Introduction
Adult-onset type II citrullinemia (CTLN2) is a
disorder caused by mutations in the solute carrier family 25
(SLC25A13) gene. CTLN2 is an autosomal recessive disorder
characterized by recurrent encephalopathy with hyperammonemia
resulting from high plasma citrulline and ammonium levels, which
are caused by a deficiency of argininosuccinate synthetase in the
liver. The literature concerning incidence rates of the disease is
limited. According to a study by Nagata et al (1) in 1993, the incidence rate of CTLN2 was
1/100,000 to 1/230,000; however, the carrier and incidence rates
differ across Japanese populations. CTLN2 is a genetic disease that
does not have clear etiology, but usage of alcohol and certain
drugs may represent risk factors. The clinical presentation of
patients with CLTN2 is characterized by a hyperammonemia
accompanied by recurrent episodes of neuropsychiatric
manifestations, including aberrant behavior, nocturnal delirium,
disorientation, consciousness disturbances, convulsive seizures and
coma (2). Typical current therapies
include liver transplantation, avoidance of protein-rich foods and
treatment with arginine. The present study reports the case of a
patient with type II citrullinemia who was misdiagnosed in the
emergency department, and was finally administered arginine upon
diagnosis of CTLN2.
Case report
A 43-year-old man was admitted to the emergency
department of Xin Hua Hospital Affiliated to Shanghai Jiao Tong
University School of Medicine (Shanghai, China) as a result of
sudden delirium and upper limb weakness. The patient was unable to
write due to upper limb weakness occurring 2 h prior to his
admittance to the hospital, and experienced memory loss subsequent
to informing his wife of the incident. The patient's wife stated
that he became confused and was unable to answer questions
appropriately. The patient had a medical history of left kidney
angiomyolipoma at 3-years-old, and repeatedly complained of chest
pain following physical activity over the previous 3–5 years, but
had not undergone treatment for this. The patient had no history of
hypertension or diabetes, is a non-drinker and had quit smoking
several years previously.
Upon admission, the patient appeared to be fully
conscious. He was aware of the time, place and his own age. He
informed the doctors that the only thing he was able to remember
was the paramedics taking him into the ambulance. The patient's
blood pressure was 146/89 mmHg. Physical and neurological
examinations revealed no abnormalities at that time. Computed
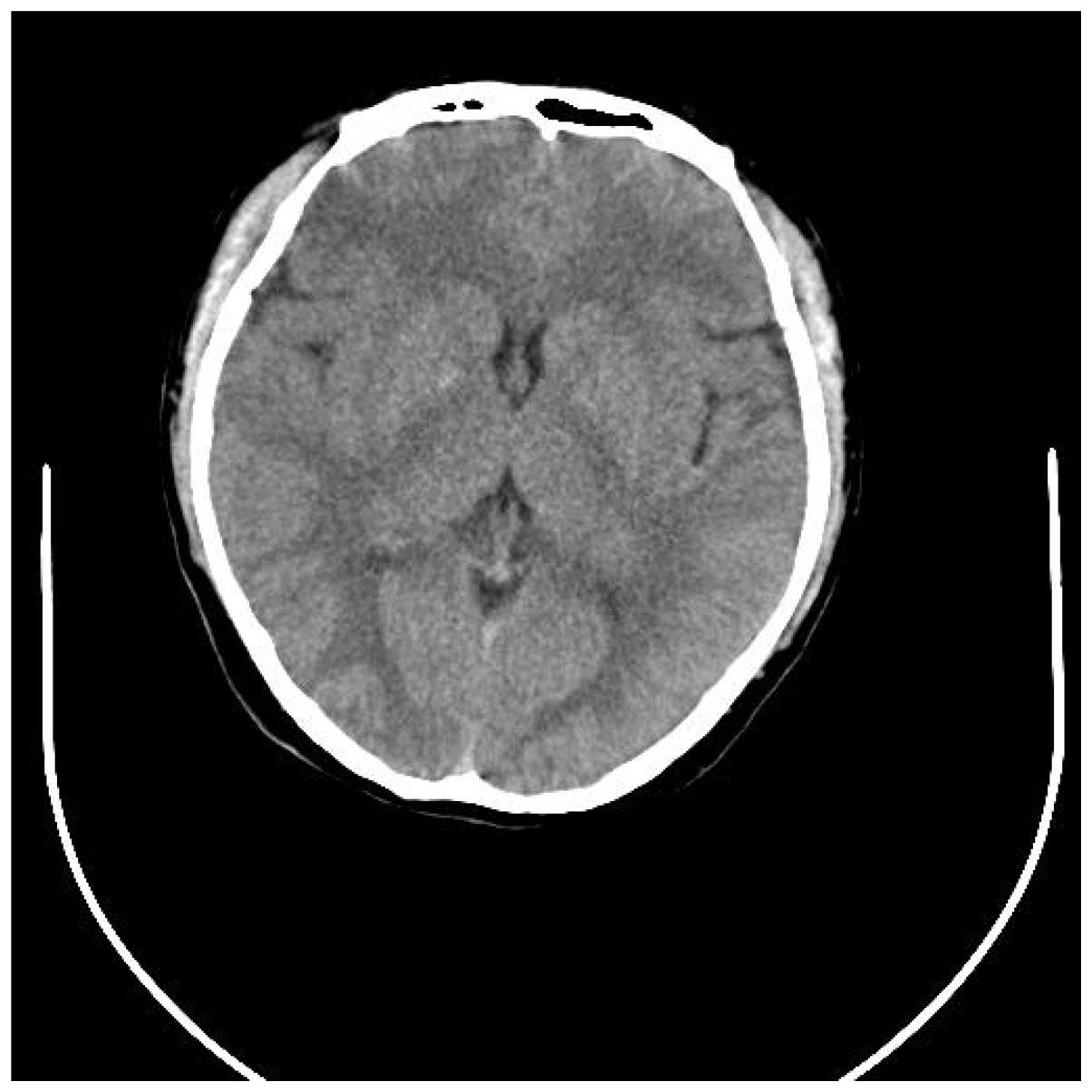
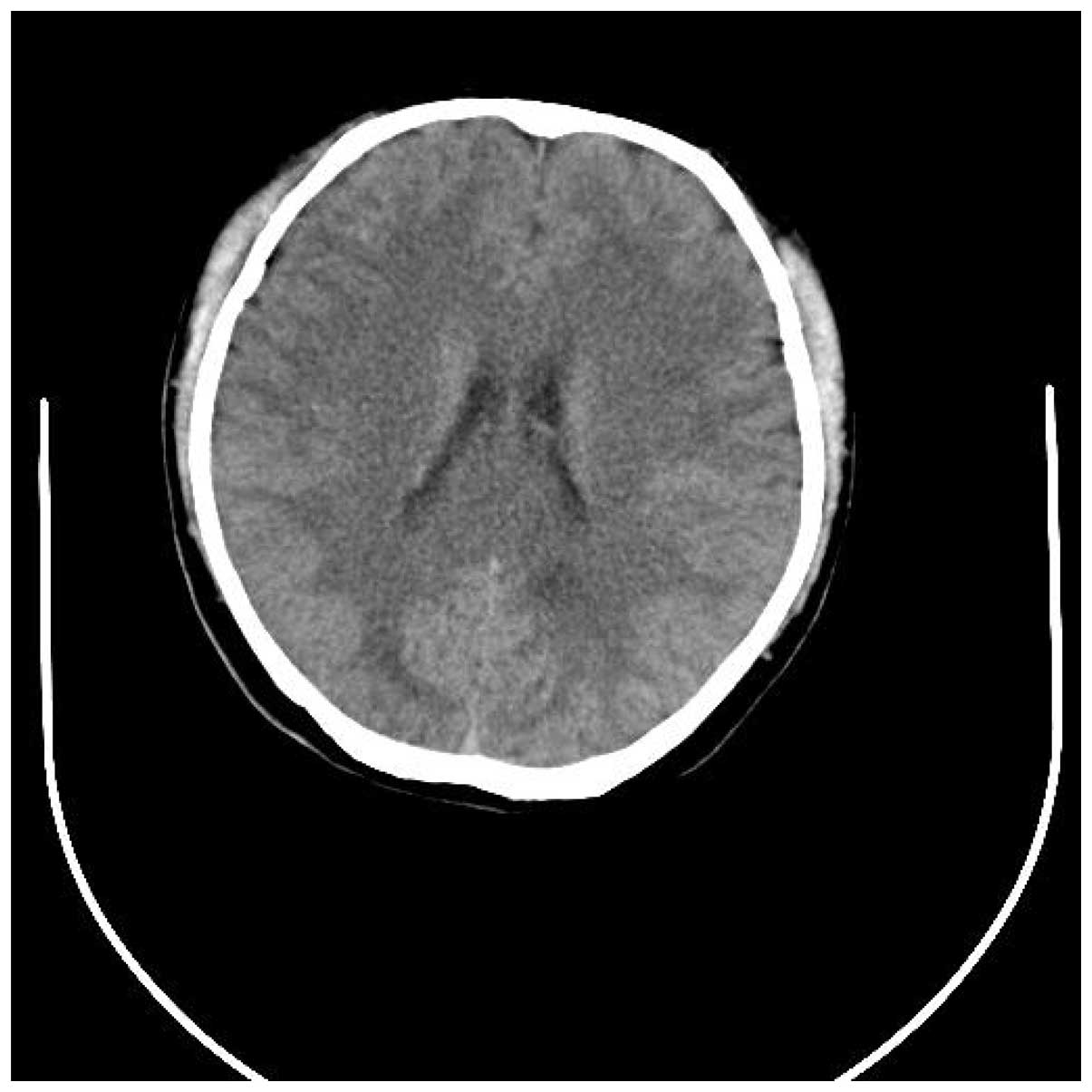
tomography (CT) of the head revealed a mild cerebral edema and a
region of minor hyperintensities in the right basal ganglia
(Figs. 1 and 2). Laboratory data was predominantly
normal. The patient was admitted to a ward of the neurosurgery
department 6 h after arrival in the emergency department.
The patient began to experience a low-grade fever on
day 2, the highest temperature of which, determined by axillary
measurement, reaching 37.2°C. The patient lost consciousness
following a sudden convulsive seizure on the 3rd day after
admission. A repeat CT head scan was performed, and magnetic
resonance imaging and magnetic resonance angiography (MRA) scans
revealed a mild cerebral edema, bilateral centrum semiovale and
subcortex discrete lacunar infarction. MRA results were normal (20%
mannitol, 125 ml IV q12h). The patient was administered mannitol
intravenously to alleviate the brain edema. The patient also
underwent a lumbar puncture on day 3. The results of cerebrospinal
fluid (CSF) analysis were abnormal (Table I). A pink color and increase of red
blood cells in the CSF was considered to be indicative of
haemorrhage during the procedure. The neurologist believed than an
infection of the central nervous system should not be eliminated as
a possible cause. Therefore, the patient received anti-epileptic
(sodium valproate), antibiotic and antiviral treatment (sulperazon,
amikacin and acyclovir). The patient's axillary temperature rose to
37.9°C on the morning of the 4th day, but the fever remitted in the
evening and the patient regained consciousness at approximately the
same time. His temperature remained normal for the following 3
days, and the patient underwent further lumbar puncture and CSF
analysis, in addition to a T-SPOT TB test. The T-SPOT TB test was
negative and the results of CSF analysis (Table II) were similar to those of the
initial analysis displayed in Table
I. The neurologist administered the patient ceftriaxone sodium,
amikacin and acyclovir.
 | Table I.Results of the cerebrospinal fluid
analysis on day 3. |
Table I.
Results of the cerebrospinal fluid
analysis on day 3.
| Parameter | Result | Reference range |
|---|
| Opening pressure | 17
cmH2O | 5–18
cmH2O |
| Glucose | 3.7 mmol/l | 3.3–4.4 mmol/l |
| Protein | 380.1 mg/l | 150–450 mg/l |
| Chloridum | 121 mmol/l | 120–132 mmol/l |
| Leukocytes |
14×106/l |
0–8×106/l |
| Erythrocytes |
20,000×106/l |
<10×106/l |
| Gross appearance | Pink | Clear and
colorless |
| Culture | Sterile | Sterile |
| Gram stain | Negative | Negative |
| Table II.Result of the repeated cerebrospinal
fluid analysis. |
Table II.
Result of the repeated cerebrospinal
fluid analysis.
| Parameter | Result | Reference range |
|---|
| Opening pressure | 16
cmH2O | 5–18
cmH2O |
| Glucose | 3.0 mmol/l | 3.3–4.4 mmol/l |
| Protein | 358.2 mg/l | 150–450 mg/l |
| Chloridum | 127 mmol/l | 120–132 mmol/l |
| Leukocytes |
21×106/l |
0–8×106/l |
| Erythrocytes |
220×106/l |
<10×106/l |
| Gross appearance | Pink | Clear and
colorless |
| Culture | sterile | Sterile |
| Gram stain | ± | Negative |
On day 8, the patient began to convulse and became
intermittently confused without displaying any indicators of a
fever, and lost consciousness after a sudden convulsive seizure on
the 9th day. An electroencephalogram performed on the 9th day
revealed that activity in the two frontal lobes and central zone
included 1–3 Ci/sec (Hz) high electric potential δ wave and
triphase waves, which were produced by increased activity in the
frontal lobes; metencephalon activity consisted of 3–5 c/s medium
electric potential δ and θ waves, and brain waves were more rapid
following the application of a stimulus. The neurologist believed
that the possibility of Creutzfeldt-Jakob disease should not be
eliminated, however, the family refused an additional lumbar
puncture intended to be used to identify the relevant proteins. The
plasma ammonium was observed to be higher than the normal range on
the 9th day (170 µmol/l; reference range, 9–33 µmol/l).
Geneticist and gastroenterologist consultations were
requested immediately. The geneticist suggested that a citrin
deficiency was possible. Tandem ammonia mass spectrometry (MS/MS)
analysis of the blood and gas chromatography/mass spectrometry
(GC/MS) profiling of urine samples from the patient were performed.
The results of the MS/MS analysis of the patient's blood revealed
high citrulline and free carnitine concentrations and a low
acylcarnitine concentration at certain points (Table III). The results of the GC/MS
analysis of a urine sample indicated that the concentrations of
tyrosine metabolites such as 4-hydroxy phenyl lactic acid-2 and
4-hydroxy phenylpyruvic acid-OX-2 were high compared with the
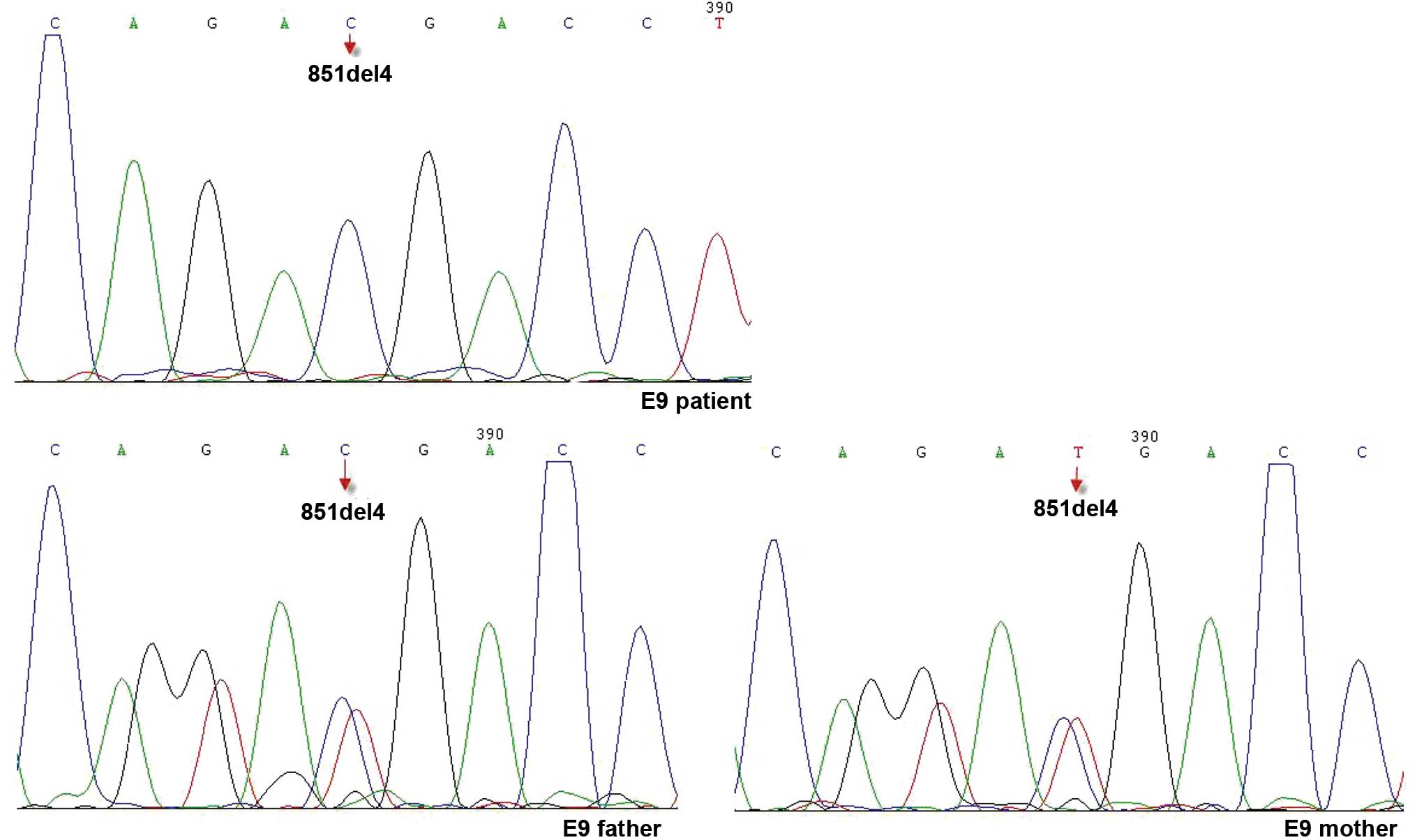
reference range (Table IV). Genomic
DNA was extracted from the peripheral blood of the patient and his
parents after obtaining written informed consent. The sequences of
the SLC25A13 gene were determined. A single mutation in 851del4
inherited from the patient's parents was identified (Fig. 3).
| Table III.MS/MS of blood. |
Table III.
MS/MS of blood.
| Assessed index | Result | Unit | Level | Reference range |
|---|
| Ala-1 | 59.38 | µM | Low | 60–300 |
| Asp-1 | 32.16 | µM |
| 10–80 |
| Glu-1 | 66.04 | µM |
| 45–200 |
| Met-1 | 12.61 | µM |
| 8–35 |
| Phe-1 | 32.39 | µM |
| 20–120 |
| Tyr-1 | 34.81 | µM |
| 20–100 |
| Leu-1 | 56.52 | µM |
| 50–250 |
| Trp-1 | 11.79 | µM |
| 10–75 |
| Val-1 | 85.9 | µM |
| 80–300 |
| Arg-1 | 8.9 | µM |
| 1.5–25 |
| Cit-1 | 59.3 | µM | High | 7–35 |
| Gly-1 | 134.65 | µM |
| 90–350 |
| Orn-1 | 28.64 | µM |
| 15–80 |
| Gln-1 | 8.33 | µM |
| 6–30 |
| His-1 | 59.44 | µM |
| 10–300 |
| Ser-1 | 28.99 | µM |
| 20–100 |
| Thr-1 | 36.48 | µM |
| 15–100 |
| Arg/Orn-1 | 0.31 |
|
| 0–0.7 |
| Cit/Arg-1 | 6.66 |
|
| 0.6–10 |
| Orn/Cit-1 | 0.48 |
| Low | 0.8–4 |
| Met/Phe-1 | 0.39 |
|
| 0.2–0.6 |
| Leu/Phe-1 | 1.73 |
|
| 1.5–4 |
| Phe/Tyr-1 | 0.93 |
|
| 0.5–2 |
| Gly/Phe-1 | 4.16 |
|
| 1.5–10 |
| C0-1 | 6.6 | µM | Low | 10–60 |
| C2-1 | 2.98 | µM | Low | 6–30 |
| C3-1 | 0.52 | µM |
| 0.5–4 |
| C3DC-1 | 0.023 | µM |
| 0–0.4 |
| C4-1 | 0.098 | µM |
| 0.06–0.5 |
| C4-OH-1 | 0.038 | µM |
| 0–0.35 |
| C4DC-1 | 0.28 | µM |
| 0.2–1.2 |
| C5-1 | 0.04 | µM |
| 0.04–0.3 |
| C5:1-1 | 0.016 | µM |
| 0–0.1 |
| C5-OH-1 | 0.16 | µM |
| 0.06–0.6 |
| C5DC-1 | 0.033 | µM |
| 0–0.2 |
| C6-1 | 0.035 | µM |
| 0.01–0.15 |
| C6:1-1 | 0.015 | µM |
| 0–0.1 |
| C6DC-1 | 0.019 | µM |
| 0–0.06 |
| C8-1 | 0.035 | µM |
| 0.01–0.3 |
| C8:1-1 | 0.016 | µM | Low | 0.03–0.5 |
| C8DC-1 | 0.018 | µM |
| 0–0.04 |
| C10-1 | 0.032 | µM |
| 0.02–0.5 |
| C10:1-1 | 0.042 | µM |
| 0.03–0.45 |
| C12-1 | 0.025 | µM |
| 0.02–0.2 |
| C12:1-1 | 0.03 | µM |
| 0–0.02 |
| C14-1 | 0.035 | µM |
| 0.02–0.25 |
| C14:1-1 | 0.053 | µM |
| 0.01–0.3 |
| C14-OH-1 | 0.01 | µM |
| 0–0.06 |
| C16-1 | 0.23 | µM | Low | 0.3–2 |
| C16:1-1 | 0.016 | µM |
| 0–0.2 |
| C16-OH-1 | 0.09 | µM |
| 0–0.05 |
| C18-1 | 0.21 | µM |
| 0.2–1.2 |
| C18:1-1 | 0.3 | µM | Low | 0.3–1.8 |
| C18-OH-1 | 0.006 | µM |
| 0–0.03 |
| Table IV.Gas chromatography/mass spectrometry
analysis of the patient's urine. |
Table IV.
Gas chromatography/mass spectrometry
analysis of the patient's urine.
| Assessed index | Result | Abnormal | Reference
range |
|---|
| PA-OX-2 | 1.5 |
| 0–24.1 |
| VPA-1 | 4.86 | Yes | 0 |
| Carbamide-2 | 8.51 |
| 104.6–763 |
|
2-Keto-3-Methylpentanoic acid-2 | 0.17 |
| 0 |
| Orthophosphoric
acid-3 | 31.59 |
| 0–43 |
| Succinic
acid-2 | 3.5 |
| 6.5–65.8 |
| Uracil-2 | 2.2 |
| 0–7 |
| Malic acid-3 | 0.22 |
| 0–0.7 |
| Hexanedioic
acid-2 | 3.98 |
| 0.5–5 |
| 2-propyl-hydroxy
glutaric acid-2 | 12.99 | Yes | 0 |
|
7-hydroxyoctanoate-2 | 0.14 | Yes | 0 |
|
2-hydroxyglutarate-3 | 0.86 |
| 0.6–5.9 |
| Phenyllactic
acid-2 | 1.54 |
| 0–4.9 |
| Pimelic acid-2 | 0.73 |
| 0–9.3 |
|
3-hydroxyphenylacetic acid-2 | 0.25 |
| 0–0.9 |
|
4-hydroxyphenylacetic acid | 25.14 |
| 8.6–73.2 |
|
2-ketoglutarate-OX-2 | 0.35 |
| 0.3–21.3 |
| N-phophonoacetyl
aspartic acid-2 | 0.31 |
| 0–3.7 |
|
2-hydroxyhexanedioic acid-3 | 3.92 | Yes | 0–2 |
| Octandioic
acid-2 | 0.69 |
| 0.4–4.7 |
| Orotic acid-3 | 0.29 |
| 0–1.5 |
| Vanillic
acid-2 | 0.18 |
| 0 |
| Azelaic acid-2 | 1.01 |
| 9–10.7 |
| Citric acid-4 | 173.21 |
| 31.4–572.3 |
| Alkapton-3 | 0.08 |
| 0–1.4 |
| Hippuric
acid-1 | 3.75 |
| 6.2–284.1 |
| VMA-3 | 13.41 |
| 11.7–84.6 |
| Sebacic acid-2 | 0.72 |
| 0.4–7 |
| Decadienoic
acid-3 | 1.08 |
| 0–2.3 |
| 4-hydroxy phenyl
lactic acid-2 | 59.1 | Yes | 0–7 |
| 4-hydroxy
phenylpyruvic acid-OX-2 | 3.96 | Yes | 0–0.9 |
| Hexadecanoic
acid-1 | 10.38 |
| 0–13.8 |
| 2-hydroxyhippuric
acid-3 | 3.96 |
| 0 |
Subsequent to the diagnosis of CTLN2, the patient
was treated with arginine and a carbohydrate-restricted high-fat
high-protein diet. The patient also received an invasive mechanical
treatment for pulmonary infection and respiratory failure, and was
then discharged. The patient returned to the emergency department
of Xin Hua Hospital Affiliated to Shanghai Jiao Tong University
School of Medicine 7 months after discharge as a result of sudden
alterations to consciousness, with no obvious extrinsic causes. The
plasma ammonium level was 40 µmol/l, which was close to normal. The
emergency physician initiated treatment with arginine after
excluding other possible diseases, which may have lead to an
alteration to the level of consciousness. The patient recovered 2 h
later and was discharged from the hospital the following day.
However, the patient returned to the emergency department again
several days later with hyperammonemia (88 µmol/l). A physician
administered arginine for several days. The patient is currently
still undergoing follow-up examination.
Discussion
The term CITRIN was coined in 1999 to represent the
protein product encoded by the SLC25A13 gene, which was localized
to chromosome 7q21.3 and determined to be the causative gene of
adult-onset CTLN2 (3,4). SLC25A13 mutations result in citrin
deficiency (CD). CTLN2 was the initial CD phenotype to be
described, occurring in adolescents or adults, and the prognosis is
typically poor (4,5). Manifestations include recurrent
hyperammonemia with neuropsychiatric symptoms including nocturnal
delirium, aggression, delusions, disorientation, restlessness, loss
of memory, convulsive seizures and coma; death can result from
brain edema. Symptoms are often induced as a result of alcohol and
sugar intake, medication, infection and/or surgery (5).
In the human body, the urea cycle is the major
pathway used for the removal of waste nitrogen, and aberrances of
this pathway exist, as in CTLN2 (6).
Citrullinemia is caused by a deficiency of the urea cycle enzyme
argininosuccinate synthetase, which catalyzes the ligation reaction
of citrulline (Cit) and aspartate to form argininosuccinate. The
loss of argininosuccinate synthetase activity results in an
accumulation of plasma Cit and ammonia in patients (7).
The current case was the first treatment of a CTLN2
patient in the emergency department of Xin Hua Hospital, although
this diagnosis was not apparent initially due to atypical symptoms,
including sudden delirium and upper limb weakness, which usually
indicate shock. CTLN2 is characterized by recurrent encephalopathy
with hyperammonemia and may be misdiagnosed as other neurological
or psychiatric diseases (8,9). The present study did not assess the
plasma ammonia levels until the 9th day, and led to a positive
diagnosis the following day. The oversight in failing to assess
plasma ammonia levels was a hindrance to a rapid diagnosis. Cases
of hyperammonemia that present without a history of hepatic disease
are usually the primary indicator of abnormal laboratory data.
CTLN2 should be considered in the event of neuropsychiatric
manifestations, even in cases presenting without hyperammonemia.
For instance, Takahashi et al (2), reported an adult-onset type II
citrullinemia case without hyperammonemia. From the current case,
it was determined that the patient may display neuropsychiatric
symptoms even with a lower plasma ammonium level than those usually
associated with CTLN2.
The most successful therapy to date for the
treatment of CTLN2 has been liver transplantation, which prevents
episodic hyperammonemic crises, corrects metabolic disturbances and
eliminates necessity for protein-rich foods (4,8,10–12).
Administration of arginine was reported to be effective in
decreasing plasma ammonium concentration. Reducing
calorie/carbohydrate intake and increasing protein intake
ameliorates hypertriglyceridemia (13). The present patient was treated with
arginine and a carbohydrate-restricted, high-fat and high-protein
diet. The patient's family would not consider liver transplantation
due to the high cost associated with this. It was appropriate for
the physician to select mannitol in preference to glycerol as
glycerol contains 5% fructose, which may affect cytosolic
nicotinamide adenine dinucleotide (NADH) production and aggravate
the symptoms (9). It is recommended
that any high-sugar solutions such as fructose and glucose should
be excluded from the diet as they may elevate the cytosolic
NADH/NAD+ ratio in the liver and exacerbate the encephalopathy
(9,14,15).
In conclusion, emergency physicians should consider
CTLN2 as a differential diagnosis in patients with sudden
neuropsychiatric symptoms, and plasma ammonium levels should not be
ignored.
References
|
1.
|
Nagata N, Matsuda I and Oyanagi K:
Estimated frequency of urea cycle enzymopathies in Japan. Am J Med
Genet. 39:228–229. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Takahashi Y, Koyama S, Tanaka H, Arawaka
S, Wada M, Kawanami T, Haga H, Watanabe H, Toyota K, Numakura C, et
al: An elderly Japanese patient with adult-onset type II
citrullinemia with a novel D493G mutation in the SLC25A13 gene.
Intern Med. 51:2131–2134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Kobayashi K, Sinasac DS, Iijima M, Boright
AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, et
al: The gene mutated in adult-onset type II citrullinaemia encodes
a putative mitochondrial carrier protein. Nat Genet. 22:159–163.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Song YZ, Zhang ZH, Lin WX, Zhao XJ, Deng
M, Ma YL, Guo L, Chen FP, Long XL, He XL, et al: SLC25A13 gene
analysis in citrin deficiency: Sixteen novel mutations in East
Asian patients, and the mutation distribution in a large pediatric
cohort in China. PLoS One. 8:e745442013. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Kobayashi K, Saheki T and Song YZ: Citrin
Deficiency. GeneReviews. Pagon RA, Bird TD, Dolan CR, Stephens K
and Adam MP: (Seattle, WA). University of Washington. 2005.
|
|
6.
|
Honda S, Yamamoto K, Sekizuka M, Oshima Y,
Nagai K, Hashimoto G, Kaneko H, Tomomasa T, Konno Y and Horiuchi R:
Successful treatment of severe hyperammonemia using sodium
phenylacetate powder prepared in hospital pharmacy. Biol Pharm
Bull. 25:1244–1246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Sinasac DS, Moriyama M, Jalil MA, Begum L,
Li MX, Iijima M, Horiuchi M, Robinson BH, Kobayashi K, Saheki T and
Tsui LC: Slc25a13-knockout mice harbor metabolic deficits but fail
to display hallmarks of adult-onset type II citrullinemia. Mol Cell
Biol. 24:527–536. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Ikeda S, Yazaki M, Takei Y, Ikegami T,
Hashikura Y, Kawasaki S, Iwai M, Kobayashi K and Saheki T: Type II
(adult onset) citrullinaemia: Clinical pictures and the therapeutic
effect of liver transplantation. J Neurol Neurosurg Psychiatry.
71:663–670. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Takahashi H, Kagawa T, Kobayashi K,
Hirabayashi H, Yui M, Begum L, Mine T, Takagi S, Saheki T and
Shinohara Y: A case of adult-onset type II
citrullinemia-deterioration of clinical course after infusion of
hyperosmotic and high sugar solutions. Med Sci Monit. 12:CS13–CS15.
2006.PubMed/NCBI
|
|
10.
|
Yazaki M, Hashikura Y, Takei Y, Ikegami T,
Miyagawa S, Yamamoto K, Tokuda T, Kobayashi K, Saheki T and Ikeda
S: Feasibility of auxiliary partial orthotopic liver
transplantation from living donors for patients with adult-onset
type II citrullinemia. Liver Transpl. 10:550–554. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Hirai I, Kimura W, Suto K, Fzjimoto H,
Watanabe T, Fuse A, Kobayashi K, Iijima M, Saheki T, Nakatsuka T,
et al: Living donor liver transplantation for type II citrullinemia
from a heterozygous donor. Hepatogastroenterology. 55:2211–2216.
2008.PubMed/NCBI
|
|
12.
|
Kobayashi K and Saheki T: Molecular basis
of citrin deficiency. Seikagaku. 76:1543–1559. 2004.(In Japanese).
PubMed/NCBI
|
|
13.
|
Imamura Y, Kobayashi K, Shibatou T,
Aburada S, Tahara K, Kubozono O and Saheki T: Effectiveness of
carbohydrate-restricted diet and arginine granules therapy for
adult-onset type II citrullinemia: A case report of siblings
showing homozygous SLC25A13 mutation with and without the disease.
Hepatol Res. 26:68–72. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Saheki T, Kobayashi K, Iijima M, Horiuchi
M, Begum L, Jalil MA, Li MX, Lu YB, Ushikai M, Tabata A, et al:
Adult-onset type II citrullinemia and idiopathic neonatal hepatitis
caused by citrin deficiency: Involvement of the aspartate glutamate
carrier for urea synthesis and maintenance of the urea cycle. Mol
Genet Metab. 81(Suppl 1): S20–S26. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Yazaki M, Takei Y, Kobayashi K, Saheki T
and Ikeda S: Risk of worsened encephalopathy after intravenous
glycerol therapy in patients with adult-onset type II citrullinemia
(CTLN2). Intern Med. 44:188–195. 2005. View Article : Google Scholar : PubMed/NCBI
|