Introduction
Acute lung injury (ALI), with acute respiratory
distress syndrome (ARDS) being its most severe manifestation, is a
common disease with hazardous effects on human health. Despite the
positive survival trends that have been reported in the past two
decades in patients with ALI/ARDS, the mortality rate remains at
30–50%, particularly among older patients who exhibit sepsis, which
is the most common predisposing factor (1). ALI/ARDS is partly characterized by
persistent, uncontrolled pulmonary inflammation, which occurs in
response to a variety of insults, including pneumonia, sepsis and
trauma (2). Epithelial cells and
macrophages comprise the primary line of defense, upon exogenous
insults in the lung. A cascade of events is triggered by the
injured cells, including acute inflammatory response, recruitment
of immune cells, including monocytes and macrophages, and release
of the cytokines, interleukin (IL)-6 and tumor necrosis factor-α
(TNF-α) (3). IL-6 and TNF-α are
transcriptional activators that is crucial to the activation of
several proinflammatory genes in human respiratory epithelial cells
(4,5). The NF-κB protein family consists of
homodimeric or heterodimeric subunits of members of the Rel family,
including p50 and p65. Functional NF-κB dimers always contain a p65
subunit and possess marked proinflammatory activity (6). Despite considerable research and an
increased understanding of the pathophysiological processes
involved in the pathogenesis of ALI (2,3), the
mechanism of the disease remains to be elucidated.
MicroRNA (miRNA or miR) molecules are small,
single-stranded, non-coding RNAs that typically bind to the 3′
untranslated region (3′UTR) of target mRNA sequences, which results
in reduced protein expression, mainly by destabilizing target mRNAs
and/or through the inhibition of translation (7,8). MiRNAs
have been found to play an important role in various biological
processes (9). Previous studies have
demonstrated that miRNAs are dynamically regulated in various human
diseases, including cardiovascular diseases (10,11) and
tumorigenesis (12,13). In addition, certain studies have
indicated that miRNAs are extensively involved in inflammation
(14–16). Changes in the expression levels of
certain miRNAs may be involved in the regulation of the
inflammatory process and tissue repair in ALI/ARDS (17). Cai et al revealed that the
levels of miR-16 were reduced in lipopolysaccharide (LPS)-induced
experimental ALI (18). In addition,
miR-16 treatment reduced the expression levels of the TNF-α and
IL-6 proinflammatory cytokines following exposure of macrophages to
LPS. Furthermore, Xie et al identified a decrease in the
pulmonary miR-127 expression in alveolar macrophages exposed to LPS
and in an animal model of noninfectious ALI (19). miR-127 treatment was also
demonstrated to reduce the IL-1β, TNF-α and IL-6 production in
macrophages that had been exposed to LPS, as well as to reduce the
lesion degree in an experimental ALI model in vivo.
Iliopoulos et al (20)
reported that, in ER-Src cells, miR-181b targets CYLD directly,
which results in an increased NF-κB activity and maintenance of the
inflammatory feedback loop underlying the epigenetic switch that
links inflammation to cancer. Therefore, the therapeutic targeting
of these miRNAs may be used as a way to suppress the inflammatory
response following ALI. However, the role of miRNAs in the
mediation of ALI has only recently been examined (18–20), and
requires further investigation.
The aim of the present study was to characterize the
regulation of the miRNA expression using LPS challenge. Through an
miRNA array-based screen, miR-181b was identified as a regulator of
BEAS-2B human bronchial epithelial cells using LPS challenge. The
study investigated the effect of LPS treatment on the expression of
miR-181b, as well as the association of miR-181b with the
expression levels of p65 and inflammation-associated cell factors,
such as IL-6, which are closely associated with ALI ARDS. In
addition, the role of miR-181b in ALI and its potential application
as a diagnostic and prognostic marker of the disease were
investigated (21).
Materials and methods
Reagents
Fetal calf serum was obtained from Gibco-BRL (Grand
Island NY, USA). The following materials were obtained from Qiagen
(Hilden, Germany): miScript miRNA Mimic syn-hsa-miR-181b; Pre-miR
miRNA negative control; QuantiTect primer assays; miScript II RT
kit; miScript SYBR Green PCR kit; and HiPerFect transfection
reagent. Pyrrolidine dithiocarbamate (PDTC), a specific inhibitor
of NF-κB, was purchased from Sigma-Aldrich (St. Louis, MO, USA).
The TNF-α and IL-6 ELISA kits were from MultiSciences Biotech Co.,
Ltd. (Hangzhou, China; cat. nos. 70-E-EK1061 and 70-E-EK1821,
respectively). The NE-PER extraction reagent and BCA protein assay
kit were from Pierce Chemical Co. (Rockford, IL, USA). Monoclonal
rabbit anti-human p65 antibodies (cat. no. 1546-s) were obtained
from Epitomics (Burlingame, CA, USA). Anti-β-actin monoclonal
antibodies (cat. no. sc-8432) were obtained from Santa Cruz
Biotechnology Inc. (Santa Cruz, CA, USA).
Cell culture
BEAS-2B cells were obtained from the Second
Affiliated Hospital of Zhejiang University School of Medicine
(Hangzhou, China) and cultured in RPMI 1640 supplemented with 10%
fetal calf serum and penicillin-streptomycin (100X; Gino Biomedical
Technology Co., Ltd., Hangzhou, China) in a humidified
CO2 incubator at 37°C. When the cells reached >85%
confluence, they were trypsinized (Gino Biomedical Technology Co.,
Ltd.) and subcultured. The cells were generally used between
passages 20–30 to avoid the generation of variation.
miRNA extraction and microarray
analysis
BEAS-2B cells were seeded into 6-well plates at a
density of 5×105 cells/well for a 24-h incubation prior
to LPS treatment. Duplicate wells were used as the controls (medium
only). The remaining wells were stimulated with 10 µg/ml LPS (the
LPS concentration was determined according to the pre-test and
references) (22) for 24 h.
Subsequently, the cells were harvested for various assays,
including RNA extraction, small RNA separation, quality control,
labeling, hybridization and scanning, which were performed by LC
Sciences LLC (Houston, TX, USA) using a Chip01_H16_070802 miRNA
array chip, based on Sanger miRBase release 16.0 (23,24)
(http://www.mirbase.org). Preliminary statistical
analysis was also performed by LC Sciences LLC on raw data
normalized using the locally-weighted regression method on the
background-subtracted data. The microarray used for the experiments
contained three probe replicates for each miRNA. A value of
P<0.01 between pixels was considered to indicate a statistically
significant difference.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA extraction and small RNA separation were
performed using the miRNeasy Mini kit (cat. no. 217004; Qiagen).
Cells and total RNA were treated as previously described. RT was
performed with 200 ng RNA using the miScript II RT kit for miRNA
transcription. RT-qPCR was performed using the miScript SYBR Green
PCR kit, according to the manufacturer's instruction. Amplification
and data analysis were performed using the 7500 Real Time PCR
system (Applied Biosystems Life Technologies, Foster City, CA,
USA). PCR cycling conditions were as follows: Inital activation at
95°C for 15 min, followed by 40 cycles of annealing at 94°C for 15
sec, annealing at 55°C for 30 sec and extension at 70°C for 35 sec.
QuantiTect primer assays were used for the determination of
miR-181b, miR-23c, miR-26b and Rnu6B expression. Values were
normalized to the level of Rnu6B expression (Qiagen).
PDTC treatment
BEAS-2B cells were seeded in 6-well plates
(3×105 cells/well). After 24 h, the cells were treated
with 50, 100 or 200 µM PDTC for 1.5 h.
Transfection
BEAS-2B cells, with or without PDTC treatment, were
transfected with miScript miRNA Mimic syn-hsa-miR-181b at 10 nmol/l
using HiPerFect transfection reagent. Negative control, LPS (10
µg/ml) were used as positive control for miR-181b overexpression.
The extent of miR-181b overexpression was measured by RT-qPCR. The
supernatants, as well as the adherent cells, were collected at 48 h
post-transfection for further analysis.
ELISA
Following transfection, the supernatants were
collected at various time points (0, 12, 24 and 48 h) by
centrifuged at 1000 × g for 10 min and then stored in −80°C. The
expression levels of TNF-α and IL-6 were measured using the
aforementioned commercial kits, according to the manufacturers'
instructions. All absorbance results were normalized according to
the standard curves.
Western blotting
For nuclear protein extraction, cells were lysed in
NE-PER extraction reagent according to the manufacturer's
instructions. Protein concentrations were determined using a BCA
protein assay kit. A total of 30 µg protein was then loaded and
electrophoresed on a 12% SDS-polyacrylamide gel, and transferred to
the nitrocellulose membrane. The membranes were subsequently probed
with anti-p65 (1:1,000 dilution) and anti-β-actin (1:1,000
dilution) monoclonal antibodies, respectively. The secondary
antibody used for detection was linked with horseradish peroxidase.
Next, an enhanced chemiluminescence method was used to detect the
conjugated horseradish peroxidase (EMD Millipore, Billerica, MD,
USA) and Image J (version 1.49) was used to analyze the
immunoblots.
Statistical analysis
Differences between groups were compared using the
Student's t-test for continuous variables. P<0.05 was considered
to indicate a statistically significant difference.
Results
Elevated levels of IL-6 in BEAS-2B
cells following LPS stimulation
LPS pretreatment has been previously used to model
inflammation in animal inhalation experiments (25). In addition, LPS is known to induce
the expression of proinflammatory cytokines, such as IL-6, in
BEAS-2B and A549 cells (22). In the
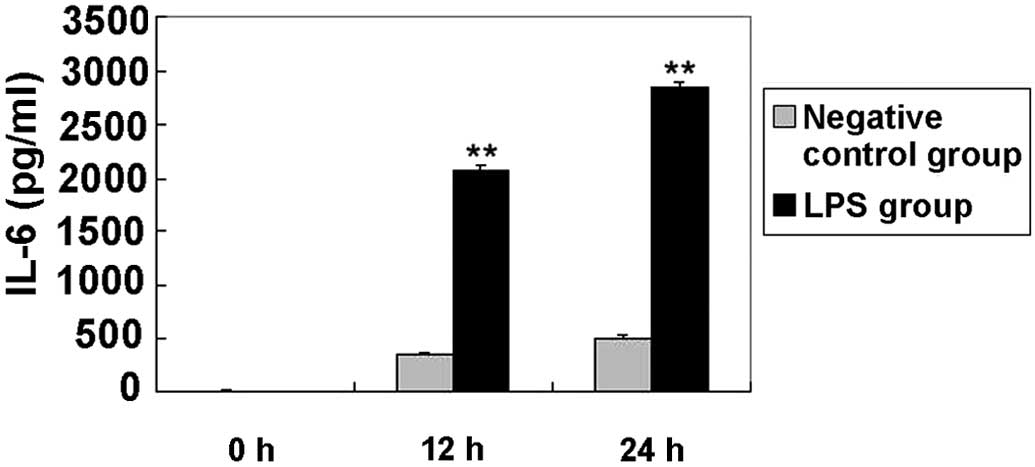
present study, the expression of IL-6 was initially examined
(Fig. 1). Compared with the negative
control, IL-6 expression was significantly increased in cells
treated with LPS at each time interval (P<0.01). Elevated levels
of this cytokine have been detected in patients with ARDS and shown
to be directly associated with the severity of lung inflammation
and mortality (26). In the current
study, TNF-α secretion was not detected in culture supernatants
from the negative control and syn-hsa-miR-181b groups. The lowest
TNF-α standards showed good reproducibility (3.8% coefficient of
variation), thus the limit of detection was <2.048 pg/ml.
Activation of miR-181b in response to
LPS stimulation in BEAS-2B cells
Although there is a poor understanding of the
underlying mechanisms of ALI, an enhanced inflammatory response is
known to be involved in this process and, at least partly,
contribute to the development of the disease (2,3).
Notably, miRNAs are emerging as new regulators of inflammatory and
immune responses (14). In order to
investigate the potential role of miRNAs in LPS-challenged BEAS-2B
cells, the miRNA expression profile was analyzed. Cells were
treated with or without 10 µg/ml LPS for 24 h and then the RNA
extraction was submitted for an miRNA chip assay
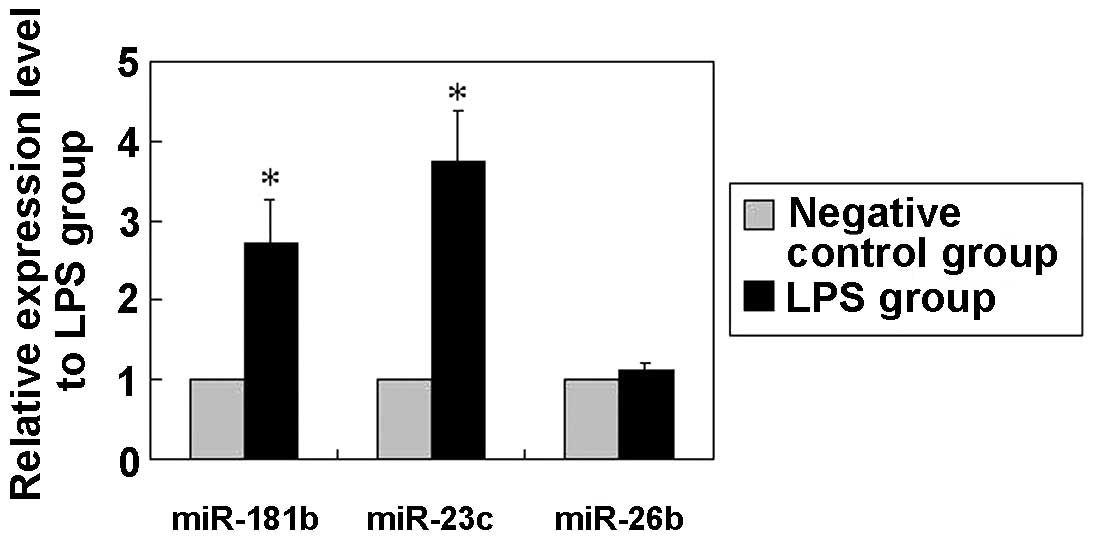
(miRHuman_16.0_070802 miRNA array). The profile analysis revealed
that the expression of 41 miRNAs, particularly that of miR-181b,
miR-23c and miR-26b, presented significant alterations in
LPS-treated cells (P<0.01; Table
I). The array results were further verified using qPCR, which
revealed that the expression of miR-181b, miR-23c and miR-26b was a
bona fide target of certain signaling pathways (Fig. 2). Of these potential candidates, the
focus was laid on miR-181b, since it was one of the most clearly
altered miRNAs and is known to be deregulated in inflammation,
although its function remains unclear (27,28). In
addition, miR-181b has recently been identified as a key player in
a positive feedback loop linking inflammation to an epigenetic
switch that controls cellular transformation in human mammary
epithelial MCF-10A cells (20). The
results of the current assay showed that the miR-181b expression
level in the BEAS-2B cells was <50% of that in the
LPS-stimulated BEAS-2B cells (Fig.
2), suggesting that miR-181b expression may be positively
correlated with the LPS-induced ALI model.
 | Table I.MicroRNA array analysis identified
that 41 miRNAs were in response to lipopolysaccharide stimulation
in human bronchial epithelial cells. |
Table I.
MicroRNA array analysis identified
that 41 miRNAs were in response to lipopolysaccharide stimulation
in human bronchial epithelial cells.
| No. | Probe_ID | Sample A signal
(prestimulation) | Sample B signal
(poststimulation) | Log2 (Sample
B/Sample A) |
|---|
| 1 |
hsa-miR-3613-3p |
206.97 |
520.07 | 1.27 |
| 2 | hsa-miR-335 |
338.68 |
853.34 | 1.25 |
| 3 | hsa-miR-26b |
638.32 | 1,490.33 | 1.20 |
| 4 | hsa-miR-23c |
778.64 | 1,834.46 | 1.19 |
| 5 | hsa-miR-181b |
380.03 |
821.69 | 1.15 |
| 6 | hsa-miR-1275 | 1,079.12 |
567.94 | −1.00 |
| 7 | hsa-miR-155 | 1,623.95 |
812.00 | −0.98 |
| 8 | hsa-miR-4324 |
450.65 |
838.80 | 0.91 |
| 9 | hsa-miR-27a | 1,120.89 | 2,037.10 | 0.86 |
| 10 | hsa-miR-15a |
338.01 |
610.37 | 0.86 |
| 11 | hsa-miR-320d | 3,035.04 | 1,678.92 | −0.82 |
| 12 | hsa-miR-224 | 1,684.58 | 2,870.00 | 0.80 |
| 13 | hsa-miR-320e | 2,374.19 | 1,410.80 | −0.75 |
| 14 | hsa-let-7e | 3,773.71 | 6,116.64 | 0.70 |
| 15 | hsa-miR-27b | 2,148.34 | 3,339.14 | 0.68 |
| 16 | hsa-let-7g | 2,219.58 | 3,507.05 | 0.67 |
| 17 | hsa-miR-320a | 4,671.19 | 3,001.94 | −0.64 |
| 18 | hsa-miR-320b | 4,521.04 | 2,934.64 | −0.62 |
| 19 | hsa-let-7b | 11,003.42 | 7,216.83 | −0.62 |
| 20 | hsa-miR-320c | 4,945.14 | 3,247.34 | −0.56 |
| 21 | hsa-miR-107 | 1,142.26 | 1,659.61 | 0.54 |
| 22 | hsa-miR-17 |
998.00 | 1,441.09 | 0.53 |
| 23 | hsa-miR-1246 | 5,445.48 | 3,735.81 | −0.51 |
| 24 | hsa-miR-181a |
626.31 |
889.46 | 0.51 |
| 25 | hsa-miR-92b | 1,925.05 | 1,373.15 | −0.49 |
| 26 | hsa-miR-21 | 15,567.78 | 21,195.97 | 0.46 |
| 27 | hsa-miR-103 | 1,464.21 | 1,991.04 | 0.44 |
| 28 | hsa-miR-222 | 6,340.95 | 4,743.61 | −0.42 |
| 29 | hsa-miR-92a | 4,248.12 | 3,181.43 | −0.41 |
| 30 | hsa-miR-25 | 2,094.70 | 2,733.08 | 0.38 |
| 31 | hsa-miR-15b | 4,844.80 | 5,967.15 | 0.34 |
| 32 | hsa-miR-26a | 5,491.24 | 6,963.52 | 0.34 |
| 33 | hsa-miR-20a | 1,251.63 | 1,569.61 | 0.33 |
| 34 | hsa-let-7a | 18,911.99 | 15,386.43 | −0.30 |
| 35 | hsa-miR-638 | 3,327.89 | 2,721.15 | −0.29 |
| 36 | hsa-let-7i | 3,071.09 | 3,725.29 | 0.28 |
| 37 | hsa-miR-23b | 12,277.94 | 15,069.06 | 0.27 |
| 38 | hsa-miR-3665 | 12,100.79 | 10,305.79 | −0.23 |
| 39 | hsa-miR-23a | 12,915.30 | 14,893.35 | 0.21 |
| 40 | hsa-miR-16 | 9,758.49 | 8,615.40 | −0.18 |
| 41 | hsa-let-7c | 13,839.79 | 12,196.52 | −0.17 |
Increased levels of IL-6 in BEAS-2B
cells following syn-hsa-miR-181b transfection
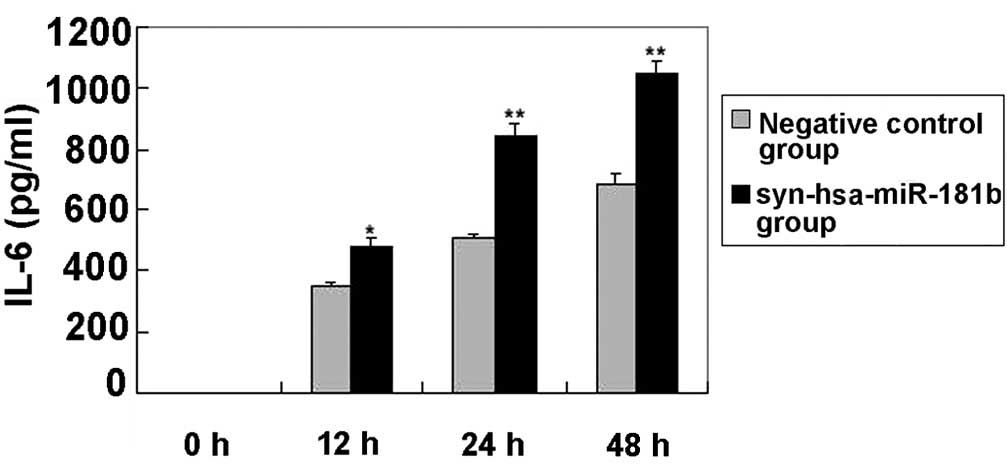
Following syn-hsa-miR-181b transfection, the levels
of IL-6 in the cultured supernatants of the BEAS-2B cells were
determined using ELISA. Fig. 3 shows
that the IL-6 levels were clearly elevated in the
syn-hsa-miR-181b-transfected cells compared with the negative
control levels. The lack of detectable TNF-α was unexpected, since
this particular cytokine has been reported to be involved in the
regulation of IL-6 and IL-8 (29).
NF-κB inhibitors abrogate upregulation
of p65 expression in response to syn-hsa-miR-181b transfection
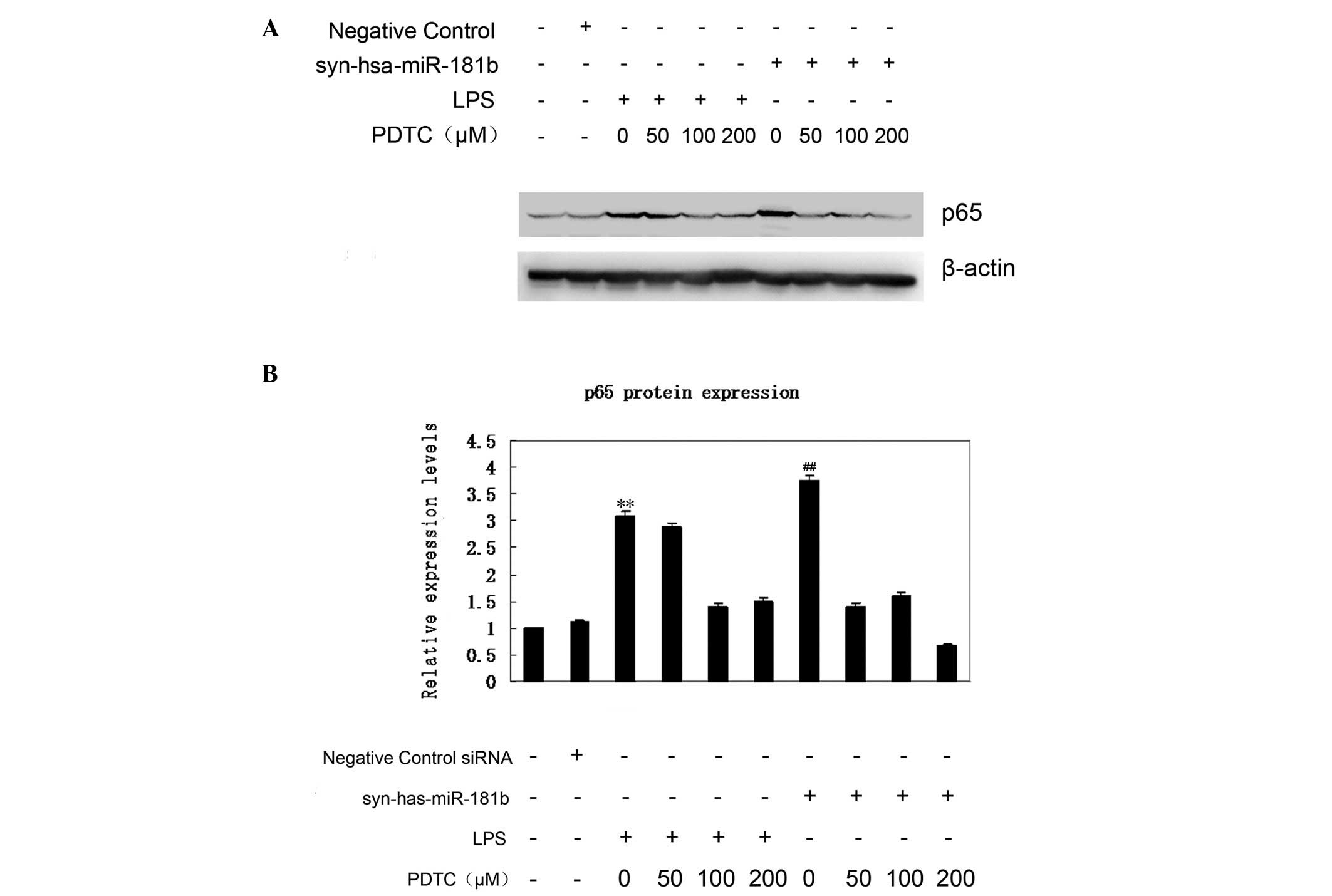
Increased nuclear p65 protein shows that the NF-κB
signaling pathway is activated. The p65 expression was therefore
first examined using western blot analysis (Fig. 4A). According to the data, an elevated
expression of p65 was observed in the syn-hsa-miR-181b-transfected
BEAS-2B cells, which was comparable with that of the LPS treatment
group. By contrast, the negative control had little effect on p65
expression (Fig. 4B). To further
confirm the effect of miR-181b on the expression of p65, BEAS-2B
cells were treated with PDTC 1.5 h prior to
syn-hsa-miR-181b-transfection. As shown in Fig. 4A, even a low dose (50 µM) of PDTC
treatment markedly abrogated the upregulation of p65 expression.
This inhibition of PDTC on p65 expression was also observed in the
LPS-treated cells. These findings demonstrated a critical link
between miR-181b and the NF-κB signaling pathway in ALI.
Discussion
miRNAs have been demonstrated to play a central role
in the regulation of the immune system development, proliferation
of monocytes and neutrophils, antibody production, differentiation
of B- and T-cells, release of inflammatory mediators (30) and certain inflammatory lung diseases.
Thus, miRNAs may also contribute to the pathogenesis of ALI/ARDS.
miR-181b has been found to be a key player in a positive feedback
loop that links inflammation to an epigenetic switch controlling
cellular transformation in MCF-10A human mammary epithelial cells
(20). Sun et al (31) revealed that miR-181b regulates
NF-κB-mediated endothelial cell activation and vascular
inflammation in response to proinflammatory stimuli. In addition,
the rescue of miR-181b expression may provide a novel target for
the treatment of critical diseases, such as diabetes, arthritis,
and other chronic inflammatory diseases, as well as for
anti-inflammatory treatment (31).
In order to identify potential miRNAs involved in ALI, the miRNAs
expression profile was analyzed in BEAS-2B cells with or without
LPS treatment. Notably, 41 miRNAs displayed significantly
differential expression levels (Table
I). The results of qPCR revealed a 2–3-fold increase in
miR-181b expression in the LPS-treated cells compared with the
non-LPS-treated BEAS-2B cells. Transfection based approaches were
further utilized in order to establish the promoting role of
miR-181b in BEAS-2B cells.
BEAS-2B cells were selected as representative airway
epithelial cell lines (32) for the
purpose of studying the LPS-induced effects in the airway
epithelium. BEAS-2B cells mimic the primary bronchial epithelial
cells considerably well (33) and
have been extensively used to investigate LPS-induced activation of
pro-inflammatory cytokines as an in vitro model based on the
first steps in the development of sepsis-induced ALI/ARDS (34,35).
E. coli LPS treatment was selected due to its use in the
majority of endotoxin-induced lung injury models (36,37).
Furthermore, LPS is a key pathogen recognition molecule for sepsis
(38), inducing apoptosis in lung
cells (39).
For the examination of the cellular function of
miR-181b, an overexpression approach in cultured BEAS-2B cells was
used to detect the levels of TNF-α and IL-6. In the present study,
it was demonstrated that the upregulation of miR-181b in BEAS-2B
cells can increase the IL-6 expression. Iliopoulos et al
(20) reported that, in human
mammary epithelial MCF-10A cells, the inhibition of miR-181b
expression, which is accomplished by treating cells with antisense
RNAs against miR-181b, results in a reduced production of IL-6, a
direct NF-κB target gene, and reduced NF-κB activity. BEAS-2B cells
are also epithelial cells and therefore the use of anti-miR-181b
was not required in the present study. IL-8 was not detected, since
the release of inflammation factor IL-8 in response to particulate
matter (≤2.5µm) and LPS treatment was qualitatively similar to the
IL-6 responses, suggesting a common or closely-associated mechanism
(40). However, besides the
secretion of inflammatory factors, other aspects, such as the
anti-apoptosis of lung cells (41)
and the promotion of immunocyte transmigration (42), may also be involved in ALI.
NF-κB, a type of multidirectional nuclear
transcriptional regulatory factor, regulates the expression of
proteins and genes associated with inflammation, immunization, and
growth regulation (43). In the
present study, it was found that the overexpression of miR-181b
leads to the upregulation of p65 expression, which is a member of
the NF-κB signaling pathway. The present findings indicated that
miR-181b acts as a proinflammatory factor through the targeting of
the NF-κB signaling pathway in vitro. This conclusion is
supported by the following evidence: First, the present study
demonstrated that the upregulation of miR-181b in BEAS-2B cells can
increase the expression of IL-6, a direct NF-κB target gene. In
addition, western blot analysis identified that p65 was upregulated
in the BEAS-2B cells following miR-181b overexpression. It was
further demonstrated that PDTC abrogated an miR-181b-mediated p65
increase. Compared with the negative control group, p65 expression
exhibited an ~3.7-fold increase following miR-181b overexpression,
whereas the inhibition of NF-κB reduced the p65 expression by 50%
following miR-181b overexpression; however the exact mechanism
remains unclear. It has been reported that PDTC potently inhibits
the activation and/or interaction of NF-κB with its upstream
regulatory binding sites, thereby preventing NF-κB-mediated
transcriptional activation (44,45).
Furthermore, PDTC restrains IκB degradation, thus specifically
inhibiting NF-κB activation (46).
The present study revealed that a collection of
miRNAs was aberrantly expressed in the LPS-treated BEAS-2B cells,
and focused on the effects of miR-181b on inflammation in BEAS-2B
cells; however, several other miRNAs, such as miR-23c, were also
dynamically regulated in LPS-induced ALI. Whether these miRNAs are
also associated with LPS-induced lung injury remains to be
elucidated. The use of bioinformatics to predict specific targets
of miR-181b and the use of luciferase assay to show whether these
genes are specific targets of miR-181b should be investigated in
future studies.
In conclusion, while >1,000 human mature miRNA
sequences are listed in the miRNA registry (47), only a handful have been characterized
as functional regulators of leukocyte or endothelial cell
inflammatory responses (48,49). The present study demonstrated that
miR-181b is involved in LPS-induced lung injury. Specifically,
miR-181b was found to serve as a potent regulator to promote
inflammation through the NF-κB signaling pathway in the BEAS-2B
cells. These findings may have important implications in the
regulation of the adaptive immune response in ALI. Thus far, there
is promising evidence supporting the potential application of
miRNAs as novel therapeutic targets, as well as biomarkers for ALI;
however, this requires further investigation prior to application
in the daily management of ALI. Further studies on the genetic
variation associated with miRNAs in real patient populations may
help achieve the ultimate goal of providing personalized medical
care for inflammatory lung disease. Considering inflammation as a
system disorder (50), it would be
interesting to examine whether miR-181b is also involved in the
inflammation in vivo.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 31201040), Science
Technology Department of Zhejiang Province (no. 2012C24005), Health
Bureau of Zhejiang Province (nos. 11-CX01 and 2013ZDA002), Zhejiang
Provincial Administration of traditional Chinese Medicine (no.
2012-XK-A04) and Natural Science Foundation of Zhejiang Province
(no. Y14H010013). The authors would like to thank all the members
of the laboratory for helpful discussions and comments on the
manuscript.
References
|
1
|
Avecillas JF, Freire AX and Arroliga AC:
Clinical epidemiology of acute lung injury and acute respiratory
distress syndrome: Incidence, diagnosis and outcomes. Clin Chest
Med. 27:549–557. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Crosby LM and Waters CM: Epithelial repair
mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol.
298:L715–L731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Papi A and Johnston SL: Rhinovirus
infection induces expression of its own receptor intercellular
adhesion molecule 1 (ICAM-1) via increased NF-kappaB-mediated
transcription. J Biol Chem. 274:9707–9720. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hoare GS, Chester AH, Yacoub MH and
Marczin N: Regulation of NF-kappaB and ICAM-1 expression in human
airway epithelial cells. Int J Mol Med. 9:35–44. 2002.PubMed/NCBI
|
|
6
|
Li Z, Zhang de K, Yi WQ, Ouyang Q, Chen YQ
and Gan HT: NF-kappaB p65 antisense oligonucleotides may serve as a
novel molecular approach for the treatment of patients with
ulcerative colitis. Arch Med Res. 39:729–734. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo H, Ingolia NT, Weissman JS and Bartel
DP: Mammalian microRNAs predominantly act to decrease target mRNA
levels. Nature. 466:835–840. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Latronico MV and Condorelli G: MicroRNAs
and cardiac pathology. Nat Rev Cardiol. 6:419–429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Port JD and Sucharov C: Role of microRNAs
in cardiovascular disease: Therapeutic challenges and potentials. J
Cardiovasc Pharmacol. 56:444–453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Buechner J, Henriksen JR, Haug BH, Tømte
E, Flaegstad T and Einvik C: Inhibition of mir-21, which is
upregulated during MYCN knockdown-mediated differentiation, does
not prevent differentiation of neuroblastoma cells.
Differentiation. 81:25–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li X, Zhang Y, Shi Y, Dong G, Liang J, Han
Y, Wang X, Zhao Q, Ding J, Wu K, et al: MicroRNA-107, an oncogene
microRNA that regulates tumour invasion and metastasis by targeting
DICER1 in gastric cancer. J Cell Mol Med. 15:1887–1895. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O'Connell RM, Rao DS, Chaudhuri AA and
Baltimore D: Physiological and pathological roles for microRNAs in
the immune system. Nat Rev Immunol. 10:111–122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oglesby IK, McElvaney NG and Greene CM:
MicroRNAs in inflammatory lung disease-master regulators or target
practice? Respir Res. 11:1482010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Roggli E, Britan A, Gattesco S, Lin-Marq
N, Abderrahmani A, Meda P and Regazzi R: Involvement of microRNAs
in the cytotoxic effects exerted by proinflammatory cytokines on
pancreatic beta-cells. Diabetes. 59:978–986. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Angulo M, Lecuona E and Sznajder JI: Role
of MicroRNAs in lung disease. Arch Bronconeumol. 48:325–330.
2012.(In English, Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cai ZG, Zhang SM, Zhang Y, Zhou YY, Wu HB
and Xu XP: MicroRNAs are dynamically regulated and play an
important role in LPS-induced lung injury. Can J Physiol Pharmacol.
90:37–43. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie T, Liang J, Liu N, Wang Q, Li Y, Noble
PW and Jiang D: MicroRNA-127 inhibits lung inflammation by
targeting IgG Fcγ receptor I. J Immunol. 188:2437–2444. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk
ML and Struhl K: STAT3 activation of miR-21 and miR-181b-1 via PTEN
and CYLD are part of the epigenetic switch linking inflammation to
cancer. Mol Cell. 39:493–506. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang YZ, Mao GX, Lv YD, Huang QD and Wang
GF: MicroRNA-181b stimulates inflammation via the NF-kappa B
signaling pathway in vitro. J Am Geriatr Soc. 62:S394.
2014.
|
|
22
|
Schulz C, Farkas L, Wolf K, Kratzel K,
Eissner G and Pfeifer M: Differences in LPS-induced activation of
bronchial epithelial cells (BEAS-2B) and type II-like pneumocytes
(A-549). Scand J Immunol. 56:294–302. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pencheva N, Tran H, Buss C, Huh D,
Dorbnjak M, Busam K and Tavazoie SF: Concergent multi-miRNA
targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis
and angiogenesis. Cell. 151:1068–1082. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma Y, Zhang P, Wang F, Zhang H, Yang J,
Peng J, Liu W and Qin H: miR-150 as a potential biomarker
associated with prognosis and therapeutic outcome in colorectal
cancer. Gut. 61:1447–1453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elder ACP, Gelein R, Finkelstein JN, Cox C
and Oberdorster G: Endotoxin priming affects the lung response to
ultrafine particles and ozone in young and old rats. Inhalation
Toxicology. 12:(Suppl 1). 85–98. 2000. View Article : Google Scholar
|
|
26
|
Meduri GU, Headley S, Kohler G, Stentz F,
Tolley E, Umberger R and Leeper K: Persistent elevation of
inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1
beta and IL-6 levels are consistent and efficient predictors of
outcome over time. Chest. 107:1062–1073. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dave RS and Khalili K: Morphine treatment
of human monocyte-derived macrophages induces differential miRNA
and protein expression: Impact on inflammation and oxidative stress
in the central nervous system. J Cell Biochem. 110:834–845. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma X, Becker Buscaglia LE, Barker JR and
Li Y: MicroRNAs in NF-kappaB signaling. J Mol Cell Biol. 3:159–166.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nelson S and Martin TR: Cytokines in
Pulmonary Disease: Infection and Inflammation (Lung Biology in
Health and Disease). Martin T: 141:(1st). Marcel Dekker. (New York,
NY). 2000.PubMed/NCBI
|
|
30
|
Pedersen I and David M: MicroRNAs in the
immune response. Cytokine. 43:391–394. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun X, Icli B, Wara AK, Belkin N, He S,
Kobzik L, Hunninghake GM, Vera MP, MICU Registry, Blackwell TS, et
al: MicroRNA-181b regulates NF-κB-mediated vascular inflammation. J
Clin Invest. 122:1973–1990. 2012.PubMed/NCBI
|
|
32
|
Koyama S, Sato E, Nomura H, Kubo K, Miura
M, Yamashita T, Nagai S and Izumi T: The potential of various
lipopolysaccharides to release monocyte chemotactic activity from
lung epithelial cells and fibroblasts. Eur Respir J. 14:545–552.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reddel RR, Ke Y, Gerwin BI, McMenamin MG,
Lechner JF, Su RT, Brash DE, Park JB, Rhim JS and Harris CC:
Transformation of human bronchial epithelial cells by infection
with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via
strontium phosphate coprecipitation with a plasmid containing SV40
early region genes. Cancer Res. 48:1904–1909. 1988.PubMed/NCBI
|
|
34
|
Boots AW, Gerloff K, Bartholomé R, van
Berlo D, Ledermann K, Haenen GR, Bast A, van Schooten FJ, Albrecht
C and Schins RP: Neutrophils augment LPS-mediated pro-inflammatory
signaling in human lung epithelial cells. Biochim Biophys Acta.
1823:1151–1162. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yeh CH, Cho W, So EC, Chu CC, Lin MC, Wang
JJ and Hsing CH: Propofol inhibits lipopolysaccharide-induced lung
epithelial cell injury by reducing hypoxia-inducible factor-1alpha
expression. Br J Anaesth. 106:590–599. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fortis S, Spieth PM, Lu WY, Parotto M,
Haitsma JJ, Slutsky AS, Zhong N, Mazer CD and Zhang H: Effects of
anesthetic regimes on inflammatory responses in a rat model of
acute lung injury. Intensive Care Med. 38:1548–1555. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mittal N and Sanyal SN: In vivo effect of
surfactant on inflammatory cytokines during endotoxin-induced lung
injury in rodents. J Immunotoxicol. 8:274–283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cheng DS, Han W, Chen SM, Sherrill TP,
Chont M, Park GY, Sheller JR, Polosukhin VV, Christman JW, Yull FE,
et al: Airway epithelium controls lung inflammation and injury
through the NF-kappa B pathway. J Immunol. 178:6504–6513. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tang PS, Mura M, Seth R and Liu M: Acute
lung injury and cell death: How many ways can cells die? Am J
Physiol Lung Cell Mol Physiol. 294:L632–L641. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Veranth JM, Reilly CA, Veranth MM, Moss
TA, Langelier CR, Lanza DL and Yost GS: Inflammatory cytokines and
cell death in BEAS-2B lung cells treated with soil dust,
lipopolysaccharide and surface-modified particles. Toxicol Sci.
82:88–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stern JB, Jaffré S, Dehoux M and Crestani
B: Keratinocyte growth factor and Hepatocyte growth factor: Their
roles in alveolar epithelial repair. Rev Mal Respir. 20:896–903.
2003.PubMed/NCBI
|
|
42
|
Hoke TS, Douglas IS, Klein CL, He Z, Fang
W, Thurman JM, Tao Y, Dursun B, Voelkel NF, Edelstein CL, et al:
Acute renal failure after bilateral nephrectomy is associated with
cytokine-mediated pulmonary injury. J Am Soc Nephrol. 18:155–164.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. 132:344–362. 2008.
|
|
44
|
Kawai M, Nishikomori R, Jung EY, Tai G,
Yamanaka C, Mayumi M and Heike T: Pyrrolidine dithiocarbamate
inhibits intercellular adhesion molecule-1 biosynthesis induced by
cytokines in human fibroblasts. J Immunol. 154:2333–2341.
1995.PubMed/NCBI
|
|
45
|
Schreck R, Meier B, Männel DN, Dröge W and
Baeuerle PA: Dithiocarbamates as potent inhibitors of nuclear
factor kappa B activation in intact cells. J Exp Med.
175:1181–1194. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang M, Zhou SH, Li XP, Shen XQ, Fang ZF,
Liu QM, Qiu SF and Zhao SP: Atorvastatin downregulates BMP-2
expression induced by oxidized low-density lipoprotein in human
umbilical vein endothelial cells. Circ J. 72:807–812. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Griffiths-Jones S: The microRNA Registry.
Nucleic Acids Res. 32:D109–D111. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fang Y, Shi C, Manduchi E, Civelek M and
Davies PF: MicroRNA-10a regulation of proinflammatory phenotype in
athero-susceptible endothelium in vivo and in vitro. Proc Natl Acad
Sci USA. 107:13450–13455. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Suárez Y, Wang C, Manes TD and Pober JS:
Cutting edge: TNF-induced microRNAs regulate TNF-induced expression
of E-selectin and intercellular adhesion molecule-1 on human
endothelial cells: Feedback control of inflammation. J Immunol.
184:21–25. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pugin J, Ricou B, Steinberg KP, Suter PM
and Martin TR: Proinflammatory activity in bronchoalveolar lavage
fluids from patients with ARDS, a prominent role for interleukin-1.
Am J Respir Crit Care Med. 153:1850–1856. 1996. View Article : Google Scholar : PubMed/NCBI
|