Introduction
The present study presents the case of a 41 year-old
male patient admitted to the Department of Prenatal Diagnosis,
Women & Children's Hospital of Sichuan Province (Chengdu,
China) in November 2012. The age of onset of the disease was 35
years, at which time the patient had difficulty in climbing stairs,
fatigue, weakness and markedly elevated serum creatine kinase
(level reached, 580 U/l; normal level, ~200U/l). An electromyogram
(EMG) revealed myogenic damage. Thus, the patient was suspected to
have progressive muscular dystrophy, and advised to undergo a
muscle biopsy and an assessment to uncover the presence of any
disease-associated genes. However, the patient declined both
options. In the following years, the patient experienced difficulty
performing squats and washes, lacked strength to walk and his
proximal muscle indicated atrophy, though the patient had no
difficulty in swallowing. No similar cases had been reported in the
patient's family. Due to pregnancy, the wife of the patient wished
to know the impact of the disease on future generations and
required a clear diagnosis. However, the patient refused a muscle
biopsy and the consulting physician only examined the
disease-associated genes of the patient and his family.
Patients and methods
Subjects
The research subjects in the present study included
4 individuals, the patient, whose father was no longer alive, and
the patient's mother, sister and wife.
Gene fragment acquisition
Venous blood was obtained (5 ml) from each of the
subjects. DNA was extracted according to standard protocol using a
QIAamp DNA Blood Mini kit (Qiagen GmbH, Hilden, Germany).
Subsequently, DNA was degraded into fragments of 200–250 bp in
length using a Covaris S2 device, (Covaris, Inc., Woburn, MA, USA)
and purified using Ampure XP Beads (Beckman Coulter, Inc., Brea,
CA, USA). Following the treatment of 1 µg purified DNA fragments
with terminal repair, and the addition of an ‘A’ by ligation
reaction, a DNA library for each patient was created. The DNA
library was combined with the gene microarray ‘Human Sequence
Capture 2.1 M Array’ (Roche Applied Science, Madison, WI, USA) at
42°C for 72 h. Following this, the gene microarray was washed and
an elution reaction was performed, followed by an amplification
step. Enrichment of the samples was assessed using an Agilent 2100
Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA) and
ABI StepOne (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Finally, the samples were treated with continuous bi-directional
sequencing for 90 cycles by an Illumina HiSeq 2000 sequencing
system (Illumina, Inc., SanDiego, CA, USA) and the original
sequencing data were revealed using Illumina Pipeline software
(Illumina, Inc; version 1.3.4). A total of 399 genes were
identified, covering all exons in addition to 10 bp on either side,
which were associated with 659 types of neuromuscular disorder,
including hypotypes.
Data analyses
The sequencing quality of raw reads were evaluated,
in order to remove reads of low quality and those containing
partial adapter sequences. Sequences were compared with Burrows
Wheeler Aligner (BWA; Multi-Vision software package; version
0.7.12-r1039) and HG19 software (1).
Concurrently, sequence capture was evaluated using SOAPsnp (version
1.03), SAOPindel (version 2.01) and Samtools software (version
0.1.18) (2,3). Initially, single nucleotide variants,
insertion sand deletions were excluded using information from
databases including NCBI dbSNP, HapMap, the 1,000 human genomes
project dataset and a database of 100 Chinese healthy adults.
Suspicious mutations were screened and non-pathogenic polymorphism
sites were excluded, such as same sense mutations (4). The experimental protocol, data
processing and analysis were completed with the assistance of the
Beijing Genomics Institute (Beijing, China).
Validation by the Sanger method
Primers were designed both upstream and downstream
for all the fragments the pathogenic point mutation was present in.
The primers were subsequently amplified by polymerase chain
reaction and products were sequenced with the Sanger method. The
thermocycling conditions were as follows: First phase, 2 min at
94°C, followed by the second phase, 15 sec at 94°C, 30 sec at 58°C
and 30 sec at 72°C for 14 cycles. The third phase was 5 min at
72°C, followed by 4°C. The results were compared with the standard
gene sequence to validate the results of the microarray and
high-throughput sequencing. Primer information for c.144+1G>A
and c.342+1G>T in the DYSF gene are displayed in Table I.
 | Table I.Primer information validated by the
Sanger method. |
Table I.
Primer information validated by the
Sanger method.
| Location | Primer | Sequence (5′-3′) |
|---|
| c.144+1 G>A | F |
TCTCACCATCGCACTCCAG |
|
| R |
GAAGGCACCTCCTCCACAA |
| c.342+1 G>T | F |
ATCTGAGTGGTGGCAGTGAG |
|
| R |
GCAGGAAGAACAACGGAGGA |
Results
Exon capture and sequence analysis
revealed 54 point mutations in the DYSF gene
Only two splice site mutations were novel
identifications, all others were also present in the general
population. Once validated by the Sanger method, the results were
consolidated by exon capture and sequence analysis. The patient
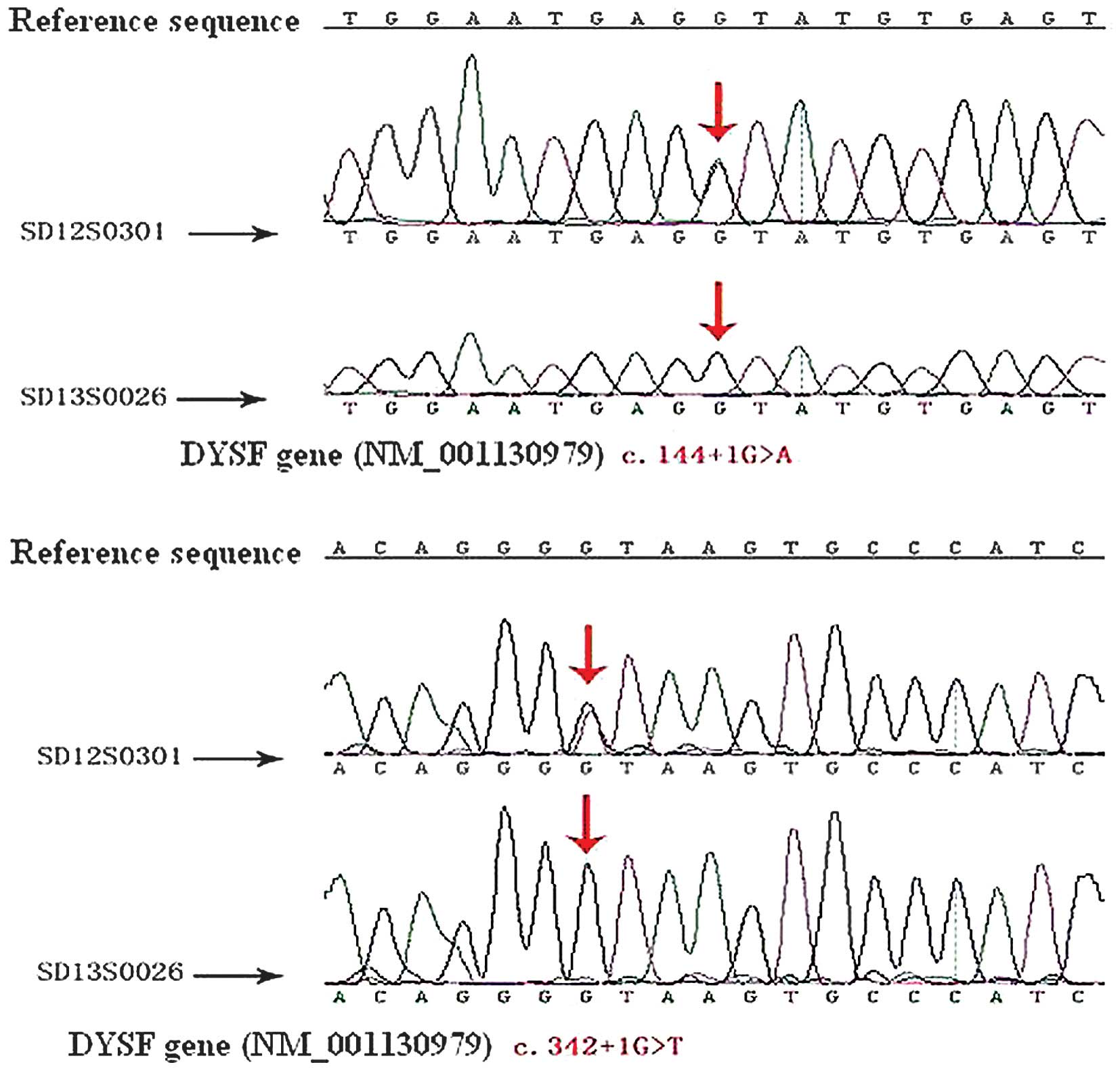
possessed two splice site mutation in the DYSF gene, c.144+1G>A
and c.342+1G>T (Fig. 1). The
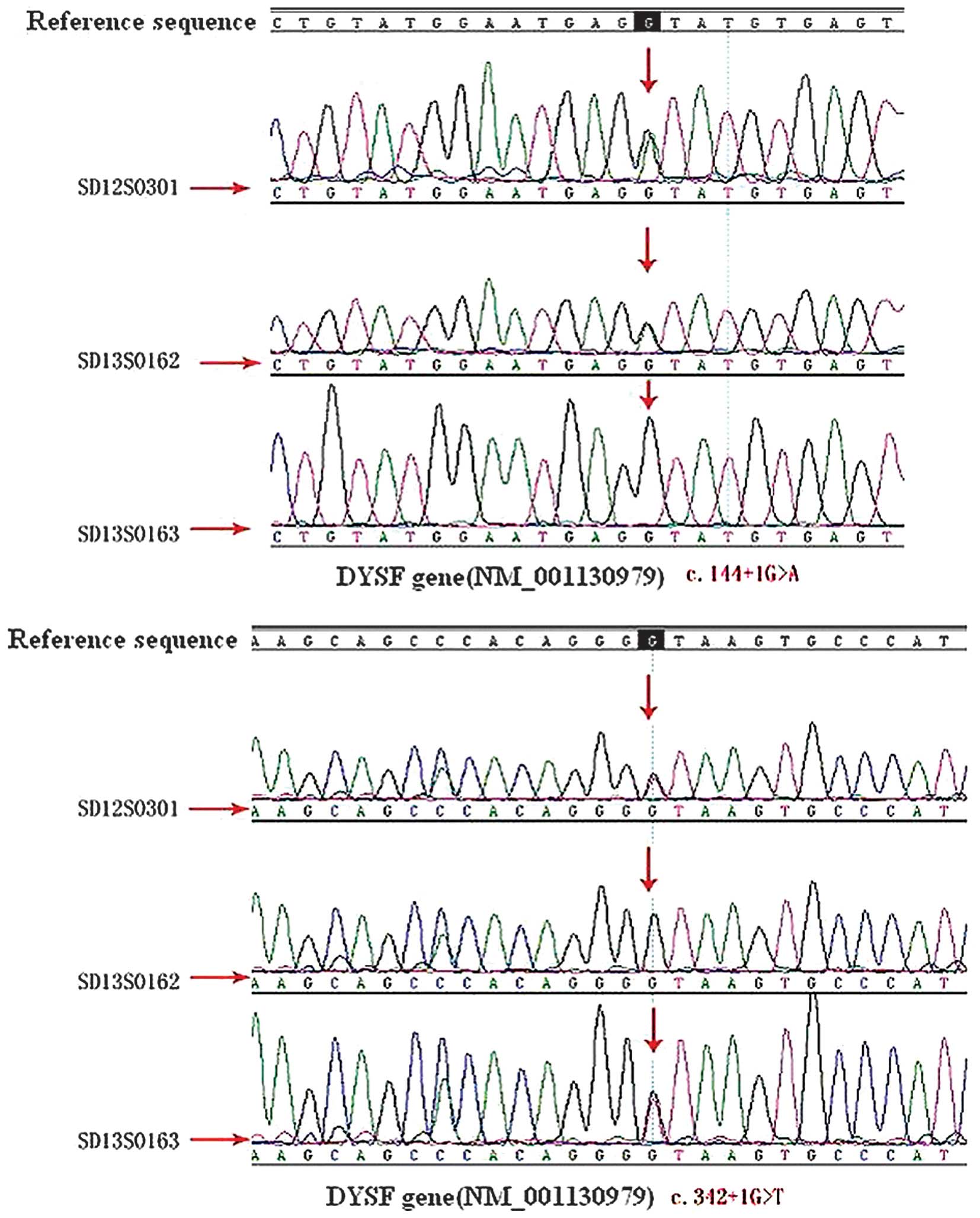
patient's mother and sister also had mutations, the heterozygotic
DYSF c.342+1G>T splice site mutation was observed in the
patient's mother, whereas the patient's sister had the
c.144+1G>A, DYST splice site mutation, also heterozygotic
(Fig. 2). Thus, the patient's father
may have been a carrier of the splice site mutation with
c.144+1G>A. In addition, the patient's wife did no carry the
gene which can cause limb-girdle muscular dystrophy type 2B
(LGMD2B) (Fig. 1).
Discussion
LGMD, also known as Erb's muscular dystrophy, is a
genetically heterogeneous group of rare muscular dystrophies, which
also display clinical diversity (5)
LGMD predominantly affects the hip and shoulder muscles and is
characterized by progressive muscle wasting (6). The LGMD type 1 family is autosomal
dominant, whereas the LGMD type 2 family is autosomal recessive.
LGMD2B arises due to mutations in the gene encoding DYSF, a
skeletal muscle protein located on chromosome 2p12-14 (7). Bashir et al (8) previously described age at onset ranged
from 15–25 years and patients typically present with fatigue,
weakness, difficulty climbing stairs, and markedly elevated serum
creatine kinase. The study also reported that EMG revealed
myopathic changes, and skeletal muscle biopsies revealed severe
myopathic changes, including variations in fiber size, fiber
splitting, an increase in connective tissue in addition to sporadic
necrotic changes (8). Disease
progression was observed to be relatively slow. Amino acid sequence
analysis of the DYSF protein has revealed 7 sites that correspond
to caveolin-3 scaffold-binding motifs, and 1 site that is a
potential target to bind the WW domain of the caveolin-3 protein.
One function of dysferlinis is to interact with caveolin-3 to
subserve signaling functions of caveolae (9). Therefore, DYSF may repair the damage to
sarcolemma caused by muscle contraction, which is why LGMD2B
patients may have slowly progressive muscular weakness of the lower
limbs, beginning in the late second decade of life. This may
explain the later age at onset of the 41 year-old male patient in
the present study. The DYSF protein is encoded by the DYSF gene
(10), which is associated with
skeletal muscle repair (2). DYSF has
predominantly been investigated for its role in a cellular process
known as membrane repair (2). It has
previously been revealed that, in patients with LGMD2B, the absence
of DYSF prevents the repair of damage to the muscle fiber membrane
(11).
Bashir et al (8) also reported that the virulence genes of
LGMD2B were associated with two sites on the short arm of
chromosome 2, D2S134 and D2S136, as revealed by linkage analysis,
and also located pathogenic genes on chromosome 2p13–16 (2,12,13).
Passos-Bueno et al (10)
further determined the location of DYSF to be on the short arm of
chromosome 2 between D2S292 and D2S286 following the study of a
Brazilian family. Furthermore, Bashir et al (12) located a virulence gene of LGMD2B
between D2S2113 and D2S2112 in 1996 by haplotyping (14). The full length of the DYSF gene is
~150 kb and consists of 55 exons and an open reading frame of 6243
bp (15). The coding sequence (CDS)
of the DYSF gene is in the 377–6712 interval of the mRNA sequence,
encompassing 6336 bp (16,17).
In the present study, when splice site mutation
c.144+1G>A on exon 2 occurred, splice site GU moved 4 bases
backward, leading to the insert size of ATAT following the 144th
base of CDS, which resulted in an abnormal translation from 49th
amino acids. In addition, when splice site mutations c.342+1G>T
on exon 4 occurred, splice site GU moved 4 bases backward, leading
to the insert size of ATAA to the position following the 342nd base
of CDS, which resulted in an abnormal translation from the 115th
amino acid. Thus, abnormal expression levels of the DYSF protein
were observed. The patient's mother and sister carried different
splice site mutations, and the patient's father was suspected to
carry the same splice site mutations as the patient's sister. The
patient had two splice site mutations, one inherited from each
parent, which led to the abnormal expression of the DYSF gene,
resulting in LGMD2B. LGMD2B displays autosomal recessive
inheritance. As the patient's wife is not a carrier of the gene,
prenatal diagnosis of the fetus was not recommended following
genetic counseling.
The characteristic features of LGMD2B are late
onset, with no typical initial symptoms and slow progression
(18). Furthermore, numerous other
genes are also associated with myogenic damage. Therefore, a
definitive diagnosis of LGMD2B is complex. Types of LGMD include
LGMD2A, LGMD2B, LGMD2G, LGMD2H, LGMD2I, LGMD2J, LGMD2C, LGMD2D,
LGMD2E, and LGMD2F, of which the first six are known as ‘light
type’, and the latter four as ‘heavy type’ (19). Pathogenic genes are associated with
coding sarcoglycan, and each type was accompanied with the mutation
of associated genes (20). With
significant genetic heterogeneity, various clinical symptoms and
the complexity of classification, the diagnosis of LGMD is
difficult (21). In addition,
several aspects of the pathogenesis have yet to be elucidated, and
excluding the mutations of possible genes one at a time is also a
convoluted process. In the present study, 399 genes were captured
for targeted microarray and detected by next-generation sequencing
technology and exon capture and sequence analysis simultaneously.
Suspicious mutations were screened and any polymorphisms were
excluded. The results were verified by the Sanger method. The
diagnosis may also be made in the absence of a muscle biopsy and
immunohistochemical examination. Exon capture and sequence analysis
greatly improved the efficiency of diagnosis, and is an efficient
and accurate method for clinical and differential diagnosis.
In conclusion, exon capture high-throughput
sequencing is able to simultaneously sequence hundreds of genes,
significantly reducing the time required for clinical and
differential diagnosis, in particular, for cases of disease caused
by inconclusive or equivocal genes. However, it may have limited
application in the clinical diagnosis of disease in which the genes
involved are clear, due to the high costs of the procedure.
Acknowledgements
All data and experiments were performed by the
authors of the present study. The authors are grateful to Professor
Xiao ming Wei (BGI-Wuhan, Wuhan, China) for technical support and
assistance in the experimental process. This investigation received
no specific grant from any funding agency in the public, commercial
or not-for-profit sectors.
References
|
1
|
Bansal D, Miyake K, Vogel SS, Groh S, Chen
CC, Williamson R, McNeil PL and Campbell KP: Defective membrane
repair in dysferlin-deficient muscular dystrophy. Nature.
423:168–172. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fuson K, Rice A, Mahling R, Snow A, Nayak
K, Shanbhogue P, Meyer AG, Redpath GM, Hinderliter A, Cooper ST and
Sutton RB: Alternate splicing of dysferlin C2A confers
Ca2+-dependent and Ca2+-independent binding
for membrane repair. Structure. 22:104–115. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lek A, Evesson FJ, Lemckert FA, Redpath
GM, Lueders AK, Turnbull L, Whitchurch CB, North KN and Cooper ST:
Calpains, cleaved mini-dysferlinC72, and L-type channels underpin
calcium-dependent muscle membrane repair. J Neurosci. 33:5085–5094.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guglieri M, Magri F, D'Angelo MG, Prelle
A, Morandi L, Rodolico C, Cagliani R, Mora M, Fortunato F, Bordoni
A, et al: Clinical, molecular, and protein correlations in a large
sample of genetically diagnosed Italian limb girdle muscular
dystrophy patients. Hum Mutat. 29:258–266. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guglieri M, Straub V, Bushby K and
Lochmüller H: Limb-girdle muscular dystrophies. Curr Opin Neurol.
21:576–584. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wenz T, Diaz F, Hernandez D and Moraes CT:
Endurance exercise is protective for mice with mitochondrial
myopathy. J Appl Physiol 1985. 106:1712–1719. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Angelini C, Nardetto L, Borsato C, Padoan
R, Fanin M, Nascimbeni AC and Tasca E: The clinical course of
calpainopathy (LGMD2A) and dysferlinopathy (LGMD2B). Neurol Res.
32:41–46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bashir R, Strachan T, Keers S, Stephenson
A, Mahjneh I, Marconi G, Nashef L and Bushby KM: A gene for
autosomal recessive limb-girdle muscular dystrophy maps to
chromosome 2p. Hum Mol Genet. 3:455–457. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsuda C, Hayashi YK, Ogawa M, Aoki M,
Murayama K, Nishino I, Nonaka I, Arahata K and Brown RH Jr: The
sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal
muscle. Hum Mol Genet. 10:1761–1766. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Passos-Bueno MR, Richard I, Vainzof M,
Fougerousse F, Weissenbach J, Broux O, Cohen D, Akiyama J, Marie SK
and Carvalho AA: Evidence of genetic heterogeneity in the autosomal
recessive adult forms of limb-girdle muscular dystrophy following
linkage analysis with 15q probes in Brazilian families. J Med
Genet. 30:385–387. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cagliani R, Fortunato F, Giorda R,
Rodolico C, Bonaglia MC, Sironi M, D'Angelo MG, Prelle A, Locatelli
F, Toscano A, et al: Molecular analysis of LGMD-2B and MM patients:
Identification of novel DYSF mutations and possible founder effect
in the Italian population. Neuromuscul Disord. 13:788–795. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bashir R, Britton S, Strachan T, Keers S,
Vafiadaki E, Lako M, Richard I, Marchand S, Bourg N, Argov Z, et
al: A gene related to Caenorhabditis elegans spermatogenesis
factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B.
Nat Genet. 20:37–42. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kerr JP, Ziman AP, Mueller AL, Muriel JM,
Kleinhans-Welte E, Gumerson JD, Vogel SS, Ward CW, Roche JA and
Bloch RJ: Dysferlin stabilizes stress-induced Ca2+ signaling in the
transverse tubule membrane. Proc Natl Acad Sci USA.
110:20831–20836. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Passos-Bueno MR, Bashir R, Moreira ES,
Vainzof M, Marie SK, Vasquez L, Iughetti P, Bakker E, Keers S and
Stephenson A: Confirmation of the 2p locus for the mild autosomal
recessive limb-girdle muscular dystrophy gene (LGMD2B) in three
families allows refinement of the candidate region. Genomics.
27:192–195. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bashir R, Keers S, Strachan T,
Passos-Bueno R, Zatz M, Weissenbach J, Le Paslier D, Meisler M and
Bushby K: Genetic and physical mapping at the limb-girdle muscular
dystrophy locus (LGMD2B) on chromosome 2p. Genomics. 33:46–52.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wenzel K, Carl M, Perrot A, Zabojszcza J,
Assadi M, Ebeling M, Geier C, Robinson PN, Kress W, Osterziel KJ
and Spuler S: Novel sequence variants in dysferlin-deficient
muscular dystrophy leading to mRNA decay and possible C2-domain
misfolding. Hum Mutat. 27:599–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Krahn M, Béroud C, Labelle V, Nguyen K,
Bernard R, Bassez G, Figarella-Branger D, Fernandez C, Bouvenot J,
Richard I, et al: Analysis of the DYSF mutational spectrum in a
large cohort of patients. Hum Mutat. 30:E345–E375. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nguyen K, Bassez G, Bernard R, Krahn M,
Labelle V, Figarella-Branger D, Pouget J, Hammouda H, Béroud C,
Urtizberea A, et al: Dysferlin mutations in LGMD2B, Miyoshi
myopathy, and atypical dysferlinopathies. Hum Mutat. 26:1652005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kong KY, Ren J, Kraus M, Finklestein SP
and Brown RH Jr: Human umbilical cord blood cells differentiate
into muscle in sjl muscular dystrophy mice. Stem Cells. 22:981–993.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Urtasun M, Poza JJ, Gallano P, Lasa A,
Sáenz A, Cobo AM, Leturcq F, de López Munain A and García-Bragado
F: Muscular dystrophy due to a mutation in the gene of
alpha-sarcoglycan subunit of dystrophin associated protein complex.
Med Clin. 110:538–542. 1998.(In Spanish).
|
|
21
|
Vainzof M, Anderson LV, McNally EM, Davis
DB, Faulkner G, Valle G, Moreira ES, Pavanello RC, Passos-Bueno MR
and Zatz M: Dysferlin protein analysis in limb-girdle muscular
dystrophies. J Mol Neuorsci. 17:71–80. 2001. View Article : Google Scholar
|