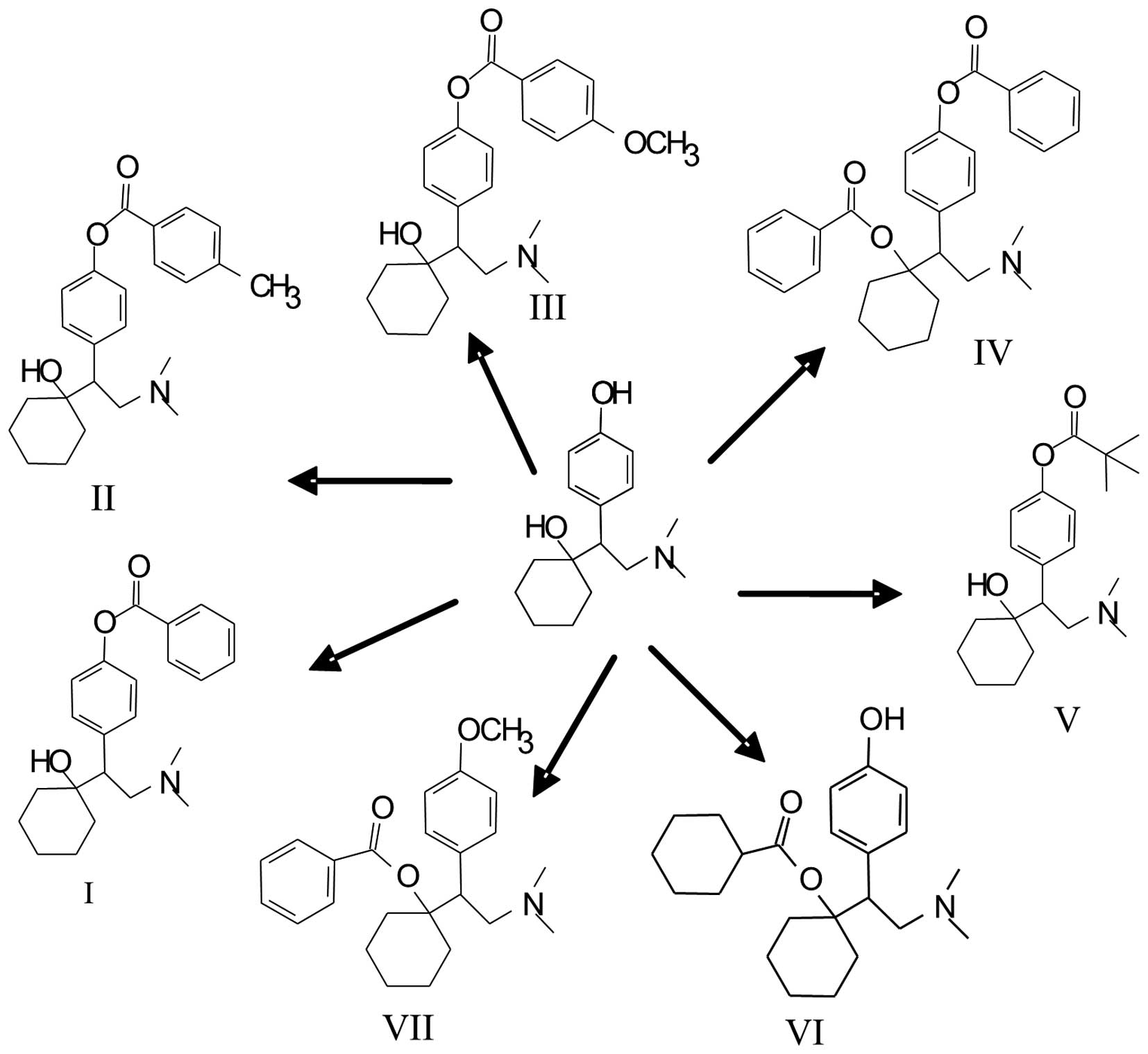
Introduction
Depression is a recurring and life-threatening
mental illness with a significant incidence in the population, and
represents a major social and economic burden (1). Although several classes of
antidepressant medications are currently prescribed to treat
depression, serious drawbacks exist that require improvement, such
as limitations in efficacy, multiple unwanted side effects, and
slow onset of the therapeutic response (2,3).
Development of new antidepressants is thus an urgent
requirement.
Venlafaxine is an antidepressant of the
serotonin-norepinephrine reuptake inhibitor (SNRI) class of drugs
that is prescribed for the treatment of major depressive disorder,
generalized anxiety disorder and comorbid indications in certain
anxiety disorders with depression (4). Venlafaxine is well absorbed in humans
and subject to extensive first-pass metabolism in the liver by
cytochrome P450 2D6 (CYP2D6) (5).
Thus, venlafaxine is susceptible to CYP2D6 polymorphism and the
associated variability in pharmacokinetics, efficacy and/or
tolerability (6–8). O-desmethylvenlafaxine (ODV), also known
as desvenlafaxine succinate, is a synthetic form of the major
active metabolite of venlafaxine with antidepressant activity
similar to that of venlafaxine but with a longer half-life
(9,10). Compared with venlafaxine, direct
intake of ODV for the treatment of diseases of the central nervous
system has the advantages of being a single compound that is
conducive to dosing adjustments and reducing the risk of
interactions with other drugs (11–15).
However, ODV contains a more exposed hydroxy group compared with
venlafaxine, and therefore it has increased hydrophilicity,
resulting in lower oral bioavailability. Accordingly, the
bioavailability of ODV was confirmed to be <40% in beagle dogs
(4). An important consequence of low
bioavailability is that direct side effects of the unabsorbed drug
in the system may be higher. Thus, it is crucial to improve the
absorption and bioavailability of ODV.
Prodrugs are usually designed to improve passive
and/or transporter-mediated intestinal absorption to increase oral
bioavailability (16–19) and/or tissue-selective delivery
(20). Between species differences
of hydrolase activity in small intestine and liver result in
different exposures to prodrug and active form, and hence different
pharmacological effects and toxicities after administration of
prodrugs in different animal species, including humans (21,22). The
ODV molecule contains two hydroxyls: A phenolic hydroxyl and an
alcoholic hydroxyl. In an attempt to increase the bioavailability
of ODV in the present study, different vectors were used to bond to
a phenolic hydroxyl or two hydroxyls, then seven ODV ester
derivatives were successfully synthesized. The purpose of this
study was to determine which chemical structure, monoester formed
on the phenolic hydroxyl of ODV and/or the diester formed both on
the alcoholic hydroxyl and the phenolic hydroxyl of ODV, could be
degraded to ODV in the blood. In addition, we aimed to identify
ester derivatives with higher bioavailability, improved
pharmacokinetics properties and more rapid penetration to further
research. Furthermore, in order to evaluate the species differences
in bioavailability of ODVP-1, ODVP-2 and ODVP-3 between rats and
beagles, although these were evaluated in rats in our previous work
(compounds If, Ij and Ik, respectively) (4,23), we
assessed the pharmacokinetic profiles of ODVP-1, ODVP-2 and ODVP-3
in beagles.
To these ends, the present study employed in
vivo pharmacokinetic analysis of ODV in beagle dogs and
pharmacokinetic screening and brain uptake studies in rats. The
results indicated that ODVP-1, ODVP-2 and ODVP-3 demonstrated
higher bioavailability, improved pharmacokinetics properties, more
rapid penetration of ODV, highlighting them as potential
development prospects.
Materials and methods
Animals
Adult male Wistar rats (weight, 250±20 g) were
purchased from the Experimental Animal Center of Jilin University
(Jilin, China). Adult male beagle dogs (weight, 12±1 kg) were
provided by Tianyao Pharmaceutical Co., Ltd. (Tianjin, China) All
animals were housed individually at 22±2°C and a relative humidity
of 50±10% with a 12-h light/dark cycle, with free access to food
and water. The present study was approved by the Ethical Committee
of Jilin University. Rats were sacrificed via an overdose of sodium
pentobarbital (150 mg/kg) by intraperitoneal injection.
Reagents and instruments
ODV succinate monohydrate, ODVP-1 hydrochloride,
ODVP-2 hydrochloride, ODVP-3 succinate, ODVP-4 hydrochloride,
ODVP-5 hydrochloride, ODVP-6 hydrochloride and ODVP-7 hydrochloride
(derivative content, >98%) were provided by Jilin Institute of
Pharmaceutical Research (Jilin, China). The chemical structures of
these compounds are shown in Fig. 1.
Ethyl ether and dichloromethane were purchased from the Jiayu
Chemical Co., Ltd., (Tianjin, China) and Beijing Chemical Factory
(Beijing, China), respectively. An Agilent 1100 high performance
liquid chromatography (HPLC) system (Agilent Technologies, Inc.,
Santa Clara, CA, USA) and QTRAP type Triple Quadrupole mass
spectrometer (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Foster City, CA, USA) were used for analysis. LD5-2A centrifuge
(Beijing Medical Centrifuge Factory, Beijing, China) and test tubes
(Nantong Haizhixing Experimental Co., Ltd., Nantong, China) were
used for sample collection.
Administration regimen for the
pharmacokinetic screening of the ODV ester derivatives in rats
The seven ODV ester derivatives were prepared in
normal saline for oral administration. A total of 21 healthy male
Wistar rats were randomly allocated into seven groups (n=3 per
group). After 12 h of fasting, with free access to water, a dose of
0.043 mmol/kg ODVP-1, ODVP-2, ODVP-3, ODVP-4, ODVP-5, ODVP-6 or
ODVP-7 in 0.5 ml normal saline was intragastrically administered to
each of the seven groups. Blood samples (1.0 ml) were extracted
from the retrobulbar venous plexus at 5, 10, 30 and 1 h after
administration. The samples were centrifuged in heparinized and
dichlorvos-treated test tubes (terminal concentration of
dichlorvos, 200 µg/ml) for 10 min at 4°C (1,570 × g). The plasma
samples were then preserved at −80°C for further analysis.
Chromatography and mass spectrometry
for the pharmacokinetic screening of the ODV ester derivatives in
rats
The chromatographic column was a ZORBAX Eclipse XDB
C8 column (I.D., 4.6×150 mm; particle diameter, 5 µm;
Agilent Technologies, Inc.) and the mobile phase was methanol: 10
mM ammonium acetate (85:15, v/v). The flow rate was 1.0 ml/min, the
injection volume was 10 µl and the column temperature was set at
20°C. The samples were determined with positive ion mode, the
scanned mode was multiple reaction monitoring and the ion reaction
used for ODV qualitative analysis was m/z
264.3→m/z 58.0.
Pretreatment of plasma samples and
HPLC-MS/MS determination for the pharmacokinetic screening of the
ODV ester derivatives in rats
Plasma samples (0.5 ml) were added into a Sep-Pak
C18 solid phase extraction column (Waters Corporation, Milford, MA,
USA) and activated by methanol and water at a pass speed of 30
drops/min. The samples were washed with 2 ml water and eluted with
2 ml methanol. The collected eluate was concentrated to ~200 µl
using a gentle stream of N2, and 10 µl was collected for
HPLC-MS/MS analysis.
Administration regimen for the
pharmacokinetic study of prodrugs in beagle dogs
Pharmacokinetic evaluation was performed in beagle
dogs. The test animals were randomly divided into five groups
(n=4). Food and water were freely available during the entire
experimental period. After a 12 h fast, each group was administered
a single dose of 0.013 mmol/kg ODV, ODVP-1, ODVP-2, ODVP-3 or
ODVP-5.
Sample collection for the
pharmacokinetic study of prodrugs in beagle dogs
Blood samples (1.0 ml) were extracted from the small
saphenous vein of the legs of beagle dogs prior to and at and
0.083, 0.167, 0.25, 0.5, 0.75, 1, 2, 3, 4, 8, 12 and 24 h after
administration. Blood samples were centrifuged in heparinized and
dichlorvos-treated test tubes (terminal concentration of
dichlorvos, 200 µg/ml) for 10 min at 4°C (1,570 × g). Plasma
samples were stored in a refrigerator protected from light at −80°C
for future analysis.
Plasma sample treatment for the
pharmacokinetic study of prodrugs in beagle dogs
Plasma samples (100 µl) were placed in a test tube
with a stopper, and 100 µl internal standard solution and 100 µl
sodium carbonate (0.1 M) were added to the test tube and allowed to
mix. A total of 3 ml ethyl ether-dichloromethane (60:40, v/v) was
then added prior to 10 min of eddy-mixing, followed by shaking for
10 min (240 times/min). The samples were then centrifuged for 5 min
at 2,136 × g. The upper organic phase was placed in a new tube, and
after blow-drying with N2 stream at 25°C, 200 µl mobile
phase was added to the residue for dissolution. After a second
round of eddy-mixing, 10 µl of the samples were collected for
LC/MS/MS analysis.
Chromatography and mass spectrometry
for the pharmacokinetic study of prodrugs in beagle dogs
The chromatographic conditions and mass spectrometer
conditions are the same as described above in the ‘Chromatography
and mass spectrometry for the pharmacokinetic screening of the ODV
ester derivatives in rats’ section. The ion reaction used for
qualitative analysis was m/z 264.1→m/z
107.0 (ODV) and m/z 256.3→m/z 167.1
(internal standard, benzhydramine).
Determination of the concentration of
ODVP-1, ODVP-2, ODVP-3 and ODV in plasma, brain, and hypothalamus
in rats
ODVP-1, ODVP-2, ODVP-3 and ODV were prepared in
normal saline for oral administration. Eighty rats were dosed
orally with ODVP-1, ODVP-2, ODVP-3 or ODV (0.043 mmol/kg in 0.5
ml). Food was restricted from 12 h predosing to 60 min postdosing.
At 0.25, 0.5, 1, 2 and 4 h postdosing, blood samples were drawn
from the sinus and collected in heparinized and dichlorvos-treated
test tubes (terminal concentration of dichlorvos, 200 µg/ml) for
plasma isolation. At 0.25, 0.5, 1, 2 and 4 h postdosing, rats were
perfused with 40 ml phosphate-buffered saline (4°C) via the left
ventricle, and the brain was removed and the hypothalamus was
dissected. The brain and hypothalamus were placed in ice-cold
methanol: water (50:50, v/v) in either 2.0 or 1.0 ml, respectively,
and maintained on ice until tissue homogenization. Plasma and brain
tissues were stored at −80°C for further processing. For
determination of the concentration of ODVP-1, ODVP-2, ODVP-3 or ODV
in tissues, three standard curves (determination of ODVP-1 and ODV,
ODVP-2 and ODV, and ODVP-3 and ODV) of plasma or brain tissue
(1.0–1,000 ng/ml) were generated from untreated animals. Samples
and standards (100 µl) were spiked with 100 µl diazepam solution
(1.0 µg/ml) in acetonitrile to serve as an internal standard. A
total of 3.5 ml N-hexane-dichloromethane-dimethyl carbinol
(300:150:15, v/v/v) was then added, and 10 min of eddy-mixing was
performed followed by shaking for 10 min (240 times/min). The
samples were then centrifuged for 5 min at 2,136 × g. The upper
organic phase was placed in a new tube, and after blow-drying with
N2 stream at 25°C, 200 µl methanol: 0.05% formic acid
(50:50, v/v) was added to the residue for dissolution. After a
second round of eddy-mixing, 20 µl sample was collected for
LC/MS/MS analysis.
Statistical analysis
BAPP2.2 software (China Pharmaceutical University,
Nanjing, China) was used to calculate the pharmacokinetic
parameters. The trapezoidal method was selected for calculation of
the AUC0-t value. Based on the semi-logarithmic
map method, the t1/2 was calculated in
accordance with the endmost four concentration points of the
elimination phase. The Cmax and
Tmax were calculated with the measured values,
the AUC0-t after ODV administration was set as
100%, and the relative bioavailability was calculated according to
the AUC0-t of ODV after the administration of
each ODV prodrug.
Results
Pharmacokinetic screening of the ODV
ester derivatives in rats
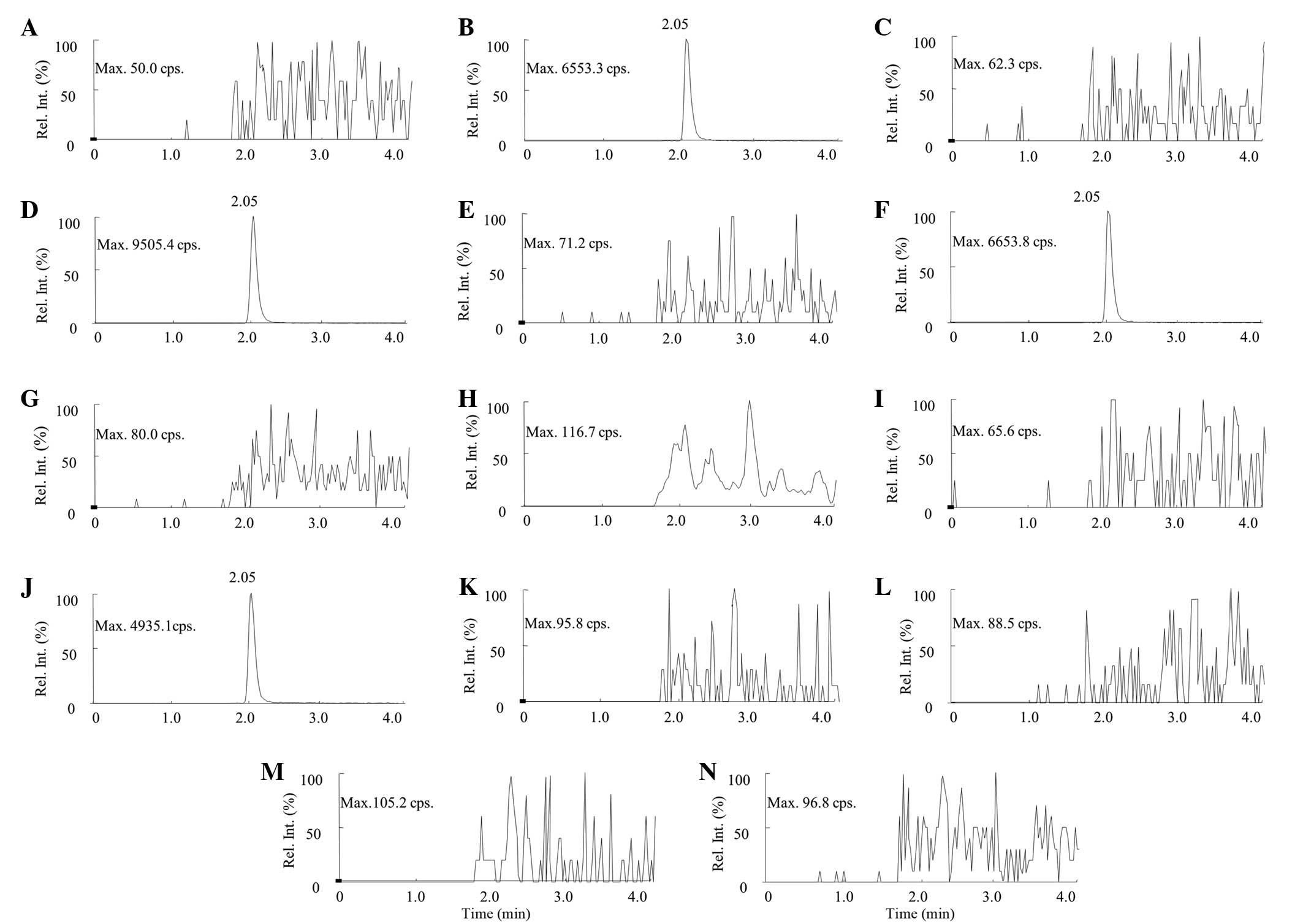
ODV was clearly detected in the rat plasma of the
ODVP-1, ODVP-2, ODVP-3 and ODVP-5 groups at 5, 10, 30 min and 1 h
after intragastric administration, while ODV was not detected in
the rat plasma of the ODVP-4, ODVP-6 and ODVP-7 groups at any time
points. The pharmacokinetic screening results showed that the
monoesters (ODVP-1, ODVP-2, ODVP-3 and ODVP-5) formed on the
phenolic hydroxyl of ODV could be degraded to ODV in blood, while
the diesters (ODVP-4, ODVP-6 and ODVP-7) formed on the alcoholic
hydroxyl and the phenolic hydroxy could not be degraded to ODV in
blood. The HPLC-MS/MS qualitative detection of ODV in blood samples
from each group 1.0 h after administration is shown in Fig. 2.
Pharmacokinetic study of prodrugs in
beagle dogs
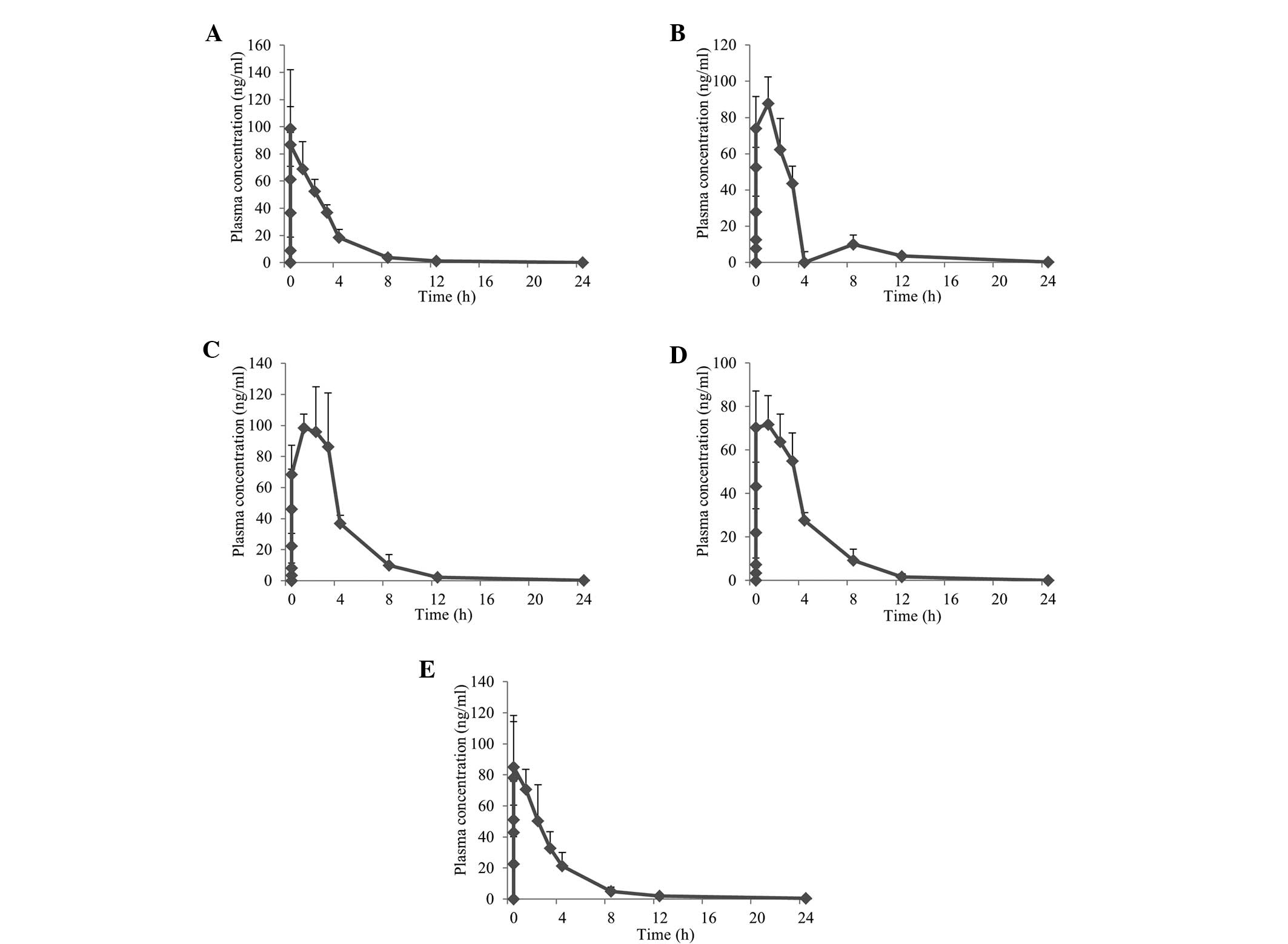
ODV, ODVP-1, ODVP-2, ODVP-3 and ODVP-5 (0.013
mmol/kg) were administered to beagle dogs. The ODV average plasma
concentration-time curve for each treatment is shown in Fig. 3. The plasma concentration data was
analyzed using the non-compartment model by the BAPP2.2 software.
The pharmacokinetic parameters are shown in Table I.
 | Table I.Pharmacokinetic parameters after
intragastric administration of 0.013 mmol/kg ODV, ODVP-1, ODVP-2,
ODVP-3 and ODVP-5 in beagle dogs (n=4), and the relative
bioavailability. |
Table I.
Pharmacokinetic parameters after
intragastric administration of 0.013 mmol/kg ODV, ODVP-1, ODVP-2,
ODVP-3 and ODVP-5 in beagle dogs (n=4), and the relative
bioavailability.
| Drug |
Cmax (ng/ml) |
Tmax (h) |
t1/2 (h) |
AUC0-t (ng·h/ml) | F (%) |
|---|
| ODV |
100±40.81 |
0.5±0.2 |
2.07±0.49 |
255±54.30 | 100 |
| ODVP-1 |
91.5±12.03 |
0.9±0.1 |
3.27±1.37 |
324±46.25 | 127 |
| ODVP-2 |
108±18.57 |
1.2±0.6 |
2.45±1.08 |
419±50.83 | 164 |
| ODVP-3 |
79.4±12.32 |
1.7±1.0 |
2.25±0.22 |
301±30.61 | 118 |
| ODVP-5 |
89.6±33.29 |
0.8±0.2 |
3.44±2.48 |
269±94.90 | 105 |
The ODV and the screened ODV prodrugs (ODVP-1,
ODVP-2, ODVP-3 and ODVP-5) were orally administered to beagle dogs
at a dose of 0.013 mmol/kg. The AUC0-t values
were calculated as 324, 419, 301 and 269 ng·h/ml, respectively.
Taking the AUC0-t of 255 ng·h/ml of ODV as 100%,
the relative bioavailabilities of ODVP-1, ODVP-2, ODVP-3 and ODVP-5
were calculated at 127, 164, 118 and 105%, respectively. The
bioavailabilities of ODVP-1, ODVP-2, ODVP-3 and ODVP-5 were all
increased compared with ODV succinate. Notably, the bioavailability
of ODVP-2 was enhanced by >60%, indicating a marked
improvement.
The Cmax of ODV was 100 ng/ml, and
the Cmax values of ODVP-1, ODVP-2, ODVP-3 and
ODVP-5 were 91.5, 108, 79.4 and 89.6 ng/ml, respectively. The
Cmax of each ODV prodrug was relatively close to
the Cmax of ODV. While ODVP-3 reached the maximum
ODV concentration at 1.7 h, ODVP-1, ODVP-2 and ODVP-5 all reached
the maximum ODV concentration at ~1.0 h. Notably, all of their peak
times were later compared with the original drug ODV (0.5 h).
The half-lives of ODV after the administration of
ODVP-1, ODVP-2, ODVP-3 and ODVP-5 were 3.27, 2.45, 2.25 and 3.44 h,
respectively. All were longer than the half-life of ODV (2.07
h).
Concentrations of ODVP-1, ODVP-2,
ODVP-3 and ODV in the plasma, brain and hypothalamus in rats after
oral administration
Concentrations of ODVP-1, ODVP-2, ODVP-3 and ODV
were determined in plasma, total brain and hypothalamus in rats for
each time point over a 4-h period (Fig.
4). Total brain represents the remainder of brain tissue after
dissecting the hypothalamus.
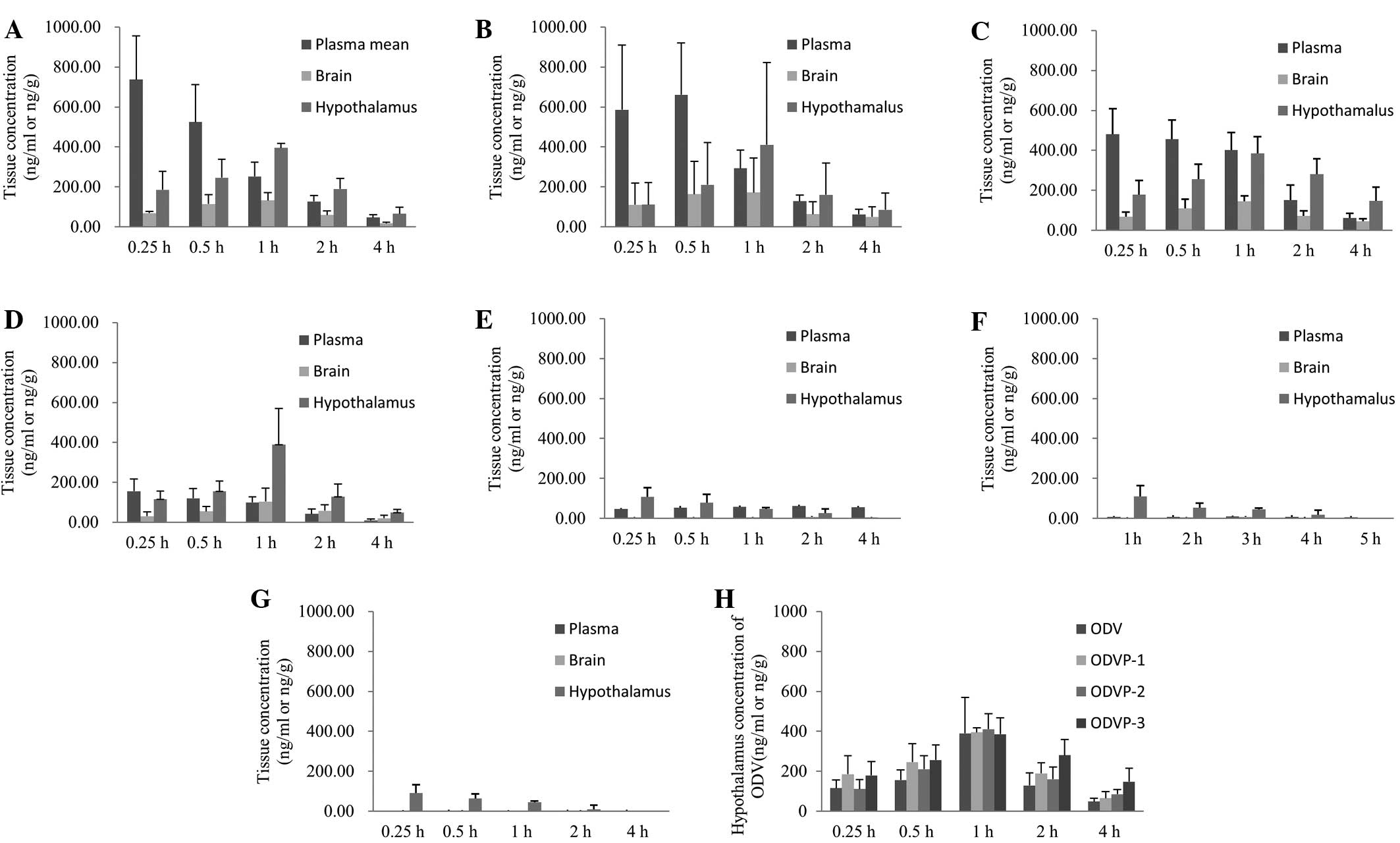 | Figure 4.Comparison of the concentrations of
ODV in plasma, brain, and hypothalamus after administration of
equimolar ODVP-1, ODVP-2, ODVP-3, and ODV (0.043 mmol/kg, mean ±
SD, n=4). ODV in plasma, brain, and hypothalamus after
administration of (A) ODVP-1, (B) ODVP-2, (C) ODVP-3 and (D) ODV,
respectively; (E) ODVP-1, (F) ODVP-2 and (G) ODVP-3 in the plasma,
brain, and hypothalamus after administration of each respective
compound. (H) ODV in hypothalamus after administration of ODV,
ODVP-1, ODVP-2 and ODVP-3. ODV, O-desmethylvenlafaxine. |
The concentration of ODV in plasma, brain and
hypothalamus after administration of ODVP-1, ODVP-2 or ODVP-3 was
higher compared with that of ODV at the same time point after
administration of ODV. Trace quantities of ODVP-1, ODVP-2 or ODVP-3
were detected in the blood and hypothalamus after administration of
ODVP-1 over a 2 h period. We clearly found that ODVP-1, ODVP-2, or
ODVP-3 were absorbed into blood and translated into ODV
rapidly.
Discussion
Prodrugs are bioreversible derivates of drug
molecules that transform by carboxylesterase in vivo to
release the active parent drug (24). The aim of this approach is to
increase the usefulness of a drug by improving the physicochemical,
biopharmaceutical or pharmacokinetic properties of the compound
(25–28). By chemically modifying an active
agent, various mitigating factors to efficacy may be overcome, such
as poor aqueous solubility, chemical instability, insufficient oral
absorption, rapid presystemic metabolism, inadequate brain
penetration, toxicity and irritation (25–28).
Prodrugs may also prolong the duration of drug action (29). The ODV molecule contains two
hydroxyls: A phenolic hydroxyl and an alcoholic hydroxyl. We used
chemical bonding to connect a phenolic hydroxyl or two hydroxyls,
and different vectors were used to successfully synthesize seven
ODV ester derivatives. In order to conduct the pharmacokinetic
evaluation of the ODV ester derivatives, equimolar quantities of
the newly synthesized ODV ester derivatives were intragastrically
administered to Wistar rats. Results showed that among the seven
newly synthesized compounds, the monoester formed on the phenolic
hydroxyl of ODV (ODVP-1, ODVP-2, ODVP-3 and ODVP-5) could be
degraded to ODV in rat plasma, confirming these as possible
prodrugs, whilst the diester formed both on the alcoholic hydroxyl
and phenolic hydroxyl of ODV (ODVP-4, ODVP 6 and ODVP-7) could not
be degraded to generate ODV in rat plasma, eliminating them for
consideration as potential prodrugs.
Carboxylesterase is an enzyme that is capable of
hydrolyzing a wide variety of carboxylic acid esters (30). The enzyme is widely distributed in
nature, being particularly common in the mammalian liver (31–33).
Blood from rodents and beagle dogs is routinely treated with
dichlorvos to inhibit esterase-catalysed ex vivo hydrolysis
of ester derivatives (34,35). In the present study, the efficacy of
this inhibitor was demonstrated in pooled samples of rat or dog
blood and plasma incubated with newly synthesized ODV ester
derivatives. The results of the present study demonstrated that
treatment of fresh blood with dichlorvos is effective in preventing
the ex vivo decomposition of the ODV ester prodrug.
Species differences of hydrolase activity in small
intestine and liver can result in different exposures to prodrug
and active form after administration of prodrugs. The relative
bioavailability of ODV after administration of ODVP-1, ODVP-2 and
ODVP-3 in beagles (127, 164 and 118%, respectively) were all higher
than those of ODV in rats, which were 98.4, 110 and 104%,
respectively, as reported in our previous paper (23). This data indicates that the
hydrolytic activity in canine small intestine is lower than in
rats, so the absorption and bioavailability of ODVP-1, ODVP-2 and
ODVP-3 are all higher in canines than in rats. These finding
coincide with the study performed by Taketani et al
(36) which reported that no
hydrolase activity was detected in dog small intestine.
Following the oral administration of ODVP-1, ODVP-2
or ODVP-3, the compounds were absorbed into the blood from the
stomach and small intestine. The majority of ODVP-1, ODVP-2 or
ODVP-3 in the blood was translated into ODV rapidly, and a trace
quantity in the blood was not degraded that rapidly entered the
brain. The present study evaluated brain concentrations of ODV,
ODVP-1, ODVP-2 or ODVP-3 over time after oral administration in
male rats. ODVP-1, ODVP-2 and ODVP-3 demonstrated rapid
hypothalamus penetration, with hypothalamus concentrations in
excess of those noted in the plasma after a single oral
administration of ODVP-1, ODVP-2 or ODVP-3. The results shown in
Table I and Fig. 3 indicate that ODVP-1, ODVP-2 and
ODVP-3 demonstrate higher bioavailability and improved
pharmacokinetics compared with equimolar ODV. In addition, ODV and
ODVP-1, ODVP-2 or ODVP-3 were all detected in the brain and
hypothalamus after oral administration of ODVP-1, ODVP-2 or ODVP-3.
The difference is that ODV has been maintained at relatively high
concentrations for 0–4 h, while ODVP-1, ODVP-2 or ODVP-3 last for
only 0–2 h to maintain a certain concentration and significantly
decreased with time.
In summary, ODVP-1, ODVP-2, and ODVP-3 demonstrated
higher bioavailability and improved pharmacokinetics properties
than ODV, penetrated the male rat brain and hypothalamus rapidly.
Collectively, the results of this study indicate that these ODV
prodrugs may be useful for further development and application.
Acknowledgements
This research was financially supported by the
Natural Science Foundation of Heilongjiang Province (grant no.
H201361), Scientific project (grant no. 12511571) of Heilongjiang
Provincial Department of Education, the National Natural Science
Foundation of China (grant nos. 30973587 and 81102383), the Science
and Technology Major Specialized Projects for ‘significant new
drugs creation’ of the 12th five-year plan (grant nos.
2012ZX09303-015 and 2014ZX09303303), the National Key Technology
R&D Program of the Ministry of Science and Technology (grant
no. 2012BAI30B00) and the China Equipment and Education Resources
System (grant no. CERS-1-70).
References
|
1
|
Wong ML and Licinio J: Research and
treatment approaches to depression. Nat Rev Neurosci. 2:343–351.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Melfi CA, Chawla AJ, Croghan TW, Hanna MP,
Kennedy S and Sredl K: The effects of adherence to antidepressant
treatment guidelines on relapse and recurrence of depression. Arch
Gen Psychiatry. 55:1128–1132. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gumnick JF and Nemeroff CB: Problems with
currently available antidepressants. J Clin Psychiatry. 61:5–15.
2000.PubMed/NCBI
|
|
4
|
Tian JW, Jiang WL, Zhong Y, Meng Q, Gai Y,
Zhu HB, Hou J, Xing Y and Li YX: Preclinical pharmacology of TP1, a
novel potent triple reuptake inhibitor with antidepressant
properties. Neuroscience. 196:124–130. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Otton SV, Ball SE, Cheung SW, Inaba T,
Rudolph RL and Sellers EM: Venlafaxine oxidation in vitro is
catalysed by CYP2D6. Br J Clin Pharmacol. 41:149–156. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klamerus KJ, Maloney K, Rudolph RL,
Sisenwine SF, Jusko WJ and Chiang ST: Introduction of a composite
parameter to the pharmacokinetics of venlafaxine and its active
O-desmethyl metabolite. J Clin Pharmacol. 32:716–724. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schweizer E, Thielen RJ and Frazer A:
Venlafaxine: A novel antidepressant compound. Expert Opin Investig
Drugs. 6:65–78. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perry R and Cassagnol M: Desvenlafaxine: A
new serotonin-norepinephrine reuptake inhibitor for the treatment
of adults with major depressive disorder. Clin Ther. 31:1374–1404.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rudorfer MV and Potter WZ: The role of
metabolites of antidepressants in the treatment of depression. CNS
Drugs. 7:273–312. 1997. View Article : Google Scholar
|
|
10
|
Ereshefsky L and Dugan D: Review of the
pharmacokinetics, pharmacogenetics, and drug interaction potential
of antidepressants: Focus on venlafaxine. Depress Anxiety. 12(Suppl
1): S30–S44. 2000. View Article : Google Scholar
|
|
11
|
Nichols AI, Focht K, Jiang Q, Preskorn SH
and Kane CP: Pharmacokinetics of venlafaxine extended release 75 mg
and desvenlafaxine 50 mg in healthy CYP2D6 extensive and poor
metabolizers: A randomized, open-label, two-period, parallel-group,
crossover study. Clin Drug Investig. 31:155–167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nichols AI, Abell M, Chen Y, Behrle JA,
Frick G and Paul J: Effects of desvenlafaxine on the
pharmacokinetics of desipramine in healthy adults. Int Clin
Psychopharmacol. 28:99–105. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reddy S, Kane C, Pitrosky B, Musgnung J,
Ninan PT and Guico-Pabia CJ: Clinical utility of desvenlafaxine 50
mg/d for treating MDD: A review of two randomized
placebo-controlled trials for the practicing physician. Curr Med
Res Opin. 26:139–150. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sproule BA, Hazra M and Pollock BG:
Desvenlafaxine succinate for major depressive disorder. Drugs Today
(Barc). 44:475–487. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nichols AI, Tourian KA, Tse SY and Paul J:
Desvenlafaxine for major depressive disorder: Incremental clinical
benefits from a second-generation serotonin-norepinephrine reuptake
inhibitor. Expert Opin Drug Metab Toxicol. 6:1565–1574. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mizuno N, Niwa T, Yotsumoto Y and Sugiyama
Y: Impact of drug transporter studies on drug discovery and
development. Pharmacol Rev. 55:425–461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baudy RB, Butera JA, Abou-Gharbia MA, Chen
H, Harrison B, Jain U, Magolda R, Sze JY, Brandt MR, Cummons TA, et
al: Prodrugs of perzinfotel with improved oral bioavailability. J
Med Chem. 52:771–778. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie Q, Wang X, Jiang Z and Qiu Z: Design,
synthesis, and bioavailability evaluation of coumarin-based prodrug
of meptazinol. Bioorg Med Chem Lett. 15:4953–4956. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kahns AH, Møss J and Bundgaard H: Improved
oral bioavailability of salicylamide in rabbits by a
1,3-benzoxazine-2,4-dione prodrug. Int J Pharm. 78:199–202. 1992.
View Article : Google Scholar
|
|
20
|
Horn AS, Kelly P, Westerink BH and
Dijkstra DA: A prodrug of ADTN: Selectivity of dopaminergic action
and brain levels of ADTN. Eur J Pharmacol. 60:95–99. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ecobichon D: Relative amounts of hepatic
and renal carboxylesterase in mammalian species. Res Commun Chem
Pathol Pharmacol. 3:629–636. 1972.PubMed/NCBI
|
|
22
|
Cook CS, Karabatsos PJ, Schoenhard Gl and
Karim A: Species dependent esterase activities for hydrolysis of an
anti-HIV prodrug glycovir and bioavailability of active SC-48334.
Pharm Res. 12:1158–64. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Yang Y, Zhao S, Yang Z, Yang H,
Fawcett JP, Li Y, Gu J and Sun T: Phenolic esters of
O-desmethylvenlafaxine with improved oral bioavailability and brain
uptake. Molecules. 18:14920–14934. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liederer BM and Borchardt RT: Enzymes
involved in the bioconversion of ester-based prodrugs. J Pharm Sci.
95:1177–1195. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Beaumont K, Webster R, Gardner I and Dack
K: Design of ester prodrugs to enhance oral absorption of poorly
permeable compounds: challenges to the discovery scientist. Curr
Drug Metab. 4:461–485. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ettmayer P, Amidon GL, Clement B and Testa
B: Lessons learned from marketed and investigational prodrugs. J
Med Chem. 47:2393–2404. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Testa B: Prodrug research: Futile or
fertile? Biochem Pharmacol. 68:2097–2106. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stella VJ and Nti-Addae KW: Prodrug
strategies to overcome poor water solubility. Adv Drug Deliv Rev.
59:677–694. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rautio J, Kumpulainen H, Heimbach T,
Oliyai R, Oh D, Järvinen T and Savolainen J: Prodrugs: Design and
clinical applications. Nat Rev Drug Discov. 7:255–270. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laizure SC1, Herring V, Hu Z, Witbrodt K
and Parker RB: The role of human carboxylesterases in drug
metabolism: Have we overlooked their importance? Pharmacotherapy.
33:210–222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pindel EV, Kedishvili NY, Abraham TL,
Brzezinski MR, Zhang J, Dean RA and Bosron WF: Purification and
cloning of a broad substrate specificity human liver
carboxylesterase that catalyzes the hydrolysis of cocaine and
heroin. J Biol Chem. 272:14769–14775. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schewer H, Langmann T, Daig R, Becker A,
Aslandis C and Schmitz G: Molecular cloning and characterization of
a novel putative carboxylesterase, present in human intestine and
liver. Biochem Biophys Res Commun. 233:117–120. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Holmes RS, Glenn JP, Vandeberg JL and Cox
LA: Baboon carboxylesterases 1 and 2: Sequences, structures and
phylogenetics relationships with human and other primate
carboxylesterases. J Med Primatol. 38:27–38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Song X and Siahaan TJ: Synthesis and
stability study of a modified phenylpropionic acid linker-based
esterase-sensitive prodrug. Bioorg Med Chem Lett. 12:3439–3442.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sakaguchi K, Nagayama M, Masaoka T,
Nishimura A, Kageyama K, Shirai M and Akahori F: Effects of
fenthion, isoxathion, dichlorvos and propaphos on the serum
cholinesterase isoenzyme patterns of dogs. Vet Hum Toxicol. 39:1–5.
1997.PubMed/NCBI
|
|
36
|
Taketani M, Shii M, Ohura K, Ninomiya S
and Imai T: Carboxylesterase in the liver and small intestine of
experimental animals and human. Life Sci. 81:924–932. 2007.
View Article : Google Scholar : PubMed/NCBI
|