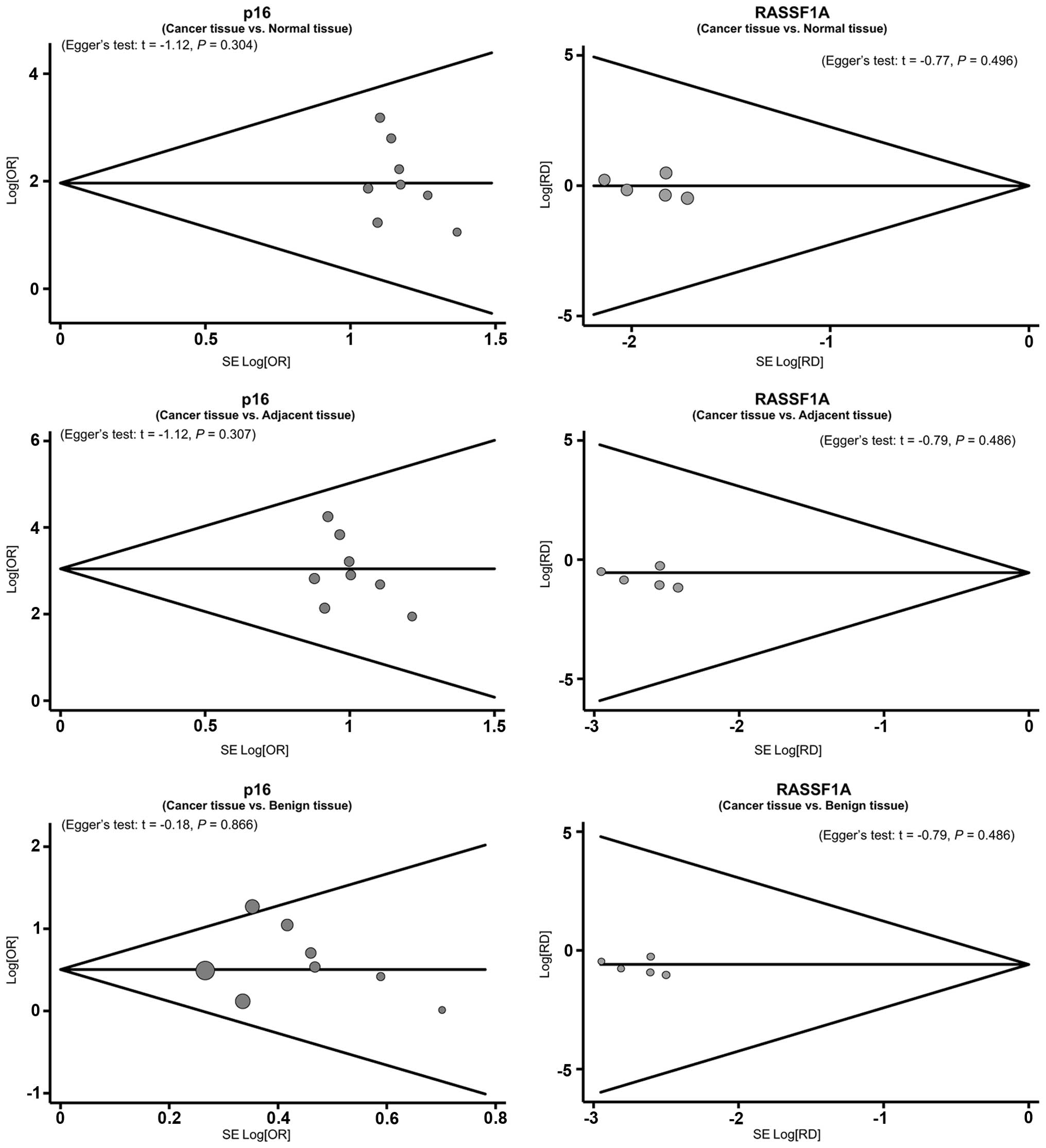
was able to influence the pooled ORs and RDs (Fig. 4). The funnel plots also demonstrated
no evidence of clear asymmetry (Fig.
5). Furthermore, Egger's test did not exhibit strong
statistical evidence for publication bias (all P>0.05).
Discussion
The present meta-analysis was carried out to
determine whether promoter methylation of p16 and RASSF1A is an
alternative mechanism for the development and progression of PTC.
In the current meta-analysis, promoter methylation of the p16 gene
was a frequent and early occurrence in benign to low/high
aggressive tumors in patients with PTC, indicating that the
methylation profile of p16 may play a significant role in the
pathogenesis of PTC. A potential explanation for this is that
promoter methylation of p16, an important tumor suppressor gene,
may result in transcriptional silencing and consequently, the loss
of p16 protein function which is believed to modulate the activity
of CDK (38). The CDK inhibitor p16
acts as a negative cell cycle regulator through binding to CDK4 and
CDK6, as well as inducing G1 phase arrest in the molecular
machinery of the cell cycle by interfering with cyclin D-CDK4
complexes (39). Therefore, it is
plausible that p16 promoter methylation may lead to its impaired
transcription, contributing to the loss of a designated regulatory
mechanism to block cell cycle progression and leading to the
uncontrolled growth of genetically damaged cells. The current
results are consistent with a previous study reporting that
functional repression of the p16 gene, resulting from promoter
methylation, plays a vital role in neoplastic progression and tumor
dedifferentiation in PTC (40).
The results of the current meta-analysis also
demonstrated that the frequency of promoter methylation of RASSF1A
in cancer tissues was significantly higher compared with that in
normal, adjacent and benign tissues. This implies that RASSF1A
promoter methylation may play a causative role in the malignancy of
PTC. Despite the precise role of RASSF1A promoter methylation in
the development and progression of PTC, its mechanism remains
unclear. It is hypothesized that the promoter methylation of
RASSF1A may contribute to its silencing and inactivation (41). RASSF1A regulates the activation of
RAS effector pathways and has been suggested to have a significant
function in the maintenance of genomic stability, cell cycle
control, the modulation of apoptosis and cell motility and invasion
(19,42). Furthermore, absent RASSF1A expression
is considered to be associated with a change in the functional
pathway through which RASSF1A may inhibit tumorigenesis (22). The results of a previous association
study also demonstrated that the RASSF1A promoter region was
methylated in 71% of patients with PTC (27). Thus, the results of the present study
are in accordance with previous studies in that they indicate that
promoter methylation of the p16 and RASSF1A genes may be implicated
in the pathogenesis of PTC. This suggests that p16 and RASSF1A
promoter methylation may be a candidate biomarker for the diagnosis
and prognosis of PTC.
The current meta-analysis has several limitations
that should be acknowledged. First, the results lacked sufficient
statistical power to assess the association between p16 and RASSF1A
promoter methylation and the pathogenesis of PTC due to the small
number of studies included. Since certain studies had small sample
sizes and standard deviations, it meant that the current
meta-analysis induced fairly wide confidence intervals that
restrained the confidence in the conclusions drawn. Furthermore,
the small number of studies may constrain the general applicability
of the results obtained and consequently the cognitive function of
the present meta-analysis should be regarded as preliminary.
Secondly, the meta-analysis was a retrospective study that may have
led to subject selection bias and thus had an impact on the
reliability of the results. Thirdly, the meta-analysis failed to
obtain original data from the included studies, which may have
limited the further evaluation of the potential role of p16 and
RASSF1A promoter methylation in the development of PTC. Although
the present study has several limitations, to the best of our
knowledge, it is the first meta-analysis focusing on the
association between p16 and RASSF1A promoter methylation and the
development of PTC. Furthermore, a highly sensitive literature
search strategy was performed of the electronic databases. A manual
search of the reference lists from the relevant studies was also
conducted to identify other potential studies. The selection
process to select eligible studies was based on strict inclusion
and exclusion criteria. Importantly, rigorous statistical analysis
of single nucleotide polymorphism (SNP) data provided a basis for
the pooling of information from individual studies.
In conclusion, promoter methylation of p16 and
RASSF1A genes may be implicated in the pathogenesis of PTC,
suggesting that the promoter methylation of p16 and RASSF1A may be
a candidate biomarker for the diagnosis and prognosis of PTC.
However, due to the limitations acknowledged above, further studies
with larger sample sizes and more detailed information are required
in order for a more representative and precise statistical analysis
to be possible.
Acknowledgements
The authors would like to acknowledge the reviewers
for their helpful comments on this study.
References
|
1
|
Sipos JA and Mazzaferri EL: Thyroid cancer
epidemiology and prognostic variables. Clin Oncol (R Coll Radiol).
22:395–404. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Swierniak M, Wojcicka A, Czetwertynska M,
et al: In-depth characterization of the microRNA transcriptome in
normal thyroid and papillary thyroid carcinoma. J Clin Endocrinol
Metab. 98:E1401–E1409. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jonklaas J, NoguerasGonzalez G, Munsell M,
et al: The impact of age and gender on papillary thyroid cancer
survival. J Clin Endocrinol Metab. 97:E878–E887. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
LiVolsi VA: Papillary thyroid carcinoma:
an update. Mod Pathol. 24 (Suppl 2):S1–S9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hughes DT, Haymart MR, Miller BS, Gauger
PG and Doherty GM: The most commonly occurring papillary thyroid
cancer in the United States is now a microcarcinoma in a patient
older than 45 years. Thyroid. 21:231–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Enewold L, Zhu K, Ron E, et al: Rising
thyroid cancer incidence in the United States by demographic and
tumor characteristics, 1980–2005. Cancer Epidemiol Biomarkers Prev.
18:784–791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Detours V, Delys L, Libert F, et al:
Genome-wide gene expression profiling suggests distinct radiation
susceptibilities in sporadic and post-Chernobyl papillary thyroid
cancers. Br J Cancer. 97:818–825. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xing M: Prognostic utility of BRAF
mutation in papillary thyroid cancer. Mol Cell Endocrinol.
321:86–93. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mohammadiasl J, Larijani B, Khorgami Z, et
al: Qualitative and quantitative promoter hypermethylation patterns
of the p16, TSHR, RASSF1A and RARβ2 genes in papillary thyroid
carcinoma. Med Oncol. 28:1123–1128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kunstman JW, Korah R, Healy JM, Prasad M
and Carling T: Quantitative assessment of RASSF1A methylation as a
putative molecular marker in papillary thyroid carcinoma. Surgery.
154:1255–1261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
AbouZeid AA, Azzam AZ and Kamel NA:
Methylation status of the gene promoter of cyclin-dependent kinase
inhibitor 2A (CDKN2A) in ovarian cancer. Scand J Clin Lab Invest.
71:542–547. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shidham VB, Mehrotra R, Varsegi G, et al:
p16INK4a immunocytochemistry on cell blocks as an
adjunct to cervical cytology: Potential reflex testing on specially
prepared cell blocks from residual liquid-based cytology specimens.
Cytojournal. 8:12011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
LutfulKabir FM, Agarwal P, Deinnocentes P,
et al: Novel frameshift mutation in the p16/INK4A tumor suppressor
gene in canine breast cancer alters expression from the
p16/INK4A/p14ARF locus. J Cell Biochem. 114:56–66. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rabien A, SanchezRuderisch H, Schulz P, et
al: Tumor suppressor p16 INK4a controls oncogenic K-Ras function in
human pancreatic cancer cells. Cancer Sci. 103:169–175. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kamb A, ShattuckEidens D, Eeles R, et al:
Analysis of the p16 gene (CDKN2) as a candidate for the chromosome
9p melanoma susceptibility locus. Nat Genet. 8:23–26. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang P, Pei R, Lu Z, Rao X and Liu B:
Methylation of p16 CpG islands correlated with metastasis and
aggressiveness in papillary thyroid carcinoma. J Chin Med Assoc.
76:135–139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tian Y, Hou Y, Zhou X, Cheng H and Zhou R:
Tumor suppressor RASSF1A promoter: p53 binding and methylation.
PLoS One. 6:e170172011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
CunhaDda S, Simões MI, Viviani DN, et al:
Carotid artery rupture following radioiodine therapy for
differentiated thyroid carcinoma. Arq Bras Endocrinol Metabol.
55:419–425. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao T, Wang S, He B, et al: The
association of RAS association domain family Protein1A (RASSF1A)
methylation states and bladder cancer risk: a systematic review and
meta-analysis. PLoS One. 7:e483002012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dreijerink K, Braga E, Kuzmin I, et al:
The candidate tumor suppressor gene, RASSF1A, from human chromosome
3p21.3 is involved in kidney tumorigenesis. Proc Natl Acad Sci USA.
98:7504–7509. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yee KS, Grochola L, Hamilton G, et al: A
RASSF1A polymorphism restricts p53/p73 activation and associates
with poor survival and accelerated age of onset of soft tissue
sarcoma. Cancer Res. 72:2206–2217. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakamura N, Carney JA, Jin L, et al:
RASSF1A and NORE1A methylation and BRAFV600E mutations in thyroid
tumors. Lab Invest. 85:1065–1075. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dammann R, Schagdarsurengin U, Seidel C,
et al: The tumor suppressor RASSF1A in human carcinogenesis: an
update. Histol Histopathol. 20:645–663. 2005.PubMed/NCBI
|
|
24
|
vanVlodrop IJ, Niessen HE, Derks S, et al:
Analysis of promoter CpG island hypermethylation in cancer:
location, location, location! Clin Cancer Res. 17:4225–4231. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Santoro A, Pannone G, Carosi MA, et al:
BRAF mutation and RASSF1A expression in thyroid carcinoma of
southern Italy. J Cell Biochem. 114:1174–1182. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang P, Li DX and Zhang Y: Methylation of
p16 gene and expression of p16 protein in human thyroid neoplasms.
J Practical Oncol. 21:49–51. 2006.(In Chinese).
|
|
27
|
Schagdarsurengin U, Gimm O, HoangVu C, et
al: Frequent epigenetic silencing of the CpG island promoter of
RASSF1A in thyroid carcinoma. Cancer Res. 62:3698–3701.
2002.PubMed/NCBI
|
|
28
|
Stang A: Critical evaluation of the
Newcastle-Ottawa scale for the assessment of the quality of
nonrandomized studies in meta-analyses. Eur J Epidemiol.
25:603–605. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zintzaras E and Ioannidis JP: HEGESMA:
genome search meta-analysis and heterogeneity testing.
Bioinformatics. 21:3672–3673. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peters JL, Sutton AJ, Jones DR, Abrams KR
and Rushton L: Comparison of two methods to detect publication bias
in meta-analysis. JAMA. 295:676–680. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dai YL, Zhang F, Jiang ZR, et al:
Association the methylation of p16 and RASSF1A with papillary
thyroid carcinoma. Chin J Int Med. 50:428–429. 2011.
|
|
32
|
Li XF, Jin YL, He ZL and Gu DK: Detecting
the abnormal methylation of plasma p16 promoter in patients with
thyroid carcinoma by nested-methylation-specific polymerase chain
reaction. Chin J Prev Contr Chronic Dis. 21:34–36. 2013.(In
Chinese).
|
|
33
|
Peng ZL, Cao RX, Wen GB, Liu JH and Wen F:
The methylation of p16 gene in papillary thyroid carcinoma. J Mod
Oncol. 14:1501–1503. 2006.(In Chinese).
|
|
34
|
Qu F and Xue WJ: RASSF1A methylation and
its clinical roles in papillary thyroid carcinoma. J Nantong Univ
(Med Sci). 32:490–492. 2012.(In Chinese).
|
|
35
|
Tang JD and Su XL: Research of CpG island
methylation status of NIS and RASSF1A gene promoters in papillary
thyroid carcinomas. China J Mod Med. 20:3282–3285. 2010.(In
Chinese).
|
|
36
|
Wang C, Shi JH and Wang WY: Expressions of
p53 and p16 and their clinical significance in thyroid papillary
carcinoma. J Bengbu Med Col. 34:871–873. 2009.(In Chinese).
|
|
37
|
Wang XH, Zhang GC, Liu YL, et al:
Detecting the abnormal methylation of RASSF1A in patients with
thyroid carcinoma. Shaanxi Med J. 38:790–792. 2009.(In
Chinese).
|
|
38
|
Barroeta JE, Baloch ZW, Lal P, et al:
Diagnostic value of differential expression of CK19, Galectin-3,
HBME-1, ERK, RET, and p16 in benign and malignant
follicular-derived lesions of the thyroid: an immunohistochemical
tissue microarray analysis. Endocr Pathol. 17:225–234. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lam AK, Lo CY, Leung P, et al:
Clinicopathological roles of alterations of tumor suppressor gene
p16 in papillary thyroid carcinoma. Ann Surg Oncol. 14:1772–1779.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Boltze C, Zack S, Quednow C, et al:
Hypermethylation of the CDKN2/p16INK4A promotor in thyroid
carcinogenesis. Pathol Res Pract. 199:399–404. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xing M, Cohen Y, Mambo E, et al: Early
occurrence of RASSF1A hypermethylation and its mutual exclusion
with BRAF mutation in thyroid tumorigenesis. Cancer Res.
64:1664–1668. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fernandes MS, Carneiro F, Oliveira C and
Seruca R: Colorectal cancer and RASSF family - a special emphasis
on RASSF1A. Int J Cancer. 132:251–258. 2013. View Article : Google Scholar : PubMed/NCBI
|