Introduction
Osteosarcoma (OS) is the most common primary bone
malignancy in children and young adults. Mortalities due to
malignant neoplasms of the bone and joint represent 8.9% of all
childhood and adolescent cancer mortalities (1).
Transcription factors (TFs) and microRNAs (miRNAs)
are prominent regulators of gene expression (2). In molecular biology, TFs are proteins
that are able to regulate the transcription of genes by the way of
promotion or suppression (3). TFs
perform this regulatory function either by binding to the upstream
regions of genes alone or through coregulation with other proteins.
miRNAs are endogenous, non-coding RNA molecules, which regulate
gene expression at a transcriptional level (4). miRNAs play a significant role in
numerous biological functions and have been shown to control key
cellular processes, including proliferation and apoptosis (5). Several miRNAs have been found to have
links with various types of cancer, and miRNAs often exhibit
aberrant expression in cancer (6,7).
The genes in which miRNAs are located are known as
host genes. The expression of the majority of miRNAs is correlated
with the expression of their host genes (8). Rodriguez et al (9) demonstrated that miRNAs are transcribed
in parallel with their host transcripts and that the two different
transcription classes of miRNA (exonic and intronic) that have been
identified may require slightly different mechanisms of
biogenesis.
The genes that are targeted by miRNAs are referred
to as target genes (targets). A number of databases supply
sufficient information to enable the association between the miRNAs
and their targets to be studied. Several transcription-profiling
studies of miRNA transfection experiments have shown that miRNAs
exert a widespread impact on the regulation of their targets
(10–13).
A number of experiments in the field of OS have been
conducted, leading to the theory that the cancer is caused by
abnormally expressed genes and miRNAs; however, the majority of
these experiments were conduced from a specific angle (gene or
miRNA), which limited the understanding of the pathogenesis of OS
from a broader perspective (14–19). The
aim of the present study was to focus on all the elements (genes,
miRNAs and TFs), instead of focusing on one or several of them.
Three types of experimentally validated associations exist between
the elements in OS, including miRNAs and their host genes, TFs and
miRNAs, and miRNAs and the corresponding targets. In the present
study, three regulatory networks consisting of an abnormally
expressed network, a related network and a global network, were
constructed in order to identify the mechanisms in OS and provide a
foundation for further research into the condition.
Materials and methods
Material collection and data
processing
The host genes of the human miRNAs were extracted
from the National Center for Biotechnology Information (NCBI) and
miRBase (20). The experimentally
validated dataset of human TFs and the miRNAs that were regulated
by them were extracted from TransmiR (21). The experimentally validated dataset
of human miRNAs and their targets were extracted from miRTarBase
(22) and Tarbase 5.0 (23). The official symbol from the NCBI
database, which can be accessed at http://www.ncbi.nlm.nih.gov/gene/, was used to
represent all miRNAs and genes.
The abnormally expressed genes of OS were collected
from the NCBI single nucleotide polymorphism database (http:www.ncbi.nlm.nih.gov/snp/), the Cancer
Genetics Web (http:www.cancerindex.org/geneweb/) and the relevant
literature. The related genes were collected from the GeneCards
database (24) and relevant
literature. In addition, the predicted TFs obtained by the P-Match
method were considered to be related genes. Only the TFs that
appeared in TransmiR were focused on. The P-Match method is a tool
to identify transcription factor binding sites (TFBSs) in
1,000-nucleotide (nt) promoter-region sequences and to map TFBSs
onto the promoter region of targets. P-Match combines
pattern-matching and weight-matrix approaches in order to provide a
higher accuracy of recognition than each of the methods alone
(25). The 1,000-nt promoter region
sequences of the targets of abnormally expressed miRNAs were
downloaded from the University of California Santa Cruz database
(26). Since P-Match uses the matrix
library as well as sets of aligned known TFBSs collected in
Transfac®, it facilitates a search for a large variety of different
TFBSs (25).
The abnormally expressed miRNAs were collected from
mir2Disease (27) and the relevant
literature. The OS-related miRNAs were also collected from the
relevant literature.
Construction of the three
networks
In this study, three regulatory networks of OS
(abnormally expressed, related and global) were constructed. The
abnormally expressed elements (genes and miRNAs) and the
associations between them were extracted to derive the abnormally
expressed network. The abnormally expressed network shows a number
of the critical data linkages in the progression of OS and acts as
a core network.
In order to construct the related network, the
related elements (genes and miRNAs) involved the pathogenesis of
OS, as well as the associations between them, were extracted. Four
subnets from the related network were selected to further elucidate
the pathogenic mechanism of OS. The abnormally expressed network
was included in the related network. All of the regulatory
associations in OS, with regard to the host genes, miRNAs, targets
and TFs, were subsequently extracted to form the global network,
which is a regulatory network that incorporates the abnormally
expressed and related networks.
Results
Abnormally expressed network of
OS
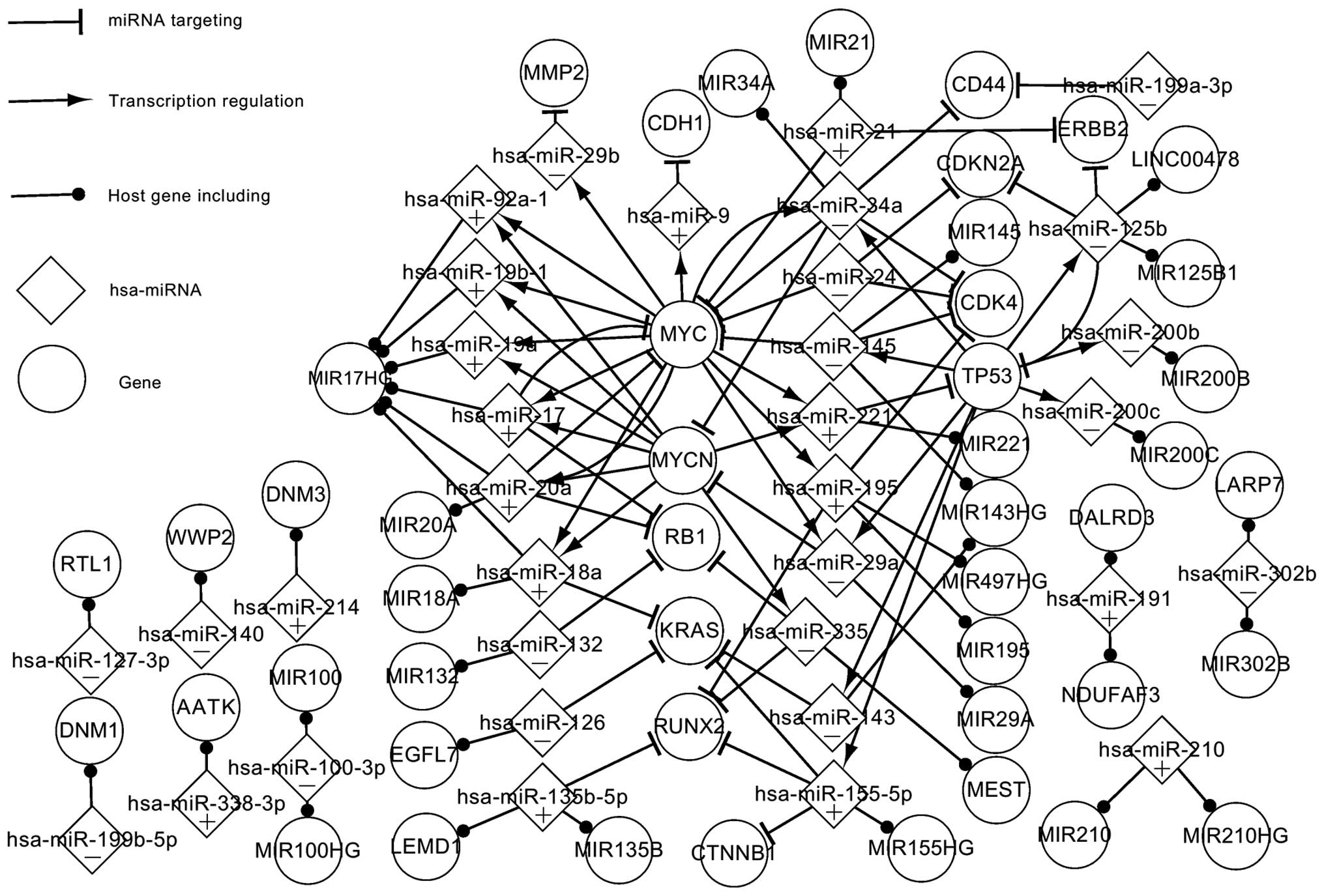
Fig. 1 shows numerous
crucial regulatory pathways and significant elements in OS. This
network is composed of 3 TFs, 13 targets, 34 miRNAs and 34 host
genes. All the TFs are part of the targets. All nodes are
abnormally expressed, with the exception of the host genes. The
main focus in Fig. 1 is on the parts
of the network that are closely related. The single nodes that do
not exhibit regulatory associations with other elements, such as
S100A4 and NRG1, have been omitted; however, the ignored elements
and pathways also have an effect on the development of OS.
The 3 TF-related pathways are the most significant
in Fig. 1. As can be seen from the
network, 6 miRNAs target MYC, which regulates 12 miRNAs; 2 miRNAs
target MYCN, which regulates 8 miRNAs; and 2 miRNAs target TP53,
which regulates 8 miRNAs. Certain data linkages exhibit special
features. TP53 and MYC, for example, regulate hsa-miR-29a and
hsa-miR-34a, which target MYCN; therefore, it is suggested that MYC
and TP53 are able to indirectly affect the expression of MYCN via
hsa-miR-29a and hsa-miR-34a. A number of regulatory circuits can
also be found in this network. For example, hsa-miR-20a targets MYC
and, in turn, hsa-miR-20a is regulated by MYC, suggesting a
self-adaptation association between the two. Notably, hsa-miR-24
targets MYC, CDK4 and CDKN2A but is not regulated by any genes,
suggesting that it may be the first element to act in the
abnormally expressed network. Similarly, MMP2, CDH1, RB1, RUNX2,
CDK4, KRAS, CD44, ERBB2, CDKN2A and CTNNB1 are targeted by certain
miRNAs but do not regulate any miRNAs, which suggests that they may
be the last active elements in the abnormally expressed network.
These elements play a critical role in cancer. For example, KRAS is
a typical proto-oncogene, which is implicated in various
malignancies, including lung adenocarcinoma, mucinous adenoma,
ductal carcinoma of the pancreas and colorectal carcinoma (28–34).
The host genes and their miRNAs exhibit certain
specific characteristics in the abnormally expressed network. The
host genes are considered as abnormally expressed genes due to the
abnormal expression of their miRNAs. Fig. 1 shows that a host gene may include
one or several miRNAs that are alone or together regulated by
specific genes. MIR17HG contains 6 miRNAs (hsa-miR-18a,
hsa-miR-19a, hsa-miR-19b-1, hsa-miR-20a, hsa-miR-92a-1 and
hsa-miR-17), which are regulated by MYC and MYCN. Among the 6
miRNAs, hsa-miR-17 and hsa-miR-20a separately form self-adaptation
associations with MYC. Furthermore, a single miRNA may be located
in several genes; hsa-miR-191, for example, is present in DALRD3
and NDUFAF3.
Fig. 1 shows certain
significant characteristics of the abnormally expressed genes and
miRNAs in OS. In theory, the adjustment of the abnormally expressed
data to a normal level could enable the control of the OS. The
abnormally expressed network partially elucidates the pathogenesis
of OS.
Related network of OS
The abnormally expressed network is contained within
the related network. In addition to the abnormally expressed data,
numerous related elements are shown. The network contains
information on an increased number of regulatory associations among
the genes and miRNAs. Four subnets were selected from the related
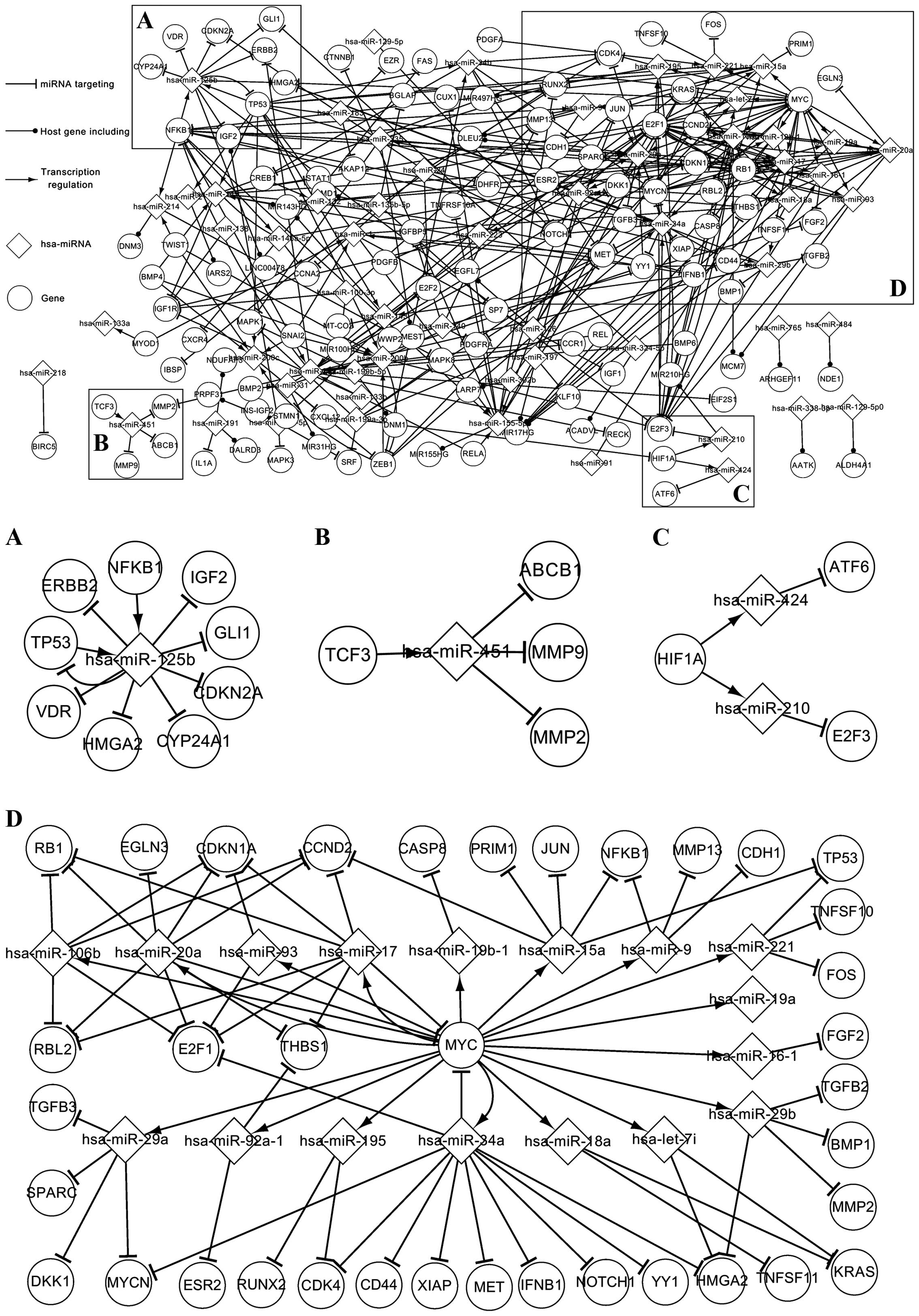
network in order to describe the network more clearly. Fig. 2A centers on the association between
hsa-miRNA-125b and other elements. As an abnormally expressed
miRNA, hsa-miRNA-125b is regulated by an abnormally expressed TF
(TP53) and a related TF (NFKB1), while hsa-miRNA-125b targets 8
genes (CDKN2A, TP53, GLI1, CYP24A1, IGF2, HMGA2, VDR and ERBB2).
Among these genes, CDKN2A, TP53 and ERBB2 are abnormally expressed
genes, whereas the others are not. TP53 and hsa-miRNA-125b form a
self-adaptation association. Fig. 2B
is composed of 1 TF, 3 targets and 1 miRNA. It can be seen that
TCF3, which is a related TF, regulates hsa-miRNA-451, targeting 3
genes (MMP2, MMP9 and ABCB1). Among these genes, MMP2 is an
abnormally expressed gene and the others are not. Fig. 2C is a subnet around the regulatory
associations between HIF1A and other elements. As can be seen from
Fig. 2C, HIF1A regulates hsa-miR-210
and hsa-miR-424; hsa-miR-210 targets E2F3 and hsa-miR-424 targets
ATF6. Fig. 2D shows MYC, which
regulates 17 miRNAs, which, in turn, target 36 genes. Three
miRNA-MYC pairs form a self-adaptation association in Fig. 2D: hsa-miR-20a and MYC, hsa-miR-17 and
MYC, and hsa-miR-34a and MYC. In addition, hsa-miR-106b,
hsa-miR-17, hsa-miR-20a and hsa-miR-93 target CDKN1A and E2F1;
hsa-miR-106b, hsa-miR-17 and hsa-miR-20a target RB1 and RBL2; and
hsa-miR-17, hsa-miR-20a and hsa-miR-92a-1 target THBS1. Fig. 2 demonstrates that the abnormally
expressed and related factors interact with each other to affect
the OS network. The related network expands the additional
topological associations of the abnormally expressed elements and
partially elucidates the mechanism of OS.
Global network of OS
The global network includes the abnormally expressed
and related networks. All the elements and associations of the
global network have been experimentally validated. Due to the
complexity and size of the network, it is difficult to provide a
clear description of the findings; therefore, the network will not
be described in detail but will instead be utilized as a tool
throughout the remainder of the study.
Transcriptional network of predicted
TFs
Predicted TFs are TFs that are believed to be
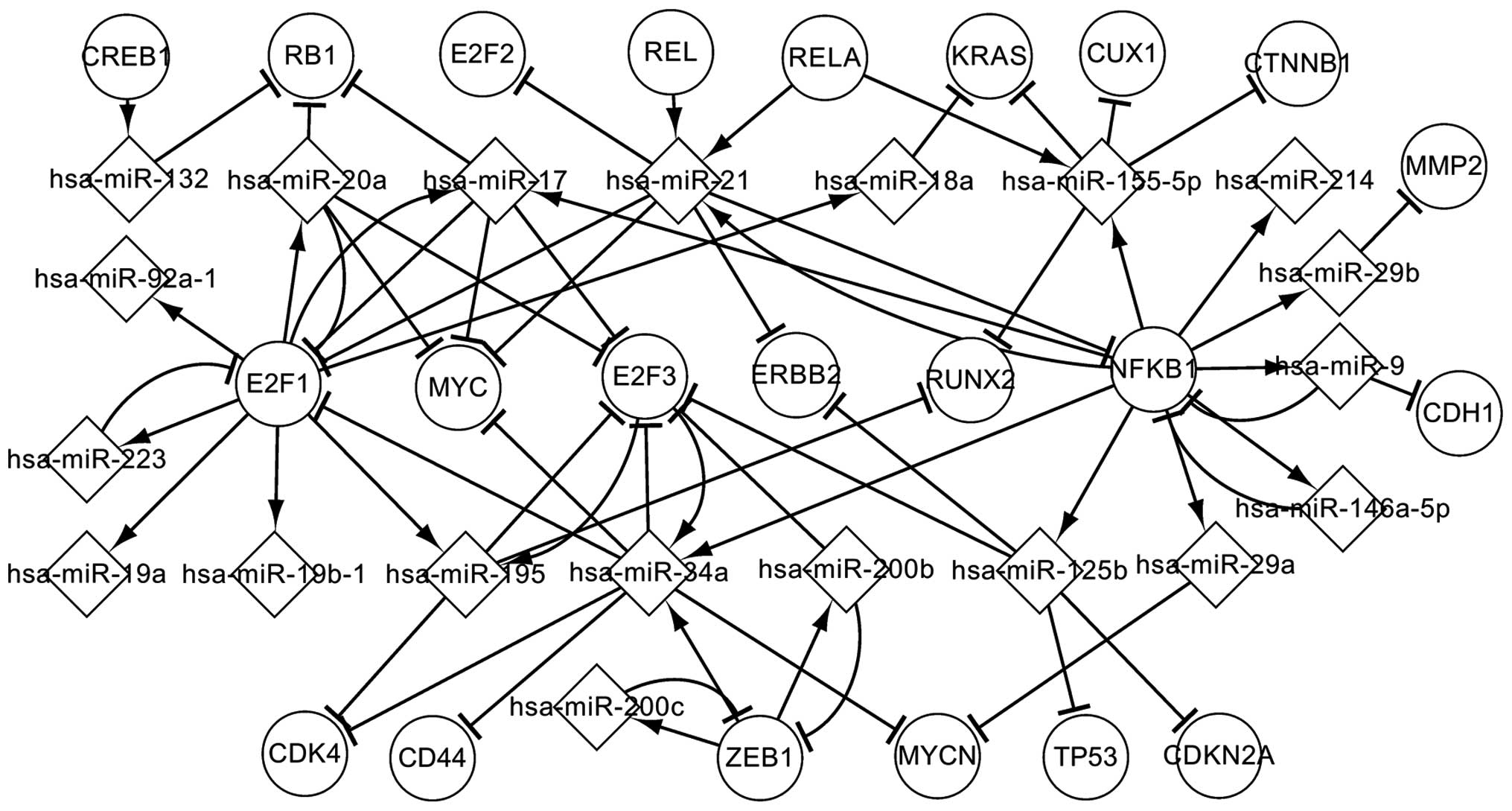
involved in the transcriptional processes of OS. Fig. 3 depicts the regulatory interactions
between these predicted TFs and the abnormally expressed miRNAs, in
addition to the targets, in OS. These factors influence their
successors by regulating or targeting them in the transcriptional
network. As shown in Fig. 3, NFKB1,
E2F1, E2F3 and ZEB1 are more significant than the other TFs.
Specifically, NFKB1, E2F3 and ZEB1 coregulate hsa-miR-34a, which
targets E2F1, CDK4, CD44, MYC, E2F3 and MYCN. E2F3 and hsa-miR-34a
form a self-adaptation association. As can be observed, NFKB1
regulates 10 miRNAs (hsa-miR-125b, hsa-miR-146a-5p, hsa-miR-155-5p,
hsa-miR-21, hsa-miR-214, hsa-miR-29a, hsa-miR-29b, hsa-miR-34a,
hsa-miR-9 and hsa-miR-17) and is targeted by 3 miRNAs
(hsa-miR-146a-5p, hsa-miR-21 and hsa-miR-9). These 3 miRNAs
separately form self-adaptation associations with NFKB1. The other
TFs are similar to NFKB1. In Fig. 3,
it can be observed that a single TF may regulate one or several
abnormally expressed miRNAs, and an abnormally expressed miRNA may
target one or several TFs. A TF indirectly affects other TFs via
specific abnormally expressed miRNAs, and an abnormally expressed
miRNA indirectly influences other miRNAs via certain TFs. The
predicted TFs may further reveal the transcriptional mechanism
associated with OS.
Regulatory associations involving
abnormally expressed genes
The upstream and downstream information of the
predicted TFs and data on the abnormally expressed genes and miRNAs
were collected to describe the network of OS more clearly. The
abnormally expressed genes were initially concentrated on.
‘Upstream’ refers to miRNAs that target abnormally expressed genes,
and ‘downstream’ refers to miRNAs that are regulated by abnormally
expressed genes in the three networks. The abnormally expressed
genes were divided into three classes by extracting and classifying
all the regulatory associations involving the abnormally expressed
genes in the three networks. The first class of gene included genes
with only upstream, but no downstream, elements, such as CD44. CD44
is targeted by a number of miRNAs in the three networks but does
not regulate any miRNAs. The second class of gene included genes
with only downstream, but no upstream, elements, such as CDKN2A.
CDKN2A regulates hsa-miR-410 in the global network but is not
targeted by any miRNAs. The third class of gene had both upstream
and downstream elements. MYC is considered as an example and is
discussed in the following section.
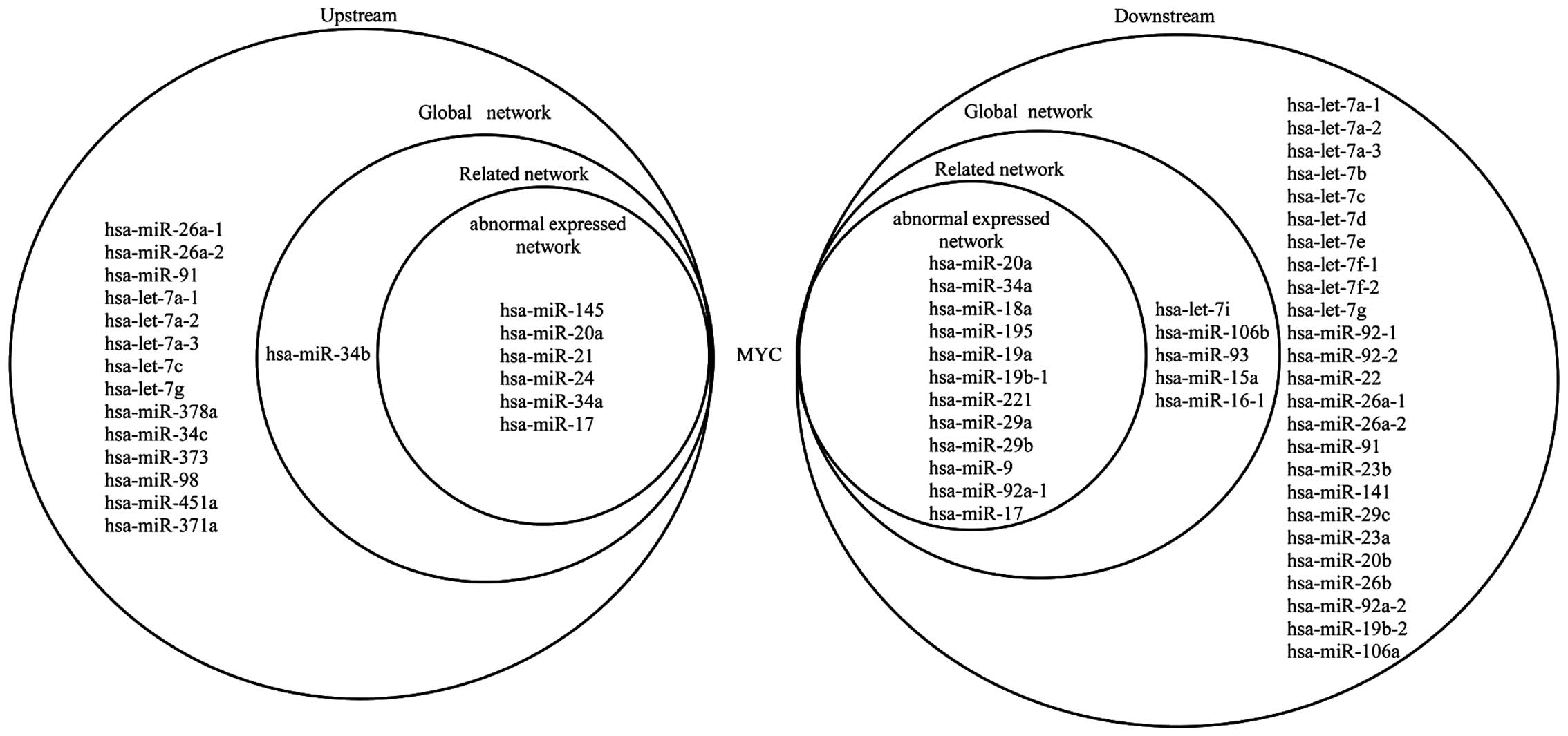
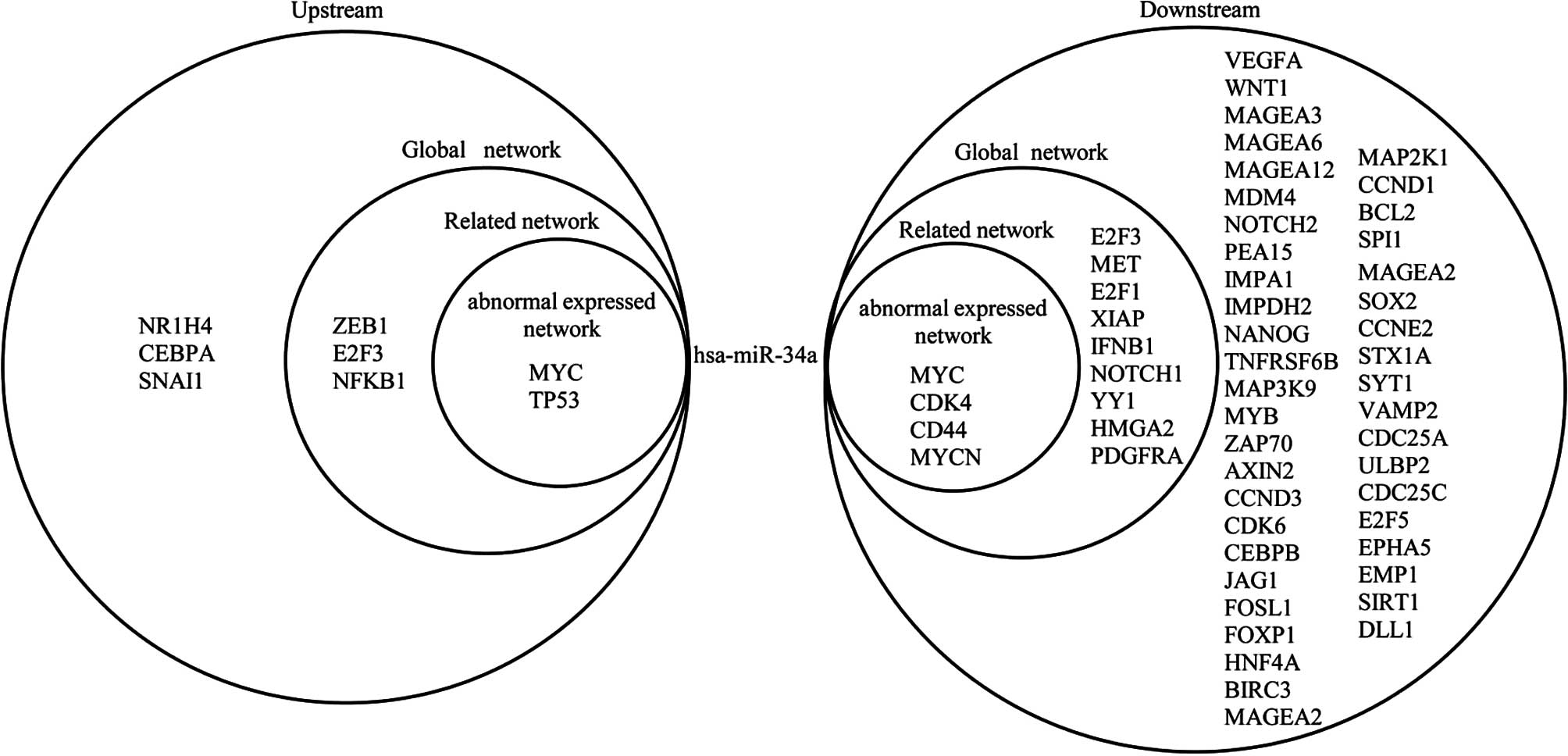
Fig. 4 shows the
upstream and downstream elements of MYC in the three networks. The
figure shows that 6 miRNAs target MYC and MYC regulates 12 miRNAs
in the abnormally expressed network; 7 miRNAs target MYC and MYC
regulates 17 miRNAs in the related network; and 21 miRNAs target
MYC and MYC regulates 42 miRNAs in the global network. The elements
upstream of MYC indirectly affect the elements downstream of MYC.
hsa-miR-20a, hsa-miR-34a and hsa-miR-17 target MYC and are
regulated by MYC in return, which forms a self-adaptation
association in the three networks. Furthermore, MYC indirectly
affects the expression of other genes via a number of miRNAs. For
example, MYC regulates hsa-miR-20a, which targets RB1. Certain
genes can also indirectly influence MYC through specific miRNAs.
For example, TP53 regulates hsa-miR-145, which targets MYC in turn.
Other genes are similar to MYC.
Regulatory associations involving
abnormally expressed miRNAs
Similar to the abnormally expressed genes, the
upstream and downstream information of abnormally expressed miRNAs
was extracted. The upstream data show genes that regulate
abnormally expressed miRNAs, and the downstream data show targets
of the abnormally expressed miRNAs in the three networks. The
abnormally expressed miRNAs were divided into two classes: i)
Downstream elements only (no upstream elements), for example
hsa-miR-376c, which targets ACVR1C in the global network and is not
regulated by any genes; ii) both upstream and downstream elements.
The subsequent experiments focused on hsa-miR-34a.
Fig. 5 shows the
upstream and downstream elements of hsa-miR-34a in the three
networks. In the abnormally expressed network, 2 genes regulate
hsa-miR-34a, which, in turn, targets 4 genes. In the related
network, 5 genes regulate hsa-miR-34a, which, in turn, targets 13
genes. In the global network, 8 genes regulate hsa-miR-34a, which
targets 56 genes. As can be noted from Fig. 5, MYC regulates hsa-miR-34a and
hsa-miR-34a targets MYC in return, which forms a self-adaptation
association in the three networks. In addition, E2F3 regulates
hsa-miR-34a and hsa-miR-34a targets E2F3 in return, which forms a
self-adaptation association in the related and global networks. In
addition to this, hsa-miR-34a indirectly affects other miRNAs via
certain genes: hsa-miR-34a, for example, targets MYC, which
regulates hsa-miR-17. A number of miRNAs also indirectly influence
hsa-miR-34a by targeting specific genes. For example, hsa-miR-221
targets TP53, but regulates hsa-miR-34a.
Regulatory associations involving
predicted TFs
The same method was applied to the predicted TFs.
The upstream data show miRNAs that target the predicted TFs in the
three networks and the downstream data show miRNAs that are
regulated by the predicted TFs in the three networks. The predicted
TFs were divided into three classes. The first class of predicted
TFs included TFs with only upstream, not downstream, elements, such
as STAT1. STAT1 is targeted by hsa-miR-145 in the three networks
but does not regulate any miRNAs. By contrast, the second class of
predicted TFs included TFs with no upstream, only downstream,
elements, such as REL. REL regulates hsa-miR-21 in the three
networks but is not targeted by any miRNAs. The third class
included TFs with both upstream and downstream elements; E2F1 is
taken as an example.
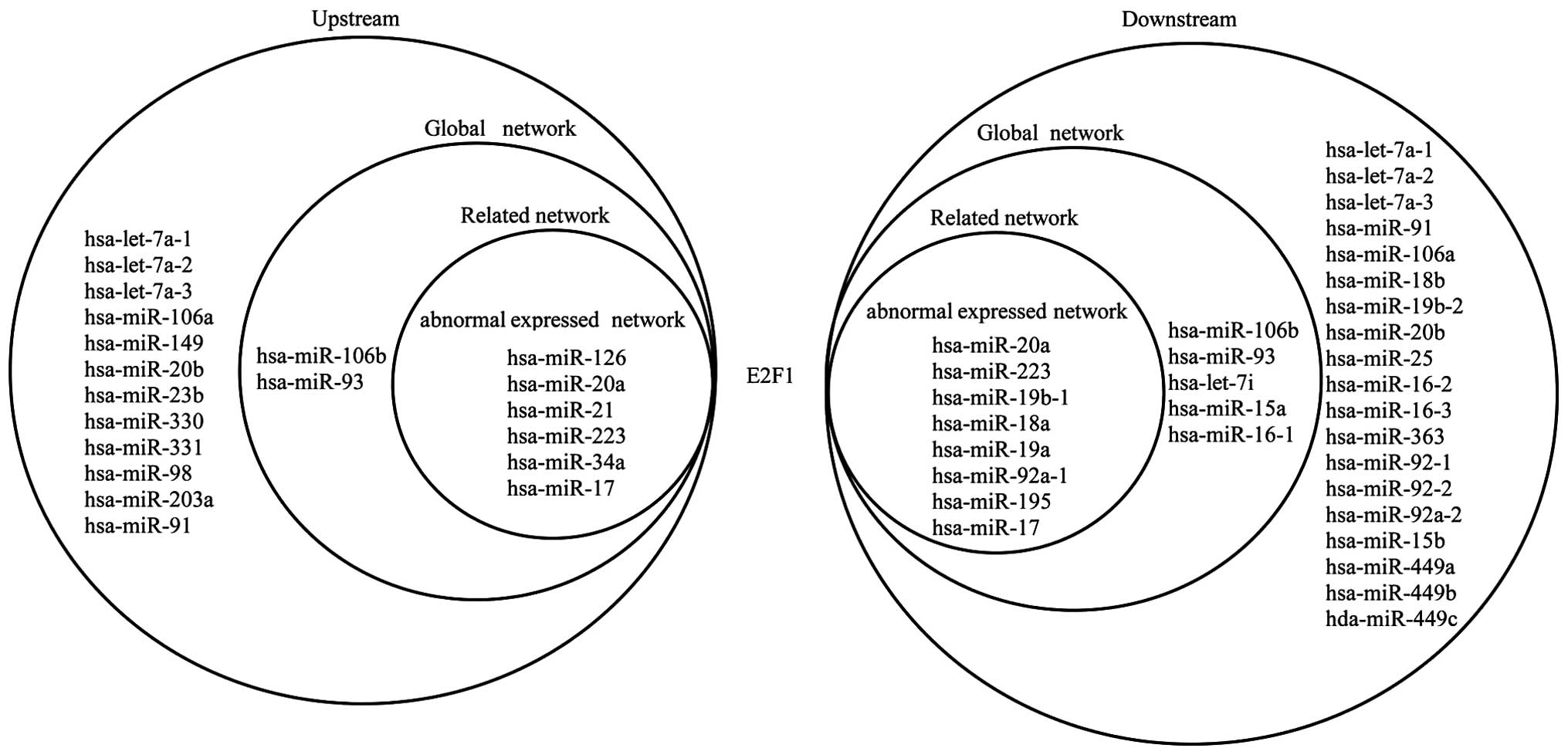
Fig. 6 shows the
upstream and downstream elements of E2F1 in the three networks. Six
miRNAs are shown to target E2F1, which regulates 8 miRNAs in the
abnormally expressed network; 8 miRNAs target E2F1, which regulates
13 miRNAs in the related network; and 20 miRNAs target E2F1, which
regulates 32 miRNAs in the global network. It can also be noted
that hsa-miR-106b and hsa-miR-93 separately form self-adaptation
associations with E2F1 in the related and global networks.
hsa-miR-20a, hsa-miR-17 and hsa-miR-223 separately form
self-adaptation associations with E2F1 in the three networks. E2F1
is not abnormally expressed in OS, but the 3 miRNAs are abnormally
expressed; therefore, these 3 miRNAs may indirectly lead to the
aberrant expression of other miRNAs via E2F1.
Discussion
By analyzing the current data associated with OS,
three regulatory networks at different levels (the abnormally
expressed, related and global networks) were derived. This study
mainly focused on the abnormally expressed network in comparison
with the related and global networks. A number of significant
regulatory associations involving the three types of elements
(abnormally expressed genes, abnormally expressed miRNAs and TFs)
were found in the abnormally expressed network. Certain data
linkages that exert key biological functions in OS were highlighted
in the abnormally expressed network, such as
MYC-hsa-miR-34a-MYC-hsa-miR-335-RUNX2. These data linkages of
elements mutated in OS are critical to the development of an
enhanced understanding of the molecular pathogenesis of this
disease.
Certain data linkages are involved in processes
associated with other types carcinoma, as well as OS. In breast
tumors, for example, TP53 drives invasion through the upregulation
of hsa-miR-155 (35), which suggests
that the interactions of genes can be expanded from one cancer to
another. Furthermore, the abnormal expression of miRNAs and genes
is considered to determine the development, metastasis and therapy
of OS. The abnormally expressed network constructed in the present
study, which is based on experimentally validated data, shows
numerous incorrect signaling pathways that occur in the human body
when OS emerges; therefore, this network represents a core
misregulation network and could be used to treat those patients
suffering from OS through the regulation of the expression of
abnormal factors to normal levels. Furthermore, the network could
be used in the prevention of OS. The adjustment of the abnormally
expressed elements and incorrect linkages formed by these elements
to a normal state could, in theory, facilitate the control or even
the cure of the cancer.
In the present study, the related network, which
contributes to the understanding of numerous processes associated
with OS, and the global network, in which all the data have been
experimentally validated, were additionally constructed.
Particularly notable are the TFs predicted from the P-Match method,
as they may reveal the pathogenesis of OS. These results and the
comprehensive data may lead biologists to the further study of the
mechanisms associated with carcinogenicity and the therapy of OS,
which may result in improvements in the prognosis, diagnosis and
therapy of OS. The present study may provide novel insights into
cancer biology.
Acknowledgements
This study was supported by the Science and
Technology Development Plan of Jilin Province (grant no.
20130101166JC).
Abbreviations:
|
OS
|
osteosarcoma
|
|
miRNA
|
microRNA
|
|
TFs
|
transcription factors
|
|
NCBI
|
National Center for Biotechnology
Information
|
|
TFBSs
|
transcription factor binding sites
|
References
|
1
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hobert O: Gene regulation by transcription
factors and microRNAs. Science. 319:1785–1786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tran DH, Satou K, Ho TB and Pham TH:
Computational discovery of miR-TF regulatory modules in human
genome. Bioinformation. 4:371–377. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yue J and Tigyi G: MicroRNA trafficking
and human cancer. Cancer Biol Ther. 5:573–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McManus MT: MicroRNAs and cancer. Semin
Cancer Biol. 13:253–258. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baskerville S and Bartel DP: Microarray
profiling of microRNAs reveals frequent coexpression with
neighboring miRNAs and host genes. RNA. 11:241–247. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rodriguez A, GriffithsJones S, Ashurst JL
and Bradley A: Identification of mammalian microRNA host genes and
transcription units. Genome Res. 14:1902–1910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Naeem H, Küffner R and Zimmer R: MIRTFnet:
Analysis of miRNA regulated transcription factors. PLoS One.
6:e225192011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Selbach M, Schwanhäusser B, Thierfelder N,
Fang Z, Khanin R and Rajewsky N: Widespread changes in protein
synthesis induced by microRNAs. Nature. 455:58–63. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lim LP, Lau NC, GarrettEngele P, Grimson
A, Schelter JM, Castle J, Bartel DP, Linsley PS and Johnson JM:
Microarray analysis shows that some microRNAs downregulate large
numbers of target mRNAs. Nature. 433:769–773. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang JC, Babak T, Corson TW, Chua G, Khan
S, Gallie BL, Hughes TR, Blencowe BJ, Frey BJ and Morris QD: Using
expression profiling data to identify human microRNA targets. Nat
Methods. 4:1045–1049. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sztán M, Pápai Z, Szendrôi M, Looij M and
Oláh E: Allelic losses from chromosome 17 in human osteosarcomas.
Pathol Oncol Res. 3:115–120. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choy E, Hornicek F, MacConaill L, Harmon
D, Tariq Z, Garraway L and Duan Z: High-throughput genotyping in
osteosarcoma identifies multiple mutations in
phosphoinositide-3-kinase and other oncogenes. Cancer.
118:2905–2914. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kansara M and Thomas DM: Molecular
pathogenesis of osteosarcoma. DNA Cell Biol. 26:1–18. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hong Q, Fang J, Pang Y and Zheng J:
Prognostic value of the microRNA-29 family in patients with primary
osteosarcomas. Med Oncol. 31:372014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Z and Cai H, Lin L, Tang M and Cai H:
Upregulated expression of microRNA-214 is linked to tumor
progression and adverse prognosis in pediatric osteosarcoma.
Pediatr Blood Cancer. 61:206–210. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lulla RR, Costa FF, Bischof JM, Chou PM,
de F Bonaldo M, Vanin EF and Soares MB: Identification of
differentially expressed microRNAs in osteosarcoma. Sarcoma.
2011:7326902011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kozomara A and Griffiths-Jones S: miRBase:
Integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39:D152–D157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Lu M, Qiu C and Cui Q: TransmiR: A
transcription factor-microRNA regulation database. Nucleic Acids
Res. 38:D119–D122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hsu SD, Lin FM, Wu WY, Liang C, Huang WC,
Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al: miRTarBase: A
database curates experimentally validated microRNA-target
interactions. Nucleic Acids Res. 39:D163–D169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Papadopoulos GL, Reczko M, Simossis VA,
Sethupathy P and Hatzigeorgiou AG: The database of experimentally
supported targets: A functional update of TarBase. Nucleic Acids
Res. 37:D155–D158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Safran M, Dalah I, Alexander J, Rosen N,
Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, et al:
GeneCards Version 3: The human gene integrator. Database (Oxford).
2010:baq0202010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chekmenev DS, Haid C and Kel AE: P-Match:
Transcription factor binding site search by combining patterns and
weight matrices. Nucleic Acids Res. 33:W432–W437. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fujita PA, Rhead B, Zweig AS, Hinrichs AS,
Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A,
et al: The UCSC genome browser database: Update 2011. Nucleic Acids
Res. 39:D876–D882. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang Q, Wang Y, Hao Y, Juan L, Teng M,
Zhang X, Li M, Wang G and Liu Y: miR2Disease: A manually curated
database for microRNA deregulation in human disease. Nucleic Acids
Res. 37:D98–D104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kranenburg O: The KRAS oncogene: Past,
present, and future. Biochim Biophys Acta. 1756:81–82.
2005.PubMed/NCBI
|
|
29
|
Marks JL, Broderick S, Zhou Q, Chitale D,
Li AR, Zakowski MF, Kris MG, Rusch VW, Azzoli CG, Seshan VE, et al:
Prognostic and therapeutic implications of EGFR and KRAS mutations
in resected lung adenocarcinoma. J Thorac Oncol. 3:111–116. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suda K, Tomizawa K and Mitsudomi T:
Biological and clinical significance of KRAS mutations in lung
cancer: an oncogenic driver that contrasts with EGFR mutation.
Cancer Metastasis Rev. 29:49–60. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hershkovitz D, Vlodavsky E, Simon E and
Ben-Izhak O: KRAS mutation positive mucinous adenocarcinoma
originating in mature ovarian teratoma: Case report and review of
literature. Pathol Int. 63:611–614. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yatsuoka T, Sunamura M, Furukawa T,
Fukushige S, Yokoyama T, Inoue H, Shibuya K, Takeda K, Matsuno S
and Horii A: Association of poor prognosis with loss of 12q, 17p,
and 18q, and concordant loss of 6q/17p and 12q/18q in human
pancreatic ductal adenocarcinoma. Am J Gastroenterol. 95:2080–2085.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Abubaker J, Bavi P, Al Haqawi W, Sultana
M, Al Harbi S, Al Sanea N, Abduljabbar A, Ashari LH, Alhomoud S, Al
Dayel F, et al: Prognostic significance of alterations in KRAS
isoforms KRAS-4A/4B and KRAS mutations in colorectal carcinoma. J
Pathol. 219:435–445. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Smakman N, Borel Rinkes IH, Voest EE and
Kranenburg O: Control of colorectal metastasis formation by K-Ras.
Biochim Biophys Acta. 1756:103–114. 2005.PubMed/NCBI
|
|
35
|
Neilsen PM, Noll JE, Mattiske S, Bracken
CP, Gregory PA, Schulz RB, Lim SP, Kumar R, Suetani RJ, Goodall GJ
and Callen DF: Mutant p53 drives invasion in breast tumors through
up-regulation of miR-155. Oncogene. 32:2992–3000. 2013. View Article : Google Scholar : PubMed/NCBI
|