Introduction
Myotonic dystrophy (DM) is an autosomal dominant
genetic disease that involves multiple systems, and is the most
common type of adult muscular dystrophy. DM can be divided into DM
type 1 (DM1) and DM type 2 (DM2) on the basis of clinical features
and genetic differences. DM1 was first described by Steinert in
1909; thus it is also known as Steinert's disease (1). Its causative gene is the dystrophia
myotonic protein kinase (DMPK) gene, located on chromosome 19. The
disease is caused by an increased number of repeat sequence copies
of trinucleotides [cytosine-thymine-guanine, (CTG)n] in
the 3′-untranslated region of the DMPK gene (2). Healthy individuals have 5–37 CTG repeat
sequences, and >37 is considered an abnormal number of CTG
repeat sequences. Individuals with 38–49 repeat sequences show no
clinical symptoms, however their offspring may have a substantial
risk of exhibiting the clinical symptoms of DM. Patients with
>50 repeat sequences would show clinical symptoms (3).
In 1994, Ricker et al (4) found that in certain DM1-like patients
proximal myotonia was observed, and the numbers of
(CTG)n repeat sequence copies in the untranslated
regions of DMPK were in the normal range; thus the term ‘proximal
myotonic myopathy’ was applied to this condition (5). In 2001, Liquori et al (6) found that the affected gene in such
patients was located on intron 1 of the chromosome 3 finger protein
9 gene, and the disease has been observed to develop as the number
of copies of the tetranucleotide CCTG repeat sequence increases
(7); thus, the condition was renamed
as DM2.
The molecular mechanisms of DM are complex. For both
DM1 and DM2, the repeatedly amplified sequences are located in the
untranslated regions of disease-causing genes, and do not change
the protein encoding of their location genes. The mechanism by
which the amplification of the repeat sequence leads to the
increased incidence of DM is debatable. Among known patients with
DM1, three mechanisms may lead to clinical symptoms (8): i) DMPK protein haplotype insufficiency.
The increased repeat sequence amplification may affect the gene
transcription of DMPK or retain the DMPK gene transcripts within
the nucleus without transporting them into the cytoplasm, thus
reducing the expression of DMPK protein, causing haplotype
insufficiency, and leading to clinical symptoms. ii) The abnormally
repeated sequence amplification could affect the structures of
chromosomes, thus affecting the expression of DMPK genes and genes
in the adjacent region. For example, the expression of SIX5 gene,
adjacent to the DMPK gene area in patients with DM1, is
downregulated. SIX5 is also similar to the eye-development gene of
Drosophila and mouse gene families that regulate distal limb
muscle development. Since cataract and distal end malnutrition are
common symptoms of DM1, it has been hypothesized that SIX5
haplotype insufficiency induces DM1 pathogenesis. SIX5-knockout
mice, however, only exhibit cataract symptoms, without multi-system
damage, so this mechanism is not sufficient to cause the
characteristic multi-system damage of DM1. iii) RNA toxicity and
abnormal shearing. The abnormal repeatedly amplified sequence may
be transcribed into mRNA that contains an amplified CUG repeated
sequence. This mRNA would be abnormally accumulated in the nucleus,
thus changing the activities of certain RNA-binding proteins, for
example, increasing the numbers of CUG-binding protein/Elav-like
family member 1, and decreasing the expression of muscle-blind-like
protein (MBNL1). MBNL1 inside the cytoplasm would then transfer to
the nucleus to compensate for the reduction of MBNL1. The reduction
of MBNL1, as well as the synergistic effects increased by the CUG
binding protein, would interfere with the normal cutting process,
resulting in error splicing of sub-transcripts and causing
multi-system damage. For example, it could cause the abnormal
expression of: Chloride channels, leading to myotonia; troponin,
generating myocardial damage; or insulin receptors, resulting in
insulin resistance (8). In addition
to these three mechanisms, various other mechanisms have also been
proposed to be involved in the pathogenesis of DM1, such as
transcription factor dysfunction, microRNA dysfunction, RNA
interference and double-stranded RNA mechanisms, cell stress and
the inhibition of translation. The synergistic effects of various
mechanisms are likely to induce the multi-system damage associated
with DM1.
The main symptoms of DM are muscle weakness, muscle
atrophy, myotonia and multi-system effects. Following repeated
muscle contractions, the myotonia phenomenon can be improved, known
as the ‘warm-up phenomenon’ (9). The
main manifestations of multi-system effects are alopecia,
cataracts, sexual dysfunction, heart block, mental retardation,
hyperthyroidism and digestive symptoms. Electrophysiological
examination by needle electromyography (EMG) demonstrates myotonia
potential and myogenic damage. The nerve conduction velocity is
generally normal. Pathological examination of muscles indicates
variations in muscle fiber diameter, showing a bimodal
distribution. Hypertrophy and atrophy may be observed. Nuclear
ingression is observed in some muscle fibers, and in certain cases,
cytoplasmic bodies in the muscle fibers, nuclear chain formation,
circular muscle fibers, selective type I muscle fiber atrophy,
non-merging necrosis and regeneration may be observed (10). Magnetic resonance imaging examination
of the muscles has shown that the affected muscles exhibit fatty
degeneration (11).
In general, southern blot analysis is used to detect
the amplified gene sequences (12).
This method has high specificity, and is able to estimate the
number of repeat units of CTG, although the sensitivity is low and
the method is prone to producing false-negative results.
Additionally, it is time-consuming, and requires the application of
radioactive probes. Furthermore, the triplet-primed polymerase
chain reaction (TP-PCR) method has been used, which is simple and
rapid (13), although it cannot
provide accurate numbers of CTG repeat sequences. In order to
eliminate false-negative results, southern blot analysis could be
performed on patients with negative TP-PCR results to ensure the
reliability of results.
The present study summarizes the clinical symptoms,
skeletal muscle pathology and genetic testing information of a
pedigree with DM1, treated in the Department of Neurology, Taiyuan
Central Hospital (Taiyuan, China) between March 2014 and April
2014, with the aim of further exploring the pathogenesis. The
clinical, pathological and genetic testing features of DM1, as well
as how to distinguish it from DM2 and other myopathies involving
myotonia, are discussed, to further deepen the understanding and
awareness of this type of disease.
Materials and methods
Clinical data
The clinical data of a pedigree with DM1, treated in
the Department of Neurology, Taiyuan Central Hospital of Shanxi
Medical University (Taiyuan, China) from March 2014 to April 2014,
were collected. The present study was conducted in accordance with
the Declaration of Helsinki and with approval from the Ethics
Committee of Shanxi Medical University. Written informed consent
was obtained from all participants.
Muscle pathology
The proband underwent an open skeletal muscle biopsy
of the left bicep brachii under local anesthesia. The sample was
then rapidly frozen in liquid nitrogenisopentane, and sliced into
7-µm frozen serial sections. Hematoxylin and eosin staining was
performed prior to pathological analysis under a Nikon Eclipse Ni-U
light microscope (Nikon Corporation, Tokyo, Japan) (10). The pathological features of DM1
patients were analyzed.
Genetic testing
DNA extraction
Venous blood for DNA extraction was sampled from the
proband and his sister, and was subjected to TP-PCR analysis to
amplify the DMPK gene (12). A whole
blood genomic DNA extraction kit (Shanghai Shenggong, Shanghai,
China) was used to extract genomic DNA for the PCR amplification.
The DNA was dissolved and stored in 10 mmol/l Tris-HCl solution (pH
7.5). The sequences of primers (Shanghai Shenggong) were: P1,
5′-GGG GCT CGA AGG GTC CTT GT-3′; P2, 5′-GTG CGT GGA GGA TGG AAC
ACG-3′; P3R, 5′-AGC GGA TAA CAA TTT CAC ACA GGA-3′; P4CAG, 5-AGC
GGA TAA CAA TTT CAC ACA GGA CAG CAG CAG CAG CAG CAG-3′. The 5′
terminus of P1 was labeled with carboxyfluorescein (14).
PCR reaction
The 25-µl reaction system contained 0.5 units KAPA
HiFi HotStart DNA Polymerase (KAPA Biosystems, Boston, MA, USA),
600 µmol/l deoxynucleotide triphosphates and 5.0 µl 5X KAPA GC
buffer (containing 1X2.0 mmol/l Mg2+; KAPA Biosystems).
The concentration of template DNA was 50–500 ng/cycle. Within the
PCR-P1P2 reaction system, the primers included P1 and P2, and their
final concentration was 0.6 µmol/l for both. Within the TP-PCR
reaction system, the primers included P1, P3R and P4CAG, with the
final concentrations of 0.8, 0.6 and 0.2 µmol/l, respectively. The
sample was pre-degenerated at 95°C for 5 min, prior to entering the
PCR cycle. The parameters of the cycle were: 98°C for 20 sec, 65°C
for 30 sec and 72°C for 2 min, for a total of 30 cycles; followed
by an extension at 72°C for 10 min. The products were stored at
4°C.
Genetic analysis
The amplification products were analyzed by
capillary electrophoresis in an ABI 3730 sequencer (Applied
Biosystems Life Technologies, Foster City, CA, USA). One microliter
of PCR products was mixed with 9 µl formamide, and capillary
electrophoresis was performed following 4 min degeneration.
GeneScan 500-LIZ dye Size Standard (Life Technologies) was used for
the reproducible sizing of fragment analysis data, and ABI
GeneMapperID version 3.2 (Applied Biosystems) was used to conduct
the analysis.
Results
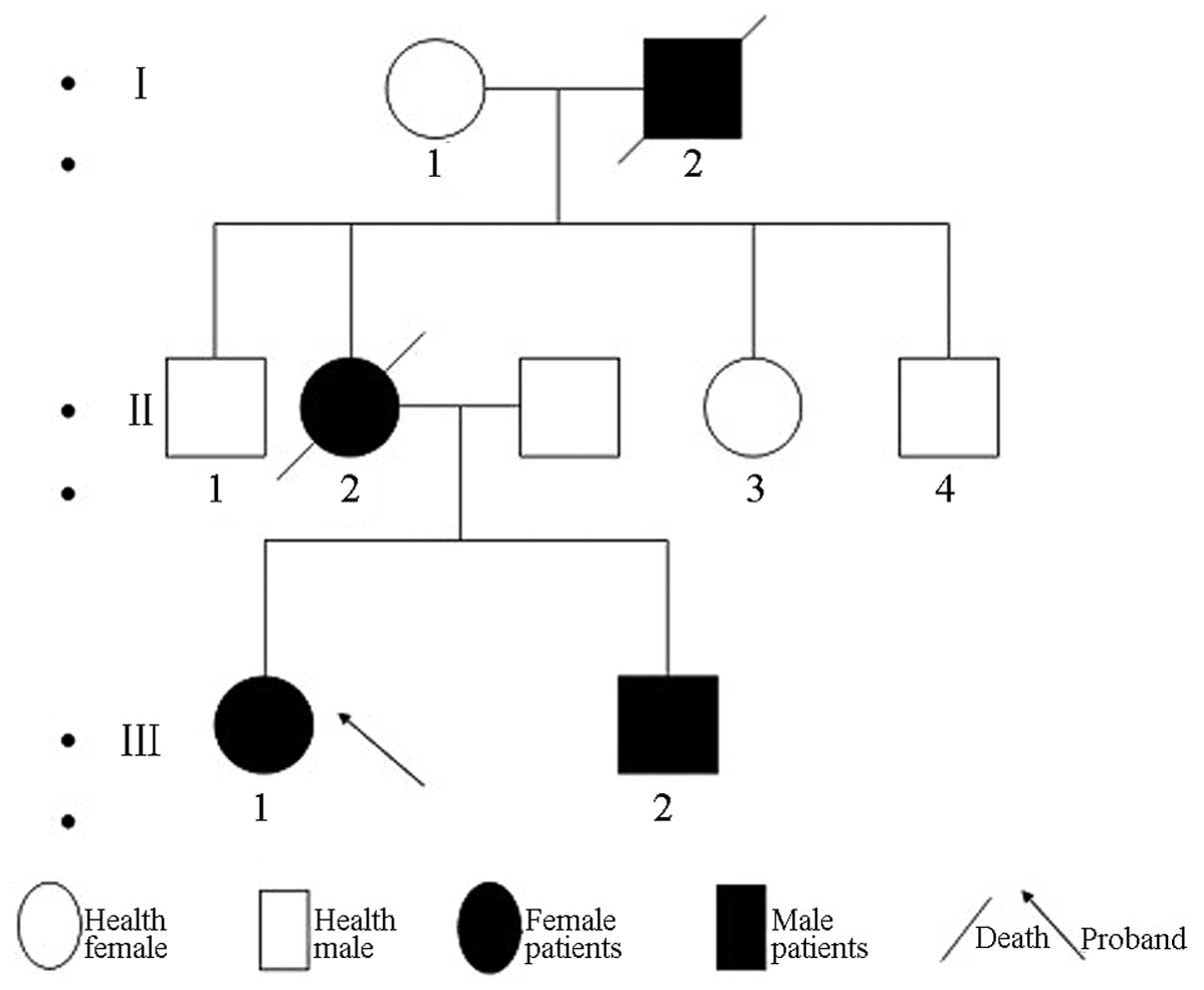
Patient III 1
The proband was a 38-year-old male, and is shown in
the pedigree chart as III 1 (Fig.
1).
Clinical manifestations
The patient had experienced upper limb weakness for
20 years, mostly in the distal upper limbs. Following hard
clenching, the patient was unable to loosen his fists. This
manifestation progressively increased, and the patient gradually
exhibited atrophy in the proximal upper and lower limbs, and
shoulder and back muscles. Lower extremity weakness appeared 6
years ago.
Medical history
For the past 20 years, the left side of the face of
the patient exhibited peripheral paralysis. In addition, hearing
was lost in the right ear, and baldness gradually developed.
Personal history
The patient was married, without children, and
sexual dysfunction was observed following the marriage.
Family history
The grandfather of the patient had symptoms of upper
limb weakness, and succumbed to stomach cancer. The mother of the
patient had symptoms of upper limb weakness, and succumbed to
pulmonary heart disease. The patient had a sister who exhibited
similar symptoms.
Admission examination
The patient had left peripheral facial paralysis,
without a typical thin and angular face. The neck flexor force was
grade 2. The muscle strength of the proximal ends of the upper
limbs was grade 4-, the finger dorsal extensor muscle strength was
grade 4+, the muscle strength of the proximal end of the lower
extremity was grade 5, the dorsal extensor strength of the foot was
grade 2 and the plantar flexor force was grade 4. The bilateral
supraspinatus, infraspinatus muscle, deltoid, biceps brachii and
triceps brachii exhibited atrophy, and the bilateral gastrocnemius
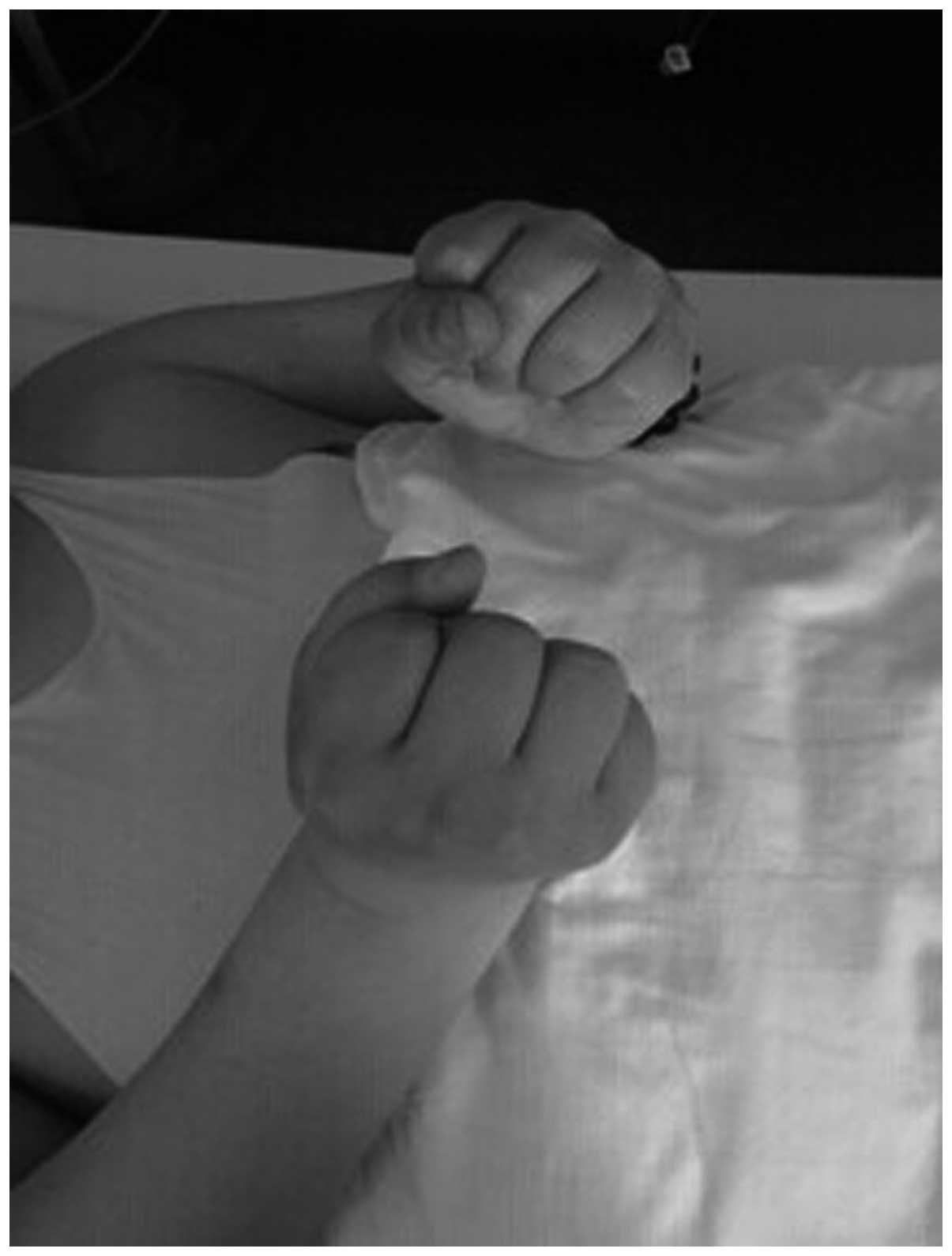
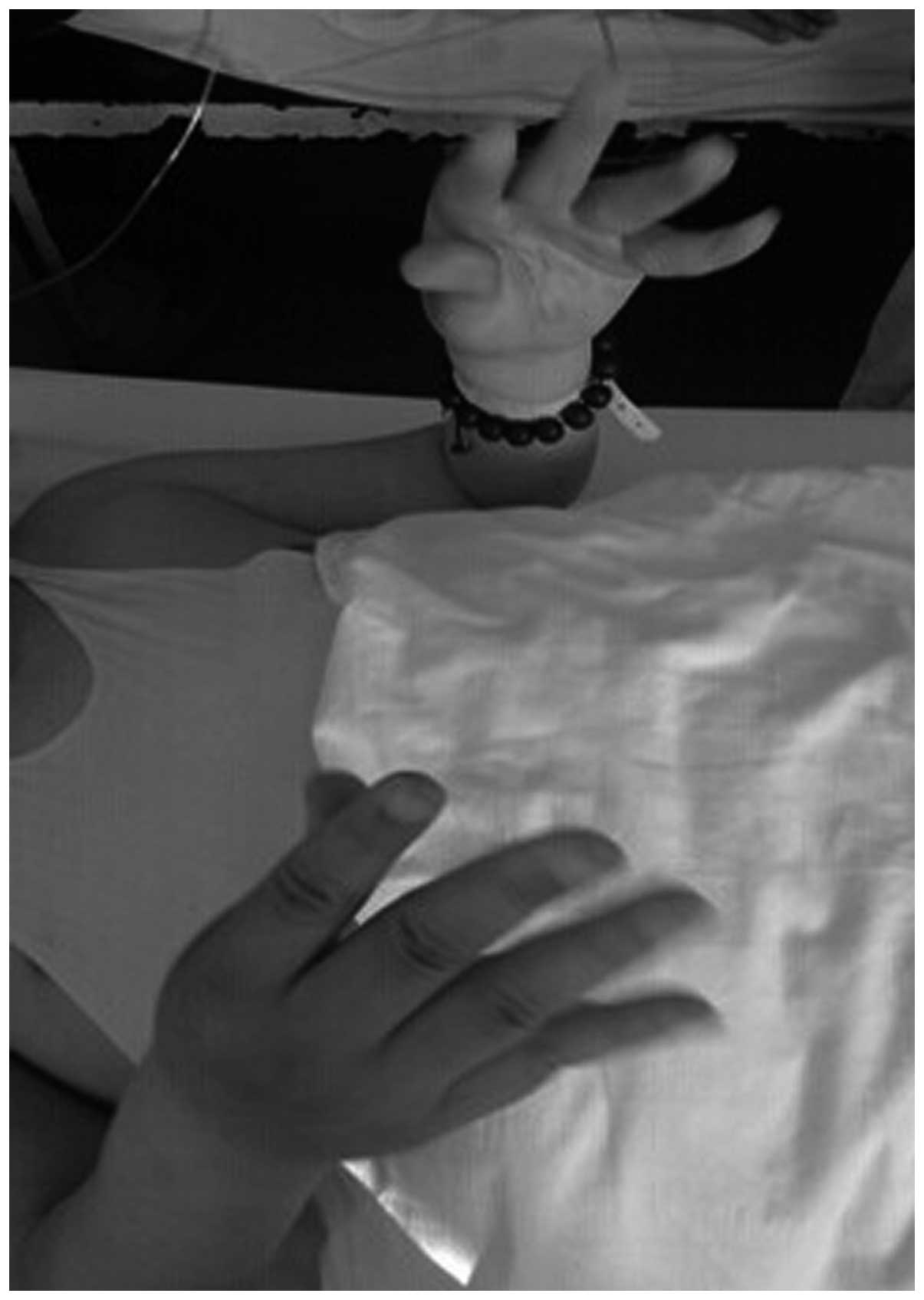
showed slight hypertrophy. The fingers exhibited the myotonic
phenomenon (Figs. 2 and 3), while the knotted muscles were not fully
released. The tendon reflex of the bilateral upper limbs was not
elicited, and the knee tendon reflex was reduced.
Laboratory information
Regarding the thyroid functions, the
thyroid-stimulating hormone level was 5.36 µIU/ml, while the
thyroxine level exhibited no evident abnormality. Biochemical
examination showed that the alanine aminotransferase activity was
62 U/l, aspartate aminotransferase activity was 56.6 U/l and uric
acid level was 421 µmol/l. In addition, the triglyceride level was
8.65 mmol/l, total cholesterol level was 5.95 mmol/l, fasting blood
glucose was 5.7 mmol/l, creatine kinase activity was 180 U/l,
lactate dehydrogenase activity was 246 U/l and the erythrocyte
sedimentation rate was 20 mm/h. The limb EMG showed myotonia, which
was suspected to be myogenic impairment. Consultation with the
Department of Ophthalmology of Taiyuan Central Hospital of Shanxi
Medical University resulted in a diagnosis of ‘bilateral cataracts
(congenital)’.
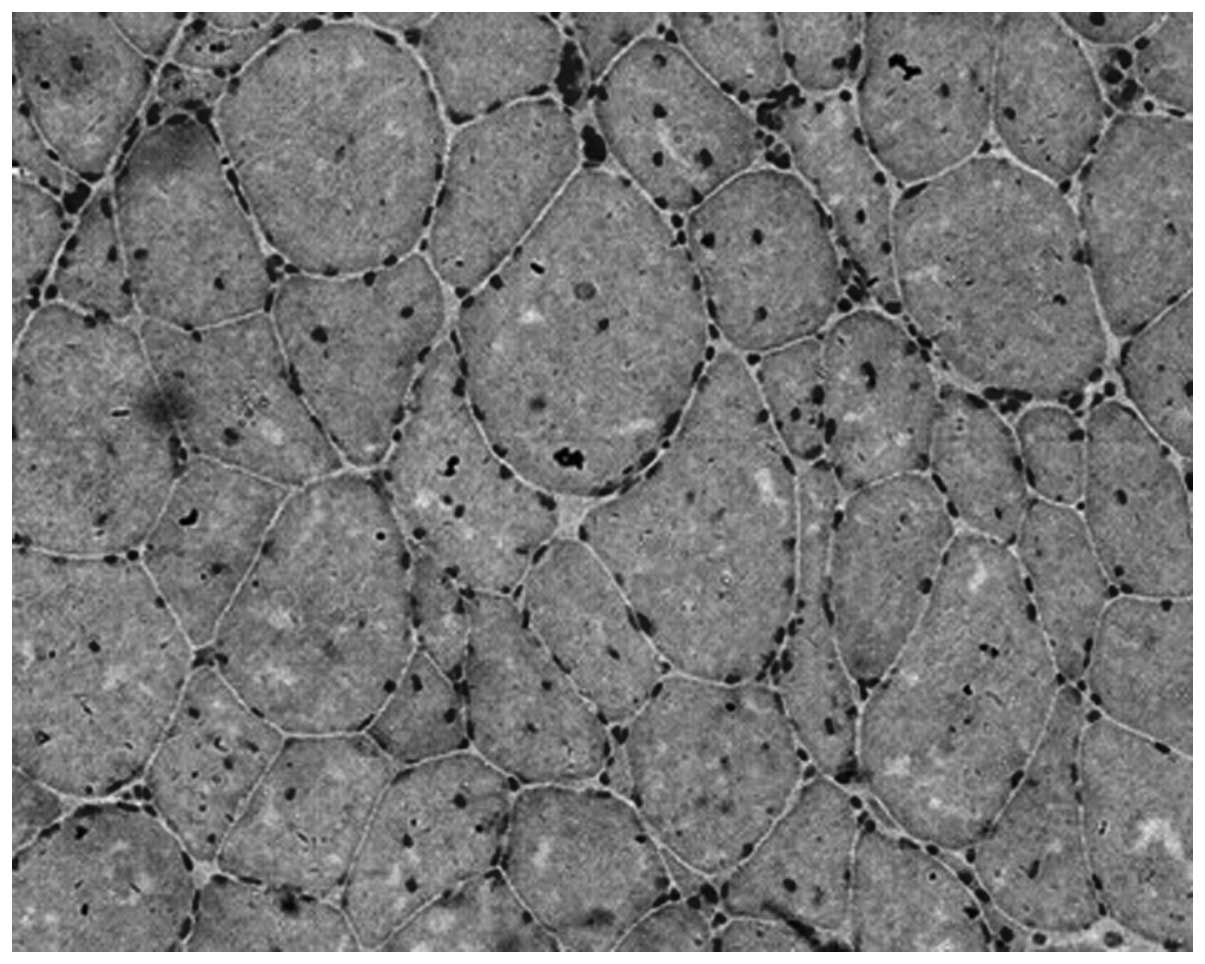
Muscle pathology
Under the light microscope, it was observed that the
muscle fibers varied in size and diameter, with a bimodal
distribution. Some of the muscle fibers were hypertrophic and
others were rounded and exhibited atrophy. A large number of muscle
fibers had undergone nuclear ingression. Nuclear aggregation was
occasionally visible, but necrosis, regeneration and whorled muscle
fibers were not evident. In addition, there were no cytoplasmic
bodies inside the muscle fibers (Fig.
4).
Patient III 2
The sister of the proband, shown as III 2 in
Fig. 1, was 30 years old and had
experienced mild weakness of the upper limbs since the age of 24
years. The patient was not able to release her fists following hard
clenching. A physical examination showed that the muscle strength
of the distal parts of the upper limbs was grade 5-, while that of
the proximal part was grade 5. The muscle strength of the lower
extremities was grade 5.
Patient II 1
The mother of the proband, shown as II 1 in Fig. 1, experienced disease onset at the age
of 40 years, and succumbed to pulmonary heart disease at the age of
65 years. The clinical manifestations in this patient were upper
limb weakness, and the inability to release the fists following
hard clenching.
Patient I 2
The grandfather of the proband, shown as I 2 in
Fig. 1, experienced disease onset at
the age of 45 years, and succumbed to stomach cancer at 72 years.
The clinical manifestations were upper limb weakness, and the fists
could not be released following hard clenching.
Genetic test results
Genetic testing of the proband and his sister found
that the DMPK gene exhibited the disease-causing mutation CTG with
>50 repeat units.
Discussion
DM is an autosomal dominant genetic disease that
normally involves multiple systems. It can be divided into DM1 and
DM2 according to its clinical features and genetic profiles.
According to the clinical manifestations, DM1 can be
further divided into four types: Asymptomatic, congenital,
child-onset and adult-onset (15).
Asymptomatic DM1 patients have no clinical symptoms. Patients with
congenital DM1 may exhibit excessive amniotic fluid or fetal
reduction prior to birth and, after birth, they may show severe
limb weakness, low muscle tone and respiratory failure. After
birth, the patients may experience muscular hypotonia of the limbs
and trunk and respiratory, facial and bulbar muscles. Due to facial
weakness, the patients may exhibit a V-shaped upper lip (also known
as a tent- or fish-shaped upper lip). Due to weakness in the
chewing muscles, there may be a difficulty in sucking, resulting in
feeding problems. The respiratory muscle weakness may cause the
patients to exhibit respiratory failure neonatally. The typical
myotonic phenomenon may not necessarily appear in the early stages.
The majority of patients succumb at an early stage; surviving
patients may suffer from mental retardation, and exhibit cerebral
atrophy and ventricular contraction (16), followed by severe respiratory and
circulatory complications in their 30s and 40s.
Patients with child-onset DM1 exhibit onset in their
childhood, when the symptoms of muscle weakness, myotonia and
muscle atrophy are not evident, so the condition can be challenging
to diagnose. Facial muscle involvement is the most common symptom,
although the typical V-shaped upper lip of congenital DM1 is not
observed. Dysarthria and hand myotonia are also typical
manifestations, and are often accompanied by a movement development
delay and mental retardation (17).
The adult-onset type generally exhibits the symptoms
of typical multi-system damage, which could involve the skeletal
muscle, heart, endocrine glands, skin, central nervous system, eyes
and digestive system. Skeletal muscle involvement may appear as
weakness in the distal parts of all four limbs, leading to
non-flexible fine motions of the upper extremities, and foot drop.
Furthermore, the levator muscle of the upper eyelid may be
involved, causing ptosis of the upper eyelid. The masticatory
muscle may be involved leading to atrophy of the temporalis and
masticatory muscles, elevated cheekbones, and thinning of the face
(the typical ‘hatchet face’). Atrophy of the sternocleidomastoid
muscle may cause a slender neck, associated with a reduction in
craning muscle strength and excessive forward protrusion, thus
forming a ‘goose-neck’. Extraocular muscle involvement is
relatively rare, and normally not accompanied by gastrocnemius
muscle hypertrophy (12). The muscle
weakness slowly progresses, and a small number of patients may
combine the symptoms of four-limb myasthenia, which is aggravated
by neuraxial peripheral neuropathy (18). Certain patients may exhibit symptoms
of myotonia. The myotonic phenomenon refers to a delayed relaxation
of the skeletal muscle following voluntary contraction. The
myotonic phenomenon of patients with DM1 normally appears in the
distal ends of limbs, and mostly happens when the fingers are
clenched, for example when using tools or rotating a doorknob. When
the muscles are repeatedly contracted, myotonia can be improved,
known as the ‘warm-up phenomenon’ (9). The sustained contraction of muscle due
to compression with a percussion hammer, where the muscles contract
and then relax a few seconds later, is known as percussion
myotonia. The eyelid muscles may also suffer myotonia, as shown
when the eyes cannot be opened immediately when closed with
force.
The proband in the present study exhibited typical
myotonia, including the warm-up phenomenon, muscle weakness mainly
in the distal ends of limbs, and muscle atrophy mostly in the
proximal ends of limbs, which were all in line with the typical
skeletal muscle performance of DM1. However, the proband also
exhibited mild hypertrophy in the bilateral gastrocnemius, which is
not a usual symptom of patients with DM1. Thus, further
accumulation of data is required to identify whether patients with
DM1 may exhibit hypertrophy of the bilateral gastrocnemius.
In addition to skeletal muscle involvement, patients
with DM1 may exhibit symptoms of multi-system involvement.
Congenital cataracts may appear, and patients may exhibit cardiac
symptoms, such as conduction block or tachyarrhythmia, sudden
cardiac mortality, myocardial disease or ischemic heart disease
(19). Cardiac pathological
examination may reveal cardiac fibrosis, cardiac hypertrophy and
the accumulation of fatty tissue in the conducting system and
sinoatrial node (20).
Electrocardiography may show a prolonged PR interval, widened QRS
wave, paroxysmal or persistent atrial fibrillation and various
malignant arrhythmias, which may often lead to sudden cardiac
mortality. Patients with DM1 may also experience endocrine
abnormalities, affecting the thyroid, pancreas, hypothalamus and
gonads. Thyroid involvement may cause thyroid dysfunction;
pancreatic involvement may cause insulin resistance resulting in
abnormal glucose tolerance; and gonad involvement may cause
testicular atrophy in male patients, leading to sexual dysfunction
and even infertility, and female patients may suffer from
spontaneous abortion and menstrual abnormalities. The involvement
of the skin may result in alopecia, which is much more common in
male patients. Central nervous system involvement may commonly be
exhibited as mental retardation, daytime sleepiness and night-time
sleep disorders. The patients may also have personality disorders,
manifested as compulsion, irritability or apathy (21). Digestive system involvement may
include cholecystitis, cholelithiasis, abnormal liver functions,
intestinal pseudo-obstruction and congenital megacolon, and also
delayed gastric emptying, diarrhea and other symptoms.
In the present study, the proband exhibited an
elevated level of thyroid-stimulating hormone, while the thyroxine
level was normal, suggesting the existence of subclinical
hypothyroidism. The increase in alanine aminotransferase and
aspartate aminotransferase prompted abnormal liver function,
associated with congenital cataracts, baldness and sexual
dysfunction, which was consistent with the multi-system involvement
of DM1. In patients with DM1, serological examination typically
shows a mild to moderate elevation of creatine kinase, an increase
in the activities of aspartate aminotransferase and alanine
aminotransferase, and a reduction in the levels of male serum
testosterone and thyroid hormone (11). The proband exhibited peripheral
facial paralysis and right-ear conductive deafness, however, this
has not been associated with DM1 previously; thus, whether a
correlation with DM1 exists requires further study.
In the present study, the proband exhibited mild
elevations of creatine kinase, aspartate aminotransferase, alanine
aminotransferase and thyroid-stimulating hormone. EMG showed
myotonic discharge, and reduction of re-contraction movement
potential, with pure to mixed phases. Muscle pathology examination
revealed variations in the diameters of muscle fibers, showing a
bimodal distribution. Some hypertrophy of the muscle fibers was
also observed. Numerous muscle fibers demonstrated nuclear
ingression, and nuclear aggregation was occasionally present.
However, there was no necrosis, regeneration or whorled muscle
fibers, and no cytoplasmic bodies were visible inside the muscle
fibers, consistent with the results of the auxiliary DM1
examinations. The TP-PCR method was able to reveal the
disease-causing mutations in the proband and his sister, with
>50 CTG repeats. Thus diagnostic confirmation was obtained and
the results were sensitive and reliable. However, it is not
possible to accurately determine the numbers of CTG repeat
sequences using this method. In order to accurately assess the
prognosis of patients and the possible severity of disease in
future generations, it may be necessary to perform southern blot
analysis to accurately determine the number of CTG repeat
sequences.
Acknowledgements
This study was supported by the Natural Science
Foundation of Shanxi Province (grant no. 2013011052-5).
References
|
1
|
Steinberg H and Wagner A: Hans Steinert:
100 years of myotonic dystrophy. Nervenarzt. 79:961–962. 2008.(In
German). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pavićević D Savić, Miladinović J,
Brkušanin M, Šviković S, Djurica S, Brajušković G and Romac S:
Molecular genetics and genetic testing in myotonic dystrophy type
1. Biomed Res Int. 2013:3918212013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Valaperta R, Sansone V, Lombardi F,
Verdelli C, Colombo A, Valisi M, Brigonzi E, Costa E and Meola G:
Identification and characterization of DM1 patients by a new
diagnostic certified assay: Neuromuscular and cardiac assessments.
Biomed Res Int. 2013:9585102013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ricker K, Koch MC, Lehmann-Horn F,
Pongratz D, Otto M, Heine R and Moxley RT: 3rd: Proximal myotonic
myopathy: A new dominant disorder with myotonia, muscle weakness,
and cataracts. Neurology. 44:1448–1452. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kamsteeg EJ, Kress W, Catalli C, Hertz JM,
Witsch-Baumgartner M, Buckley MF, van Engelen BG, Schwartz M and
Scheffer H: Best practice guidelines and recommendations on the
molecular diagnosis of myotonic dystrophy types 1 and 2. Eur J Hum
Genet. 20:1203–1208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liquori CL, Ricker K, Moseley ML, Jacobsen
JF, Kress W, Naylor SL, Day JW and Ranum LP: Myotonic dystrophy
type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science.
293:864–867. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kurosaki T, Ueda S, Ishida T, Abe K, Ohno
K and Matsuura T: The unstable CCTG repeat responsible for myotonic
dystrophy type 2 originates from an AluSx element insertion into an
early primate genome. PLoS One. 7:e383792012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Machuca-Tzili L, Brook D and Hilton-Jones
D: Clinical and molecular aspects of the myotonic dystrophies: A
review. Muscle Nerve. 32:1–18. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Logigian EL, Blood CL, Dilek N, Martens
WB, Moxley RTIV, Wiegner AW, Thornton CA and Moxley RT III:
Quantitative analysis of the ‘warm-up’ phenomenon in myotonic
dystrophy type 1. Muscle Nerve. 32:35–42. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meola G: Clinical aspects, molecular
pathomechanisms and management of myotonic dystrophies. Acta Myol.
32:154–165. 2013.PubMed/NCBI
|
|
11
|
Udd B, Meola G, Krahe R, Wansink DG,
Bassez G, Kress W, Schoser B and Moxley R: Myotonic dystrophy type
2 (DM2) and related disorders report of the 180th ENMC workshop
including guidelines on diagnostics and management 3–5 December
2010, Naarden, The Netherlands. Neuromuscul Disord. 21:443–450.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li M, Wang Z, Cui F, Yang F, Chen Z, Ling
L, Pu C and Huang X: Investigation of molecular diagnosis in
Chinese patients with myotonic dystrophy type 1. Chin Med J (Engl).
127:1084–1088. 2014.PubMed/NCBI
|
|
13
|
Addis M, Serrenti M, Meloni C, Cau M and
Melis MA: Triplet-primed PCR is more sensitive than southern
blotting-long PCR for the diagnosis of myotonic dystrophy type 1.
Genet Test Mol Biomarkers. 16:1428–1431. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Singh S, Zhang A, Dlouhy S and Bai S:
Detection of large expansions in myotonic dystrophy type 1 using
triplet primed PCR. Front Genet. 5:942014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Turner C and Hilton-Jones D: The myotonic
dystrophies: Diagnosis and management. J Neurol Neurosurg
Psychiatry. 81:358–367. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ashizawa T: Myotonic dystrophy as a brain
disorder. Arch Neurol. 55:291–293. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Steyaert J, Umans S, Willekens D, Legius
E, Pijkels E, de Die-Smulders C, Van den Berghe H and Fryns JP: A
study of the cognitive and psychological profile in 16 children
with congenital or juvenile myotonic dystrophy. Clin Genet.
52:135–141. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krishnan AV and Kiernan MC: Axonal
function and activity-dependent excitability changes in myotonic
dystrophy. Muscle Nerve. 33:627–636. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lund M, Diaz LJ, Ranthe MF, Petri H, Duno
M, Juncker I, Eiberg H, Vissing J, Bundgaard H, Wohlfahrt J and
Melbye M: Cardiac involvement in myotonic dystrophy: A nationwide
cohort study. Eur Heart J. 35:2158–2164. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Phillips MF and Harper PS: Cardiac disease
in myotonic dystrophy. Cardiovasc Res. 33:13–22. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Douniol M, Jacquette A, Cohen D, Bodeau N,
Rachidi L, Angeard N, Cuisset JM, Vallée L, Eymard B, Plaza M, et
al: Psychiatric and cognitive phenotype of childhood myotonic
dystrophy type 1. Dev Med Child Neurol. 54:905–911. 2012.
View Article : Google Scholar : PubMed/NCBI
|