Introduction
Ischemic brain injury occurs when cerebral blood
flow is reduced to a low level, which results in deprivation of
oxygen and glucose in brain tissues. These events activate multiple
processes that lead to cell death signaling, including
excitotoxicity, oxidative stress and caspase activation (1–3). The
hippocampus is vulnerable to temporary imperforation of blood flow,
and impairment in learning ability and memory function is a
remarkable behavioral consequence of transient global ischemia
(4,5).
Apoptosis is the major pathway of cell death
following cerebral ischemic injury (6). It is a process of programmed cell death
triggered by a variety of stimuli, and it is a part of common
mechanisms in tissue remodeling, cell replacement and the removal
of damaged cells (7). Excessive or
inappropriate apoptosis, however, is involved in several types of
neurodegenerative disorders, including stroke (4,8).
Apoptotic cell death can be assessed using terminal
deoxynucleotidyl transferase-mediated deoxyuridine triphosphate
nick end labeling staining, which detects DNA fragmentation
(9). In addition, apoptosis can also
be assessed by examining the expression of proteins involved in the
regulation of apoptosis. For example, the caspases, a family of 14
cysteine proteases, are crucial players in apoptotic cell death
both as initiators (caspase-2, −8, −9 and −10) and as executioners
(caspase-3, −6 and −7) (10).
Furthermore, activation of caspase-3 is an important feature of
apoptosis following ischemic brain insults (11). The cell-damaging mechanisms that are
activated by ischemia are countered by cell-survival mechanisms
that include upregulation of the anti-apoptotic molecules, B-cell
lymphoma 2 (Bcl-2) and Bcl-2XL (12). In addition, the anti-apoptotic
protein Bcl-2 suppresses apoptosis by preventing the release of
cytochrome c (13) whereas
the pro-apoptotic protein Bcl-2-associated X protein (Bax) promotes
apoptosis by enhancing cytochrome c release (14). Cytosolic cytochrome c binds to
apoptotic protease activating factor-1 (Apaf-1), which then forms
the apoptosome and leads to the subsequent activation of caspase-9.
Caspase-9 is important in neuronal cell death following ischemia
(15).
The α2-adrenoreceptor agonist
dexmedetomidine is a sedative and analgesic agent used for
postoperative intensive care sedation, which reduces the
requirement for opioid analgesia (16). It stimulates the pre-synaptic
α2-adrenoceptors, which subsequently inhibit the release
of noradrenaline at the sympathetic synapses and reduce the
surgical stress response and central sympathetic outflow (17). Dexmedetomidine has been suggested to
have neuroprotective effects by stimulating
α2-adrenoreceptors (18,19). In
addition, it has also been demonstrated to improve
histomorphological and neurological outcomes following cerebral
ischemia (20). Furthermore, the
dexmedetomidine-activated signaling pathway was shown to prevent
cellular apoptosis when neuronal cells were subjected to ischemic
insults (21,22).
In the present study, the effect of dexmedetomidine
on short-term memory was investigated following transient global
ischemia in gerbils. In addition, the modulatory effect of
dexmedetomidine on the expression of apoptosis-regulating proteins
in the hippocampus was evaluated.
Materials and methods
Experimental animals and
treatments
A total of 50 adult male Mongolian gerbils (12 weeks
of age; Orient Bio Co., Seoul, Korea) were used for this
experiment. The experimental procedures were performed in
accordance with the Animal Care Guidelines of the National
Institutes of Health and the Korean Academy of Medical Sciences,
and the study was approved by the Kyung Hee University
Institutional Animal Care and Use Committee (Seoul, Korea). Gerbils
were housed under controlled temperature (23±2°C) and lighting
(08:00 to 20:00 h) conditions with food and water made available
ad libitum.
Gerbils were randomly divided into five groups (n=10
per group): Sham-operated, ischemia-induction, ischemia-induction
and 0.1 µg/kg dexmedetomidine-treated, ischemia-induction and 1
µg/kg dexmedetomidine-treated and the ischemia-induction and 10
µg/kg dexmedetomidine-treated groups. Dexmedetomidine was procured
from Hospira, Inc. (Rocky Mount, NC, USA). The animals in the
dexmedetomidine-treated groups received the respective dose of
dexmedetomidine intraperitoneally, once per day for 14 consecutive
days, starting 1 day after surgery. In addition, the animals in the
Sham-operated group received an equivalent dose of saline
intraperitoneally, once per day for the same duration.
Induction of transient global
ischemia
Transient global ischemia was induced according to a
previously described surgical procedure (4). Gerbils were anesthetized with Zoletil
50® [10 mg/kg, intraperitoneally (i.p.); Virbac
Laboratories, Carros, France]. Following bilateral neck incisions,
both common carotid arteries were exposed and occluded with
aneurysm clips for 5 min. The clips were then removed to restore
cerebral blood flow. The body and rectal temperature was maintained
at 36±0.5°C during surgery using a homeothermic blanket control
unit (Harvard Apparatus, Holliston, MA, USA) that enveloped the
body and the head. Following recovery, the animals were monitored
for an additional 2 h to prevent hypothermia. The animals in the
Sham-operated group were treated identically except that the common
carotid arteries were not occluded after the neck incisions.
Step-down avoidance task
Latency in the step-down avoidance task was
determined to evaluate short-term memory according to a previously
described method (8,23). Gerbils were trained in a step-down
avoidance task 15 days after the induction of ischemia. At 1 h
after training, the latency in each group was determined. The
gerbil was then placed on a 7×25-cm platform that was 2.5 cm in
height, and the platform faced a 45×25-cm grid of parallel
stainless steel bars, 0.1 cm in caliber and spaced 1 cm apart. In
the training session, the animal received a 0.3 mA scramble foot
shock for 2 sec immediately upon stepping down. The time that
elapsed until the gerbil stepped down and placed all four paws on
the grid was defined as the latency period. A latency time over 180
sec was counted as 180 sec.
Tissue preparation
Gerbils were sacrificed immediately after
determining the latency of the step-down avoidance task. The
gerbils were anesthetized with Zoletil 50® (10 mg/kg,
i.p.), transcardially perfused with 50 mM phosphate-buffered saline
and fixed with a freshly prepared solution consisting of 4%
paraformaldehyde in 100 mM phosphate buffer (PB, pH 7.4). Brains
were dissected and stored overnight in the same fixative and were
then transferred to 30% sucrose for cryoprotection. For
immunohistochemistry, 40-µm slices were coronally sectioned by the
use of a cryostat (Leica Biosystems, Nussloch, Germany). In total
ten slice sections in the hippocampus were collected on average
from each gerbil. The sections ranging between 2.5 and 2.7 mm
posterior from the bregma were used for immunohistochemistry.
Terminal deoxynucleotidyl
transferase-mediated deoxyuridine triphosphate nick end labeling
(TUNEL) staining
In order to visualize DNA fragmentation, a marker of
apoptosis, TUNEL staining was performed using an In Situ Cell Death
Detection kit® (Roche Diagnostics GmbH, Mannheim,
Germany) according to the manufacturer's instructions (4,24). The
sections were post-fixed in ethanol-acetic acid (2:1), rinsed, then
incubated with proteinase K (100 µg/ml). They were then rinsed,
incubated in 3% H2O2, permeabilized with 0.5%
Triton X-100, rinsed again and incubated in the TUNEL reaction
mixture. Finally, the sections were rinsed once more and visualized
using Converter-POD with 0.03% 3,3′-diaminobenzidine (DAB). Mayer's
hematoxylin (Dako, Glostrup, Denmark) was used as a counterstain,
and the sections were mounted onto gelatin-coated slides. The
slides were then air-dried overnight at room temperature, and
coverslips were mounted using Permount® (Thermo Fisher
Scientific, Inc., Waltham, MA, USA).
Caspase-3 immunohistochemistry
In order to visualize the caspase-3 expression,
caspase-3 immunohistochemistry was performed as previously
described (4,24). The sections were selected from each
brain and incubated overnight with caspase-3 antibody (cat. no.
SC-7272; 1:500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
then with biotinylated mouse secondary antibody (cat. no. BA-2000;
1:200; Vector Laboratories, Inc., Burlingame, CA, USA) for 1 h. The
secondary antibody was amplified using a Vector Elite ABC
kit® (1:100; Vector Laboratories, Inc.).
Antibody-biotin-avidin-peroxidase complexes were visualized using
0.03% DAB, and the sections were mounted onto gelatin-coated
slides. The slides were air-dried overnight at room temperature,
and coverslips were mounted using Permount® (Thermo
Fisher Scientific, Inc.).
Western blot analysis
Western blotting was performed as previously
described (8,23). The hippocampal tissues were collected
and then frozen immediately at −70°C. The hippocampal tissues were
homogenized on ice and lysed in a lysis buffer containing 50 mM
HEPES (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM
phenylmethylsulfonyl fluoride, 1 mM EGTA, 1.5 mM MgCl2 ×
6H2O, 1 mM sodium orthovanadate and 100 mM sodium
fluoride. The protein content was measured using a colorimetric
protein assay kit (cat. no. 5000002; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). For the detection of cytochrome c,
tissues were fractionated into the cytosol using the mitochondrial
fraction kit (cat. no. 40015; Active Motif, Carlsbad, CA, USA)
according to the manufacturer's instructions. In total, 30 µg
protein was separated on 12% SDS-polyacrylamide gels and
transferred onto a nitrocellulose membrane. The membranes were
incubated with 5% skim milk in Tris-buffered saline containing 0.1%
Tween-20 (TBST) and then incubated for 1 h at room temperature. The
membrane was washed with TBST for 10 min, 3 times at room
temperature. Mouse β-actin antibody (cat. no. SC-47778; 1:1,000),
mouse Bax antibody (cat. no. SC-7480; 1:1,000), mouse Bcl-2
antibody (cat. no. SC-7382; 1:1,000), goat Apaf-1 antibody (cat.
no. SC-7232 1:1,000), rabbit Bid antibody (cat. no. SC-11423;
1:1,000), rabbit caspase-9 antibody (cat. no. SC-7885; 1:1,000) and
rabbit cytochrome c antibody (cat. no. SC-7159; 1:1,000) all
from Santa Cruz Biotechnology, Inc. were used at 4°C ovenight.
Horseradish peroxidase-conjugated anti-rabbit antibody for Bid,
caspase-9 and cytochrome c (cat. no. PI-1000; 1:2,000;
Vector Laboratories, Inc.); horseradish peroxidase-conjugated
anti-mouse antibody for β-actin, Bax and Bcl-2 (cat. no. PI-2000;
1:2,000; Vector Laboratories, Inc.); and horseradish
peroxidase-conjugated anti-goat antibody for Apaf-1 (cat. no.
PI-9500; 1:2,000; Vector Laboratories, Inc.) were used as the
secondary antibodies for 1 h at room temperature.
The experiments were performed in normal laboratory
conditions and at room temperature, except for the transferred
membranes. Transferred membranes were performed at 4°C with a cold
pack and pre-chilled buffer. The bands were detected using an
enhanced chemiluminescence detection kit (cat. no. 170-5061; Santa
Cruz Biotechnology, Inc.). In order to compare the relative
expression of proteins, the detected bands were calculated
densitometrically using Molecular Analyst, version 1.4.1 (Bio-Rad
Laboratories, Inc.,).
Data analysis
The numbers of TUNEL-positive and caspase-3-positive
cells in the selected hippocampal areas (CA1, CA2-3 and dentate
gyrus) were counted hemilaterally under an Olympus-BX51 light
microscope (Olympus Corporation, Tokyo, Japan) and were expressed
as the number of cells per mm2 in the selected
hippocampal areas. The areas of hippocampal CA1, CA2-3 and dentate
gyrus were measured using the Image-Pro® Plus
computer-assisted image analysis system version 4.5.1.22 (Media
Cybernetics Inc., Rockville, MD, USA). In order to confirm the
expression of Bcl-2, Bax, Bid, cytochrome c, Apaf-1 and
caspase-9, the detected bands were calculated densitometrically
using the Image-Pro Plus image analysis system (Media Cybernetics
Inc.). Statistical analysis was performed using one-way analysis of
variance followed by Duncan's post-hoc test, and the results
were expressed as the mean ± standard error of the mean. P<0.05
was used to indicate a statistically significant difference.
Results
Effect of dexmedetomidine on
short-term memory in the step-down avoidance task
A step-down avoidance task was performed in order to
evaluate the effect of dexmedetomidine on short-term memory. The
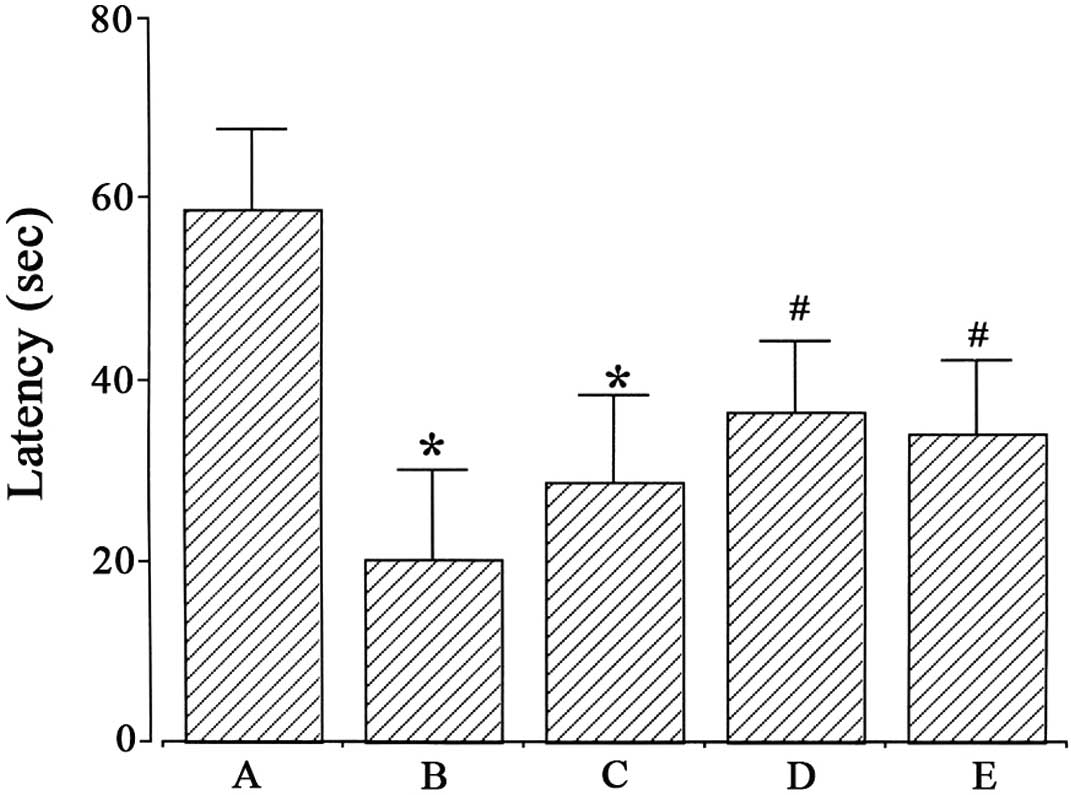
latencies in the step-down avoidance task are presented in Fig. 1. Latency was 58.92±8.83 sec in the
Sham-operated, 20.08±10.06 sec in the ischemia-induction, 28.7±9.58
sec in the ischemia-induction and 0.1 µg/kg
dexmedetomidine-treated, 36.5±7.92 sec in the ischemia-induction
and 1 µg/kg dexmedetomidine-treated and 34.0±7.81 sec in the
ischemia-induction and 10 µg/kg dexmedetomidine-treated groups.
Induction of ischemia disturbed short-term memory
(P<0.05), whereas dexmedetomidine treatment with 1 and 10 µg/kg
alleviated ischemia-induced short-term memory impairment in gerbils
(P<0.05). In the Sham-operated gerbils, dexmedetomidine
treatment exerted no significant effect on short-term memory
(Fig. 2).
Effect of dexmedetomidine on the
number of TUNEL-positive cells in the hippocampus
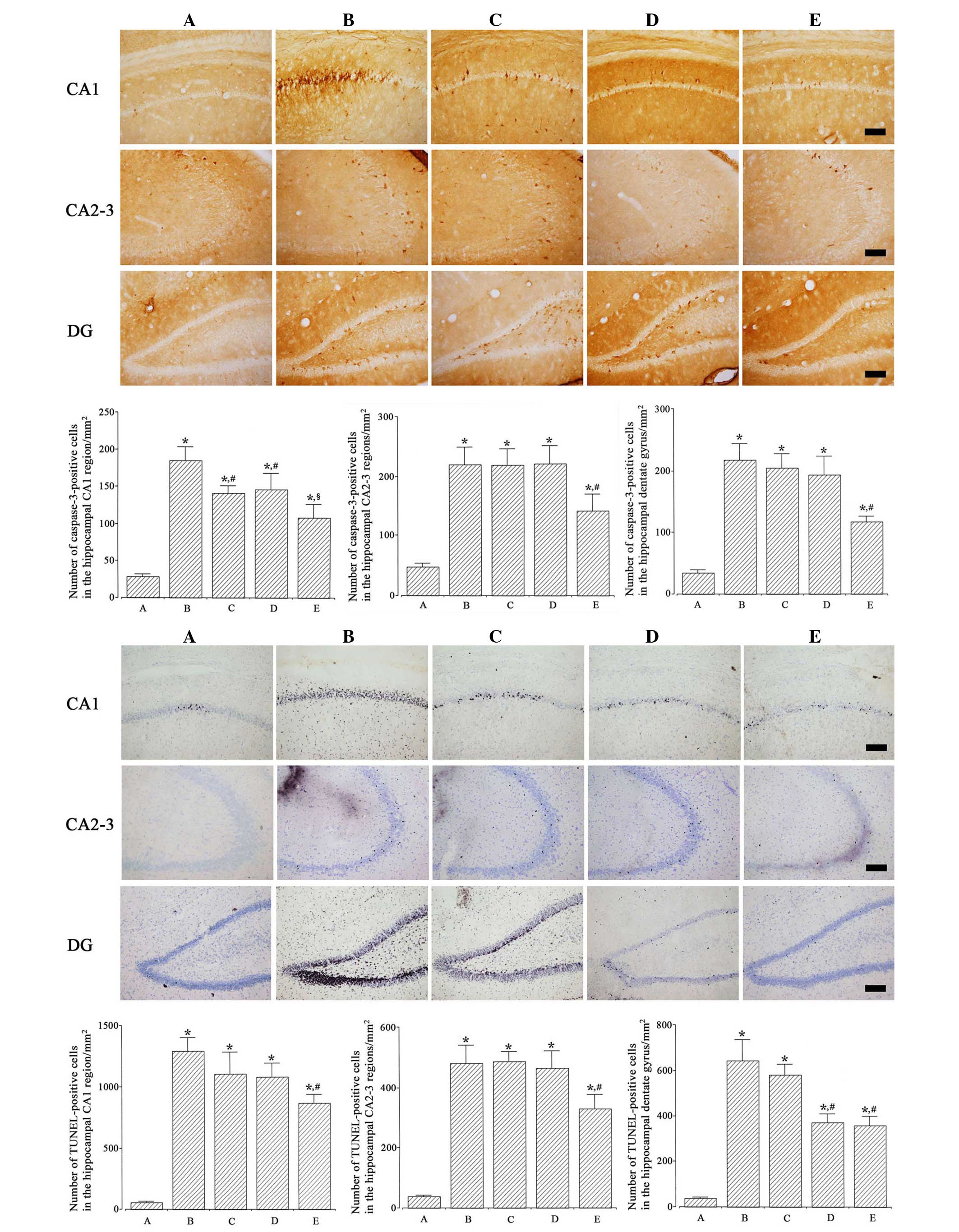
Photomicrographs of TUNEL-positive cells in the
hippocampus are presented in Fig. 3
(lower). The numbers of TUNEL-positive cells in the CA1, CA2-3 and
dentate gyrus regions were 57.30±7.85, 38.28±2.90 and
36.67±5.52/mm2 in the Sham-operated; 1,290.59±106.45,
481.59±58.90 and 643.21±91.05/mm2 in the
ischemia-induction; 1,103.63±176.84, 486.66±33.06 and
580.02±46.85/mm2 in the ischemia-induction and 0.1 µg/kg
dexmedetomidine-treated; 1,077.96±108.16, 465.17±56.67 and
372.78±37.36/mm2 in the ischemia-induction and 1 µg/kg
dexmedetomidine-treated; and 864.04±70.76, 329.95±46.93 and
357.87±40.98/mm2 in the ischemia-induction and 10 µg/kg
dexmedetomidine-treated groups, respectively.
Induction of ischemia enhanced DNA fragmentation in
the hippocampus (P<0.05), whereas dexmedetomidine treatment
suppressed ischemia-induced DNA fragmentation in gerbils
(P<0.05). In the Sham-operated gerbils, dexmedetomidine
treatment exerted no significant effect on DNA fragmentation
(Fig. 4, left).
Effect of dexmedetomidine on the
number of caspase-3-positive cells in the hippocampus
Photomicrographs of caspase-3-positive cells in the
hippocampus are represented in Fig.
3 (upper). The numbers of caspase-3-positive cells in the CA1,
CA2-3 and dentate gyrus regions were 28.78±3.56, 47.60±5.19 and
34.42±4.25/mm2 in the Sham-operated; 184.35±18.81,
219.92±28.75 and 217.42±25.93/mm2 in the
ischemia-induction; 141.26±9.92, 218.56±27.45 and
204.21±23.68/mm2 in the ischemia-induction and 0.1 µg/kg
dexmedetomidine-treated; 146.32±21.72, 220.88±29.74 and
193.81±30.05/mm2 in the ischemia-induction and 1 µg/kg
dexmedetomidine-treated, and 108.35±17.42, 141.77±28.10 and
116.96±9.05/mm2 in the ischemia-induction and 10 µg/kg
dexmedetomidine-treated groups, respectively.
Induction of ischemia enhanced caspase-3 expression
in the hippocampus (P<0.05) and dexmedetomidine treatment
suppressed ischemia-induced caspase-3 expression in gerbils
(P<0.05). In the Sham-operated gerbils, dexmedetomidine
treatment exerted no significant effect on caspase-3 expression
(Fig. 4, right).
Effect of dexmedetomidine on the
expression of Bcl-2 and Bax proteins
The expression of the anti-apoptotic protein Bcl-2
and the pro-apoptotic protein Bax in the hippocampus was analyzed
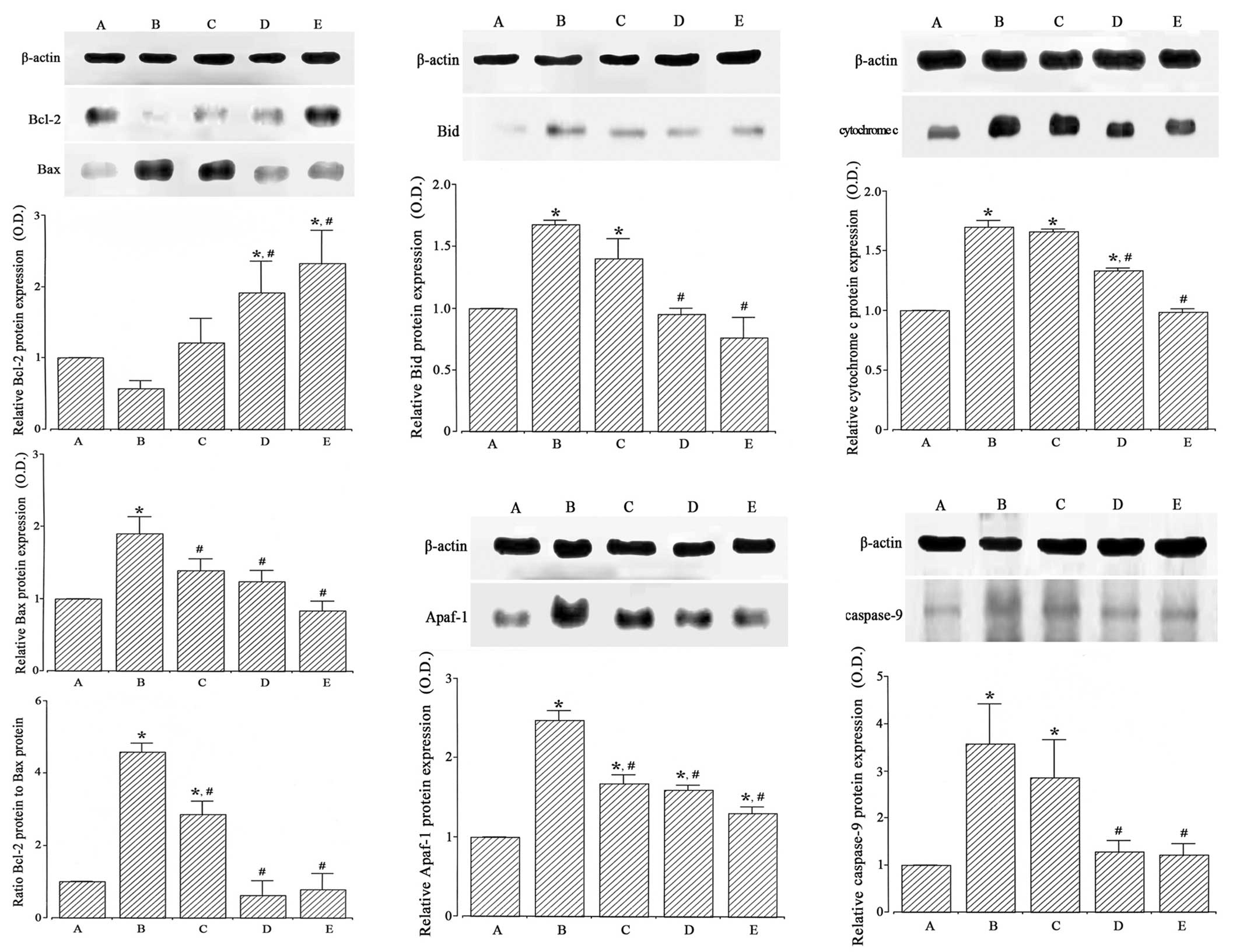
by western blotting (Fig. 5, left).
When the level of Bcl-2 (26–29 kDa) in the Sham-operated group was
set at 1.00, the level of Bcl-2 was 0.57±0.11 in the
ischemia-induction, 1.22±1.01 in the ischemia-induction and 0.1
µg/kg dexmedetomidine-treated, 1.92±1.26 in the ischemia-induction
and 1 µg/kg dexmedetomidine-treated and 2.33±1.32 in the
ischemia-induction and 10 µg/kg dexmedetomidine-treated groups.
When the level of Bax (23 kDa) in the Sham-operated
group was set at 1.00, the level of Bax was 1.90±0.23 in the
ischemia-induction, 1.39±0.16 in the ischemia-induction and 0.1
µg/kg dexmedetomidine-treated, 1.24±0.15 in the ischemia-induction
and 1 µg/kg dexmedetomidine-treated and 0.84±0.13 in the
ischemia-induction and 10 µg/kg dexmedetomidine-treated groups.
When the ratio of Bax to Bcl-2 in the Sham-operated
group was set at 1.00, the ratio of Bax to Bcl-2 was 4.59±2.58 in
the ischemia-induction group, 2.86±2.37 in the ischemia-induction
and 0.1 µg/kg dexmedetomidine-treated, 0.62±0.40 in the
ischemia-induction and 1 µg/kg dexmedetomidine-treated and
0.78±0.44 in the ischemia-induction and 10 µg/kg
dexmedetomidine-treated groups.
These results revealed that induction of ischemia
suppressed Bcl-2 expression (P<0.05) and enhanced Bax expression
in the hippocampus (P<0.05). Dexmedetomidine treatment enhanced
Bcl-2 expression (P<0.05) and suppressed Bax expression in the
ischemic gerbils (P<0.05). As a result, the ratio of Bax to
Bcl-2 was increased by ischemic insult (P<0.05), and
dexmedetomidine treatment inhibited the ratio of Bax to Bcl-2 in
the ischemic gerbils (P<0.05). In the Sham-operated gerbils,
dexmedetomidine treatment exerted no significant effect on Bcl-2 or
Bax expression (Fig. 6, left).
Effect of dexmedetomidine on the
expression of Bid protein
The expression of the pro-apoptotic protein Bid in
the hippocampus was analyzed by western blotting (Fig. 5, middle upper). When the level of Bid
(25 kDa) in the Sham-operated group was set at 1.00, the level of
Bid was 1.68±0.04 in the ischemia-induction, 1.40±0.05 in the
ischemia-induction and 0.1 µg/kg dexmedetomidine-treated, 0.95±0.05
in the ischemia-induction and 1 µg/kg dexmedetomidine-treated and
0.77±0.17 in the ischemia-induction and 10 µg/kg
dexmedetomidine-treated groups.
Induction of ischemia enhanced Bid expression in the
hippocampus (P<0.05), and dexmedetomidine treatment decreased
Bid expression in the ischemic gerbils (P<0.05). In the
Sham-operated gerbils, dexmedetomidine treatment exerted no
significant effect on Bid expression (Fig. 6, middle upper).
Effect of dexmedetomidine on the
expression of cytochrome c protein
The expression of cytochrome c in the
hippocampal cytosol fraction was analyzed by western blotting
(Fig. 5, upper right). When the
level of cytochrome c (15 kDa) in the Sham-operated group
was set at 1.00, the level of cytochrome c was 1.70±0.56 in
the ischemia-induction, 1.66±0.02 in the ischemia-induction and 0.1
µg/kg dexmedetomidine-treated, 1.33±0.03 in the ischemia-induction
and 1 µg/kg dexmedetomidine-treated and 0.99±0.04 in the
ischemia-induction and 10 µg/kg dexmedetomidine-treated groups.
Induction of ischemia enhanced cytochrome c
expression in the hippocampus (P<0.05), and dexmedetomidine
treatment decreased cytochrome c expression in the ischemic
gerbils (P<0.05). In the Sham-operated gerbils, dexmedetomidine
treatment exerted no significant effect on cytochrome c
expression (Fig. 6, upper
right).
Effect of dexmedetomidine on the
expression of Apaf-1 protein
The expression of the pro-apoptotic protein Apaf-1
in the hippocampus was analyzed by western blotting (Fig. 5, lower middle). When the level of
Apaf-1 (130 kDa) in the Sham-operated group was set at 1.00, the
level of Apaf-1 was 2.48±0.12 in the ischemia-induction, 1.67±0.11
in the ischemia-induction and 0.1 µg/kg dexmedetomidine-treated,
1.59±0.06 in the ischemia-induction and 1 µg/kg
dexmedetomidine-treated and 1.30±0.08 in the ischemia-induction and
10 µg/kg dexmedetomidine-treated groups.
Induction of ischemia enhanced Apaf-1 expression in
the hippocampus (P<0.05), and dexmedetomidine treatment
decreased Apaf-1 expression in the ischemic gerbils (P<0.05). In
the Sham-operated gerbils, dexmedetomidine treatment exerted no
significant effect on Apaf-1 expression (Fig. 6, lower middle).
Effect of dexmedetomidine on the
expression of caspase-9 protein
The expression of caspase-9 in the hippocampus was
analyzed by western blotting (Fig.
5, right lower). When the expression of caspase-9 (46 kDa) in
the Sham-operated group was set at 1.00, the level of caspase-9 was
3.58±0.84 in the ischemia-induction, 2.86±0.80 in the
ischemia-induction and 0.1 µg/kg dexmedetomidine-treated, 1.29±0.24
in the ischemia-induction and 1 µg/kg dexmedetomidine-treated and
1.23±0.23 in the ischemia-induction and 10 µg/kg
dexmedetomidine-treated groups.
Induction of ischemia enhanced caspase-9 expression
in the hippocampus (P<0.05), and dexmedetomidine treatment
decreased caspase-9 expression in the ischemic gerbils (P<0.05).
In the Sham-operated gerbils, dexmedetomidine treatment exerted no
significant effect on caspase-9 expression (Fig. 6, right lower).
Discussion
Hypoxic ischemia injury induces incapacitation of
short-term memory in a step-down avoidance task (4). The decrease of latency in the step-down
avoidance task represents memory loss; by contrast, the recovery of
latency near to the normal level represents alleviation of memory
loss (8). Dexmedetomidine has been
revealed to have neuroprotective effects and to facilitate recovery
from various brain insults (18,25,26). An
ameliorating effect of dexmedetomidine on intracerebral
hemorrhage-induced memory impairment was also reported (8). In the present study, the latency period
in the step-down avoidance task was shortened by the induction of
cerebral ischemia, and dexmedetomidine treatment lengthened the
latency in the ischemic gerbils. Thus, the results of the present
study revealed that dexmedetomidine alleviated ischemia-induced
short-term memory impairment in gerbils.
Apoptosis has some morphological characteristics,
which include cell shrinkage, membrane blebbing, chromatin
condensation, formation of apoptotic bodies and internucleosomal
DNA fragmentation (9). Caspase-3
activation is a significant hallmark of apoptosis following
ischemic and hemorrhagic brain insults (5,27). An
increment in the numbers of TUNEL-positive and caspase-3-positive
cells in the hippocampus represents acceleration of apoptotic cell
death (5,28). In oxygen-glucose deprivation
conditions, pre-administration of dexmedetomidine reduced neuronal
death and cleaves caspase-3 expression (21). Furthermore, Eser et al
(25) reported that dexmedetomidine
treatment decreased the number of TUNEL-positive cells in the
hippocampus following transient global cerebral
ischemia/reperfusion injury in rats. Dexmedetomidine also inhibited
isoflurane-induced caspase-3 expression (26). In the present study, the numbers of
caspase-3-positive and TUNEL-positive cells in the hippocampus were
increased following cerebral ischemia, and dexmedetomidine
treatment suppressed these numbers in the ischemic gerbils. Thus,
the results of the present study revealed that dexmedetomidine had
a suppressive effect on ischemia-induced apoptosis in the
hippocampus of gerbils.
Bax and Bcl-2 are two separate members of a gene
family that is important in the regulation of apoptosis (7,8).
Although they share partial nucleotide sequence homology, their
individual products possess opposing functions. Bcl-2 is
functionally characterized as an apoptosis-suppressing factor,
whereas Bax is considered as an apoptosis-promoting factor
(8,10). Furthermore, several inhibitors of Bax
suppress neuronal apoptosis (29).
In the study by Engelhard et al (30), induction of cerebral
ischemia/reperfusion injury, the relative protein concentration of
Bax was increased, in contrast, dexmedetomidine treatment
upregulated Bcl-2 expression. The balance and the location of
anti-apoptotic proteins versus pro-apoptotic proteins are closely
associated with cell death or survival following focal cerebral
ischemia (31). Moreover, the ratio
of Bax to Bcl-2 is an important factor that determines whether
cells undergo apoptosis (32), and
ischemic brain injury disrupts this ratio (33). In addition, Bid is a cytosolic
membrane protein of the Bcl-2 family that is pro-apoptotic. It is
translocated from the cytoplasm into the mitochondria when the cell
receives a death signal (34).
Moreover, it is also known to be a critical mediator for inducing
ischemic cell death in neurons (35). In the present study, the expression
of Bcl-2 in the hippocampus was decreased following cerebral
ischemia; in contrast, the expression of Bax and Bid in the
hippocampus was increased. In addition, dexmedetomidine treatment
enhanced Bcl-2 expression and suppressed Bax and Bid expression in
the ischemic gerbils. Thus, the results of the present study
indicate that the anti-apoptotic effect of dexmedetomidine was
achieved through upregulation of Bcl-2 with downregulation of Bax
and Bid in the hippocampus of ischemic gerbils.
Furthermore, Bcl-2 can inhibit apoptosis by
preventing the release of cytochrome c from the
mitochondria, and Bax appears as the dominant pro-apoptotic protein
involved in cytochrome c release (13,14).
Cytochrome c release that is induced by calcium in neurons
during ischemia involves mitochondrial outer membrane rupture, and
can be blocked by inhibitors of the mitochondrial permeability
transition (13). Thus, such
inhibitors have demonstrated efficacy in animal models of ischemia
(29). Continuous mitochondrial
fission and fusion are involved in the apoptotic neuronal cell
death in the ischemic penumbra following transient middle cerebral
artery occlusion in mice (36). In
the present study, cytochrome c expression in the
hippocampus was increased following cerebral ischemia, and
dexmedetomidine treatment suppressed cytochrome c expression
in the hippocampus of ischemic gerbils. Thus, in the present study,
dexmedetomidine had a suppressive effect on ischemia-induced
cytochrome c expression in the hippocampus of gerbils.
Once cytochrome c is released, it activates
the cytosolic protein Apaf-1 as well as procaspase-3 and
procaspase-9, and together with ATP, they from an apoptosome
(15). Cytochrome c-initiated
activation of Apaf-1 is a crucial step in the
mitochondrial-signaling pathway for the activation of
death-executing caspases in apoptosis. Since this signaling pathway
has been implicated in the pathophysiology of various neurological
disorders including ischemic brain injury, molecular targeting of
the Apaf-1-caspase-9 signaling pathway may be a possible
neuroprotective strategy for enhancing the endogenous threshold for
caspase activation and prevent neuronal loss in stroke and related
disorders (37). Apaf-1 expression
was demonstrated to be increased by permanent focal ischemia in
rats, and downregulation of Apaf-1 could have a potential clinical
value for the treatment of stroke (38). In the present study, caspase-9 and
Apaf-1 expression in the hippocampus was decreased, and
dexmedetomidine treatment inhibited caspase-9 and Apaf-1 expression
in the hippocampus of ischemic gerbils. These observations indicate
that the anti-apoptotic effect of dexmedetomidine was also achieved
through downregulation of the Apaf-1-caspase-9 signaling pathway in
the hippocampus of ischemic gerbils.
In the present study, dexmedetomidine suppressed the
expression of apoptosis-related molecules under ischemic
conditions, resulting in short-term memory improvement. In the
previous study, dexmedetomidine did not induce apoptosis in the
brachial plexus of rats (39). The
present study also revealed that dexmedetomidine did not exert any
effects on the apoptosis-related molecules under the normal (Sham
operation) conditions. Therefore, the α2-adrenoceptor
agonist dexmedetomidine is suggested to be capable of overcoming
cerebral ischemia-induced neuronal apoptotic cell death, which thus
facilitates memory recovery following ischemic cerebral injury.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
founded by the Ministry of Education, Science and Technology (grant
no. 2011-0013878).
References
|
1
|
González RG: Imaging-guided acute ischemic
stroke therapy: From ‘time is brain’ to ‘physiology is brain’. AJNR
Am J Neuroradiol. 27:728–735. 2006.PubMed/NCBI
|
|
2
|
McLean C and Ferriero D: Mechanisms of
hypoxic-ischemic injury in the term infant. Semin Perinatol.
28:425–432. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pereira LO, Arteni NS, Petersen RC, da
Rocha AP, Achaval M and Netto CA: Effects of daily environmental
enrichment on memory deficits and brain injury following neonatal
hypoxia-ischemia in the rat. Neurobiol Learn Mem. 87:101–108. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ko IG, Shin MS, Kim BK, Kim SE, Sung YH,
Kim TS, Shin MC, Cho HJ, Kim SC, Kim SH, et al: Tadalafil improves
short-term memory by suppressing ischemia-induced apoptosis of
hippocampal neuronal cells in gerbils. Pharmacol Biochem Behav.
91:629–635. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Suh HJ, So SM, Na YG, Ko IG, Kim SE, Sung
YS, Shin MS, Kim CJ, Cho YS and Kim KH: Neuroprotective effects of
tamsulosin on intracerebral hemorrhage. Neural Regen Res.
6:2505–2510. 2011.
|
|
6
|
Mattson MP, Duan W, Pedersen WA and
Culmsee C: Neurodegenerative disorders and ischemic brain diseases.
Apoptosis. 6:69–81. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thompson CB: Apoptosis in the pathogenesis
and treatment of disease. Science. 267:1456–1462. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hwang L, Choi IY, Kim SE, Ko IG, Shin MS,
Kim CJ, Kim SH, Jin JJ, Chung JY and Yi JW: Dexmedetomidine
ameliorates intracerebral hemorrhage-induced memory impairment by
inhibiting apoptosis and enhancing brain-derived neurotrophic
factor expression in the rat hippocampus. Int J Mol Med.
31:1047–1056. 2013.PubMed/NCBI
|
|
9
|
Li Y, Chopp M, Jiang N, Zhang ZG and
Zaloga C: Induction of DNA fragmentation after 10 to 120 minutes of
focal cerebral ischemia in rats. Stroke. 26:1252–1257; discussion
1257–1258. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reed CJ: Apoptosis and cancer: Strategies
for integrating programmed cell death. Semin Hematol. 37(4): Suppl
7. 9–16. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Benchoua A, Guégan C, Couriaud C, Hosseini
H, Sampaïo N, Morin D and Onténiente B: Specific caspase pathways
are activated in the two stages of cerebral infarction. J Neurosci.
15:7127–7134. 2001.
|
|
12
|
Kato H and Kogure K: Biochemical and
molecular characteristics of the brain with developing cerebral
infarction. Cell Mol Neurobiol. 19:93–108. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zamzami N, Susin SA, Marchetti P, Hirsch
T, Gómez-Monterrey I, Castedo M and Kroemer G: Mitochondrial
control of nuclear apoptosis. J Exp Med. 183:1533–1544. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Narita M, Shimizu S, Ito T, Chittenden T,
Lutz RJ, Matsuda H and Tsujimoto Y: Bax interacts with the
permeability transition pore to induce permeability transition and
cytochrome c release in isolated mitochondria. Proc Natl Acad Sci
USA. 95:14681–14686. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Love S: Apoptosis and brain ischaemia.
Prog Neuropsychopharmacol Biol Psychiatry. 27:267–282. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Venn RM and Grounds RM: Comparison between
dexmedetomidine and propofol for sedation in the intensive care
unit: Patient and clinician perceptions. Br J Anaesth. 87:684–690.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gertler R, Brown HC, Mitchell DH and
Silvius EN: Dexmedetomidine: A novel sedative-analgesic agent. Proc
(Bayl Univ Med Cent). 14:13–21. 2001.PubMed/NCBI
|
|
18
|
Laudenbach V, Mantz J, Lagercrantz H,
Desmonts JM, Evrard P and Gressens P: Effects of alpha(2)-agonist
adrenoceptor agonists on perinatal excitotoxic brain injury:
Comparison of clonidine and dexmedetomidine. Anesthesiology.
96:134–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma D, Hossain M, Rajakumaraswamy N, Arshad
M, Sanders RD, Franks NP and Maze M: Dexmedetomidine produces its
neuroprotective effect via the alpha 2A-adrenoceptor subtype. Eur J
Pharmacol. 502:87–97. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Y and Kimelberg HK: Neuroprotection
by alpham 2-adrenergic agonists in cerebral ischemia. Curr
Neuropharmacol. 3:317–323. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dahmani S, Rouelle D, Gressens P and Mantz
J: Effects of dexmedetomidine on hippocampal focal adhesion kinase
tyrosine phosphorylation in physiologic and ischemic conditions.
Anesthesiology. 103:969–977. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rajakumaraswamy N, Ma D, Hossain M,
Sanders RD, Franks NP and Maze M: Neuroprotective interaction
produced by xenon and dexmedetomidine on in vitro and in vivo
neuronal injury models. Neurosci Lett. 409:128–133. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim SE, Ko IG, Park CY, Shin MS, Kim CJ
and Jee YS: Treadmill and wheel exercise alleviate
lipopolysaccharide-induced short-term memory impairment by
enhancing neuronal maturation in rats. Mol Med Rep. 7:31–36.
2013.PubMed/NCBI
|
|
24
|
Jeon JW, Lee JI, Shin HP, Cha JM, Joo KR,
Kim SH, Ko IG, Jin JJ, Kim SE and Kim CJ: Adenosine
A2A-receptor agonist polydeoxyribonucleotide promotes
gastric ulcer healing in Mongolian gerbils. Anim Cells Sys.
18:399–406. 2014. View Article : Google Scholar
|
|
25
|
Eser O, Fidan H, Sahin O, Cosar M, Yaman
M, Mollaoglu H, Songur A and Buyukbas S: The influence of
dexmedetomidine on ischemic rat hippocampus. Brain Res.
1218:250–256. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sanders RD, Sun P, Patel S, Li M, Maze M
and Ma D: Dexmedetomidine provides cortical neuroprotection: Impact
on anaesthetic-induced neuroapoptosis in the rat developing brain.
Acta Anaesthesiol Scand. 54:710–716. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Choi JH, Kim TS, Park JK, Sim YJ, Kim K
and Lee SJ: Short-term treadmill exercise preserves sensory-motor
function through inhibiting apoptosis in the hippocampus of hypoxic
ischemia injury rat pups. J Exerc Rehabil. 9:457–462. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim M, Cho KH, Shin MS, Lee JM, Cho HS,
Kim CJ, Shin DH and Yang HJ: Berberine prevents nigrostriatal
dopaminergic neuronal loss and suppresses hippocampal apoptosis in
mice with Parkinson's disease. Int J Mol Med. 33:870–878.
2014.PubMed/NCBI
|
|
29
|
Jemmerson R, Dubinsky JM and Brustovetsky
N: Cytochrome c release from CNS mitochondria and potential for
clinical intervention in apoptosis-mediated CNS diseases. Antioxid
Redox Signal. 7:1158–1172. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Engelhard K, Werner C, Eberspächer E,
Bachl M, Blobner M, Hildt E, Hutzler P and Kochs E: The effect of
the alpha 2-agonist dexmedetomidine and the N-methyl-D-aspartate
antagonist S(+)-ketamine on the expression of apoptosis-regulating
proteins after incomplete cerebral ischemia and reperfusion in
rats. Anesth Analg. 96:524–531. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hu XL, Olsson T, Johansson IM, Brännström
T and Wester P: Dynamic changes of the anti- and pro-apoptotic
proteins Bcl-w, Bcl-2 and Bax with Smac/Diablo mitochondrial
release after photothrombotic ring stroke in rats. Eur J Neurosci.
20:1177–1188. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chung JW, Seo JH, Baek SB, Kim CJ and Kim
TW: Treadmill exercise inhibits hippocampal apoptosis through
enhancing N-methyl-D-aspartate receptor expression in the
MK-801-induced schizophrenic mice. J Exerc Rehabil. 10:218–224.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dave KR, Bhattacharya SK, Saul I, DeFazio
RA, Dezfulian C, Lin HW, Raval AP and Perez-Pinzon MA: Activation
of protein kinase C delta following cerebral ischemia leads to
release of cytochrome c from the mitochondria via bad pathway. PLoS
One. 6:e220572011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sugawara T, Fujimura M, Noshita N, Kim GW,
Saito A, Hayashi T, Narasimhan P, Maier CM and Chan PH: Neuronal
death/survival signaling pathways in cerebral ischemia. NeuroRx.
1:17–25. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Broughton BR, Reutens DC and Sobey CG:
Apoptotic mechanisms after cerebral ischemia. Stroke. 40:e331–e339.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu W, Tian F, Kurata T, Morimoto N and
Abe K: Dynamic changes of mitochondrial fusion and fission proteins
after transient cerebral ischemia in mice. J Neurosci Res.
90:1183–1189. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cao G, Xiao M, Sun F, Xiao X, Pei W, Li J,
Graham SH, Simon RP and Chen J: Cloning of a novel
Apaf-1-interacting protein: A potent suppressor of apoptosis and
ischemic neuronal cell death. J Neurosci. 24:6189–6201. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu XH, Hua YN, Zhang HL, Wu JC, Miao YZ,
Han R, Gu ZL and Qin ZH: Greater stress protein expression enhanced
by combined prostaglandin A1 and lithium in a rat model of focal
ischemia. Acta Pharmacol Sin. 28:1097–1104. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Han JH, Kim DO, Yi JW, Park SW, Kang WJ,
Choi YK, Kim SH, Ko IG, Jin JJ, Kim SE and Kim CJ: Dexmedetomidine,
α2-adrenoceptor agonist, does not induce apoptosis in
the brachial plexus of rats. Anim Cells Sys. 18:407–415. 2014.
View Article : Google Scholar
|