Introduction
Renal ischemia/reperfusion (I/R) injury typically
occurs after renal transplantation and surgery, and is a leading
cause of acute renal failure (1).
Although reperfusion is a necessary approach to improve the
survival probability of ischemic tissue, reperfusion itself can
lead to additional cell injury, largely through the production of
excessive reactive oxygen species (ROS) (2). ROS are a class of chemically reactive
metabolites, including superoxide anions, hydroxyl radicals and
hydrogen peroxide. Overproduction of ROS directly triggers the
oxidation of biological macromolecules, such as DNA, proteins and
lipids, and modulates numerous signaling pathways, particularly
those involving mitogen-activated protein kinases (MAPKs), which
ultimately leads to cell death (3).
MAPKs are a family of serine/threonine protein kinases that
principally consists of extracellular signal-regulated kinase
(ERK), c-Jun N-terminal kinase (JNK) and p38 mitogen-activated
protein kinase (p38 MAPK). Previous results indicate that
modulation of MAPK signaling pathways may have a role in
ROS-induced cell apoptosis during I/R injury (4), suggesting that reducing the generation
of ROS may be a useful therapeutic strategy in the prevention of
I/R injury.
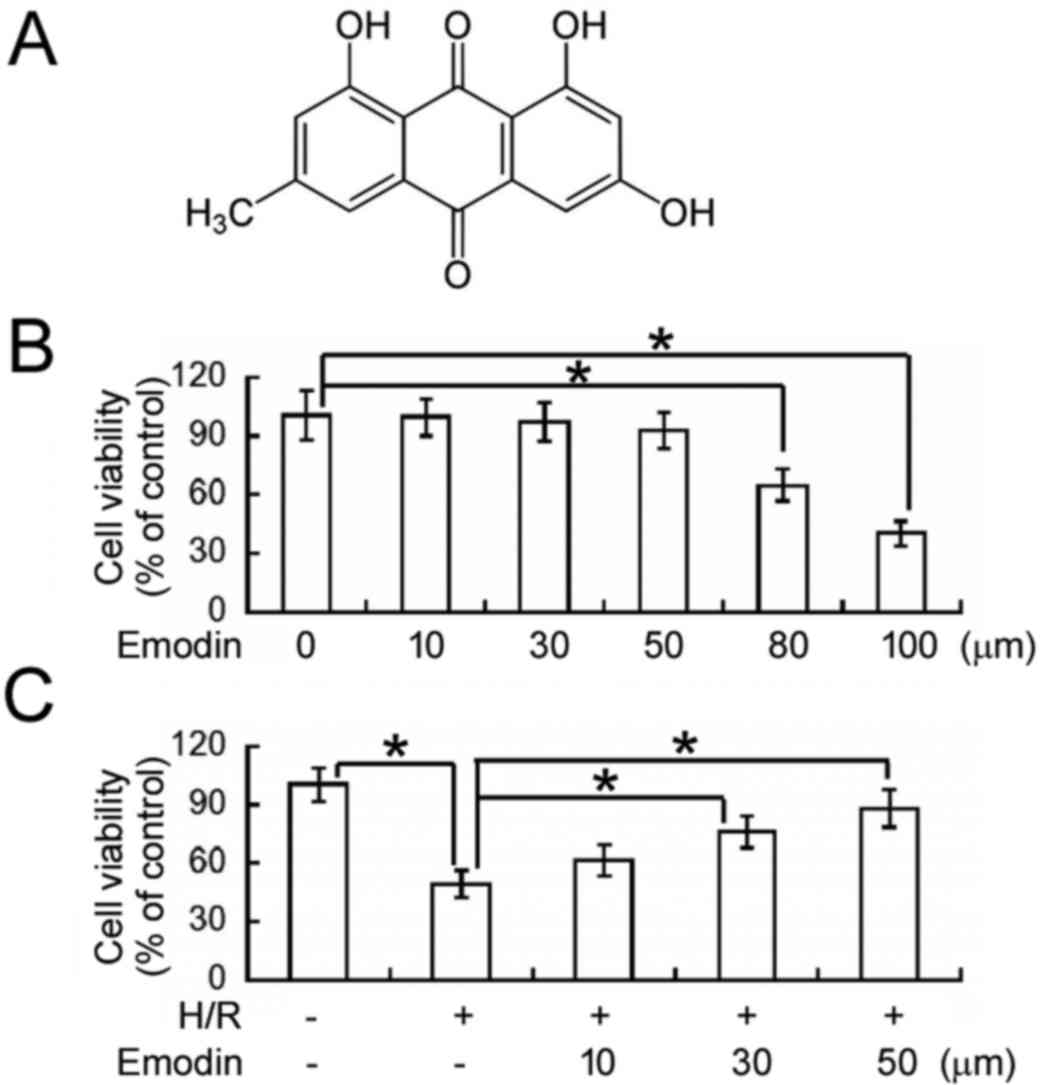
Emodin (1, 3, 8-trihydroxy-6-methyl-anthraquinone;
Fig. 1A) is an active compound of
various traditional Chinese herbs, including Rheum
officinale and Rheum palmatum (5). Emodin has been demonstrated to have
multiple biological activities, including anti-cancer,
anti-diabetic, anti-inflammatory and antioxidant effects (6). It has also been documented that emodin
protects against acute myocardial infarction through the inhibition
of inflammation and apoptosis (7).
In addition, emodin treatment may alleviate I/R injury in rat
hearts (8). However, the effects of
emodin on renal I/R injury are currently unknown.
In the present study, an in vitro model of
renal I/R injury was established in human HK-2 renal tubular cells.
HK-2 cells were treated with different concentrations of emodin
prior to hypoxia/reoxygenation (H/R), and the viability and
apoptotic rates of HK-2 cells were subsequently evaluated using MTT
and annexin-V/propidium iodide (PI) staining assays, respectively.
In addition, levels of intracellular ROS were measured using a
2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA) probe, and
western blot analysis was performed to determine the levels of MAPK
activation.
Materials and methods
Cell culture
HK-2, an immortalized proximal tubule epithelial
cell line isolated from normal adult human kidney, was purchased
from American Type Culture Collection (ATCC, Manassas, VA, USA).
Cells were maintained in Dulbecco's modified Eagle medium
(DMEM)/nutrient mixture F12 supplemented with 10% fetal bovine
serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at
37°C and were subcultured every 3–4 days after reaching 80%
confluence.
H/R protocol and drug treatment
An HK-2 cell-based H/R model was established to
simulate in vivo I/R injury, as described previously
(9). To induce hypoxia, confluent
HK-2 cells were incubated for 24 h in serum-free DMEM in a hypoxia
chamber containing 95% N2 and 5% CO2 at 37°C. Following exposure to
hypoxic conditions, cell medium was replaced with fresh oxygenated
DMEM and cells were reoxygenated for 12 h in normoxic conditions
(5% CO2, 21% O2 and 74% N2) at 37°C.
To evaluate the effect of emodin on H/R injury,
different concentrations (10–50 µM) of emodin (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) in 0.1% dimethyl sulfoxide were added to
cell cultures 2 h before the induction of H/R and incubated at
37°C. Prior to H/R experiments, the cytotoxicity of emodin against
normoxic HK-2 cells was evaluated by exposing cells to different
concentrations of emodin (10, 30, 50, 80 and 100 µM) in DMEM for 48
h at 37°C and measuring changes in cell viability. HK-2 cells
cultured in DMEM under normoxic conditions were used as a vehicle
control. Based on the results of the normoxic HK-2 cell viability
assay, ≤50 µM emodin was used in all subsequent experiments. A
negative control group (no H/R or emodin treatment) and positive
control group (H/R-exposed cells lacking emodin treatment) were
included.
Cell viability assay
Following the aforementioned treatments, HK-2 cells
were seeded into 96-well plates at a density of 2,000 cells/well.
Cells were then subjected to an MTT assay. In brief, 100 µl DMEM
containing 10% FBS was replaced with 100 µl fresh DMEM containing 1
mg/ml MTT (Sigma-Aldrich; Merck KGaA). After 4 h of incubation at
37°C, media was replaced with 150 µl dimethyl sulfoxide to dissolve
the formazan crystals. Absorbance was measured at 570 nm with a
microplate reader (Model 3550; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Apoptosis assay
The ratio of apoptotic cells was examined using an
Annexin V-fluorescein isothiocyanate/PI kit (BD PharMingen, San
Diego, CA, USA) according to the manufacturer's protocol. Briefly,
following the aforementioned drug treatments, cells were
trypsinized, harvested and incubated in the dark at room
temperature with Annexin V and PI for 15 min. Apoptotic cells
(Annexin V+/PI− and Annexin
V+/PI+) were analyzed with a FACSCalibur flow
cytometer (BD Biosciences, San Jose, CA, USA) using CellQuestPro
5.2 software (BD Biosciences).
Measurement of caspase-3 activity
As an indicator of apoptosis, the activity of
caspase 3 enzyme was measured using a Caspase 3 Colorimetric Assay
kit (Sigma-Aldrich; Merck KGaA), according to the manufacturer's
protocol. Briefly, HK-2 cells were lysed in ice-cold lysis buffer
(50 mM HEPES, pH 7.4, 5 mM CHAPS, 5 mM dithiothreitol;
Sigma-Aldrich; Merck KGaA) for 15 min, then centrifuged at 14,000 ×
g for 10 min at 4°C. Protein concentrations were quantified using a
bicinchoninic acid (BCA) protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Cell lysates (200 µg) from each sample were used
to determine caspase-3 activity. The assay was based on the release
of chromophore molecules by the enzymatic cleavage of DEVD
(Asp-Glu-Val-Asp, a caspase-specific peptide substrate conjugated
to reporter ρ-nitroanaline molecules). Caspase 3 activity was
determined by measuring absorbance of the released chromophore at
405 nm with a microplate reader (Model 3550; Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Western blot analysis
The protein content of HK-2 cells was isolated using
a Total Cell Protein Extraction kit (EMD Millipore, Billerica, MA,
USA). Protein concentrations were determined using a BCA Protein
Assay kit (Pierce; Thermo Fisher Scientific, Inc.). An equivalent
amount of protein (40 µg/lane) from each sample was separated by
12% SDS-PAGE and transferred onto nitrocellulose membranes.
Following blocking in 5% non-fat milk at room temperature for 1 h,
membranes were incubated with primary antibodies (Table I) at 4°C overnight. Membranes were
washed three times with Tris-buffered saline with 0.1% Tween-20 (5
min/wash) and incubated with secondary antibodies (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA; Table I) for 1 h at room temperature.
Protein bands were developed with an enhanced chemiluminescence
detection kit (Santa Cruz Biotechnology, Inc.) and quantified by
densitometric analysis using Quantity One software, version 4.6.2
(Bio-Rad Laboratories, Inc.).
 | Table I.Antibodies. |
Table I.
Antibodies.
| Primary antibody | Supplier | Dilution | Catalogue no. |
|---|
| Anti-Bcl-2 | SCB | 1:800 | sc-7382 |
| Anti-Bax | SCB | 1:800 | sc-7480 |
| Anti-ERK | CST | 1:300 | 4348 |
| Anti-p-ERK | CST | 1:300 | 4376 |
| Anti-JNK | CST | 1:300 | 9252 |
| Anti-p-JNK | CST | 1:300 | 9251 |
| Anti-p38 | CST | 1:500 | 9212 |
| Anti-p-p38 | CST | 1:500 | 9211 |
| Anti-β-actin | SCB | 1:1,000 | sc-47778 |
| Secondary
antibody |
|
|
|
| HRP-conjugated goat
anti-mouse IgG | SCB | 1:3,000 | sc-2005 |
| HRP-conjugated goat
anti-rabbit IgG | SCB | 1:3,000 | sc-2004 |
Measurement of ROS levels
Briefly, HK-2 cells in DMEM containing 10% FBS were
incubated for 30 min at 37°C with 5 µM DCF-DA (Molecular Probes;
Thermo Fisher Scientific, Inc.) and washed with phosphate-buffered
saline (PBS). The fluorescence intensity of DCF was measured using
a FACSCalibur flow cytometer (BD Biosciences).
Measurement of malondialdehyde (MDA)
levels and superoxide dismutase (SOD), catalase (CAT) and
glutathione peroxidase (Gpx) activities
To evaluate the extent of oxidative stress in HK-2
cells following H/R and emodin treatment, the levels of MDA and the
activities of SOD, CAT and Gpx in cells were measured using assay
kits for MDA (A003-1), Total SOD (A001-1), CAT (A007-1) and Gpx
(A005; all from Nanjing Jiancheng Bioengineering Institute,
Nanjing, China), respectively.
Statistical analysis
Data are expressed as mean ± standard deviation.
Each assay was performed in triplicate and was repeated three
times. Statistical differences between groups were determined using
one-way analysis of variance followed by a Tukey's post hoc test.
All statistical analyses were performed using SPSS 16.0 software
(SPSS, Inc., Chicago, IL, USA) and P<0.05 was considered to
indicate a statistically significant difference.
Results
Emodin alleviates H/R-induced cell
damage in HK-2 cells
The cytotoxicity of emodin (Fig. 1A) was evaluated in HK-2 cells using
an MTT cell viability assay. As depicted in Fig. 1B, exposure to ≤50 µM emodin for 48 h
did not significantly affect the viability of normoxic HK-2 cells.
However, emodin concentrations of 80 and 100 µM lead to ~35 and 60%
reductions in cell viability, respectively, relative to
vehicle-treated cells (P<0.05). Therefore, ≤50 µM emodin was
used in all proceeding experiments unless otherwise stated.
Using an MTT assay, the effects of emodin on
H/R-induced cellular injury were subsequently determined (Fig. 1C). It was observed that the viability
of HK-2 cells was significantly decreased following H/R exposure,
relative to control cells under normoxic conditions (P<0.05). In
turn, pre-treatment with 30 and 50 µM emodin significantly
alleviated the inhibitory effects of H/R on cell viability
(P<0.05) in a concentration-dependent manner.
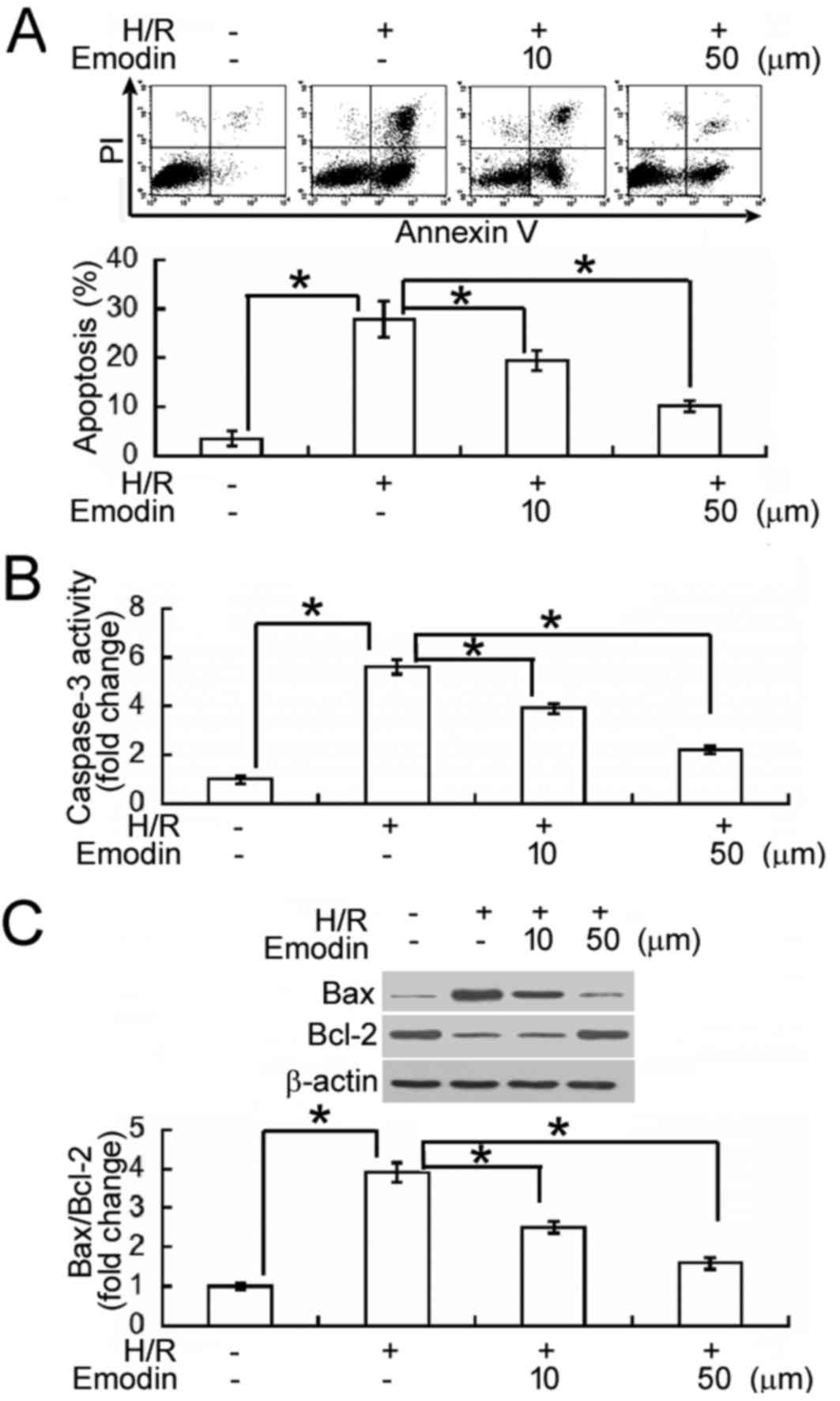
Emodin inhibits HK-2 cell apoptosis
following H/R
Annexin-V/PI staining and flow cytometry analysis
were subsequently performed to determine whether emodin inhibited
H/R-induced apoptosis in HK-2 cells (Fig. 2A). Following H/R exposure, an 8-fold
increase in the percentage of Annexin V+ apoptotic cells
was observed, indicating that the apoptotic rate of HK-2 cells was
significantly increased, relative to normoxic controls (P<0.05).
In turn, emodin pre-treatment (10 and 50 µM) significantly
alleviated the pro-apoptotic effects of H/R, when compared to H/R
treatment alone (P<0.05). To verify the effects of emodin on
H/R-induced apoptosis, levels of caspase-3 activity were also
measured, as an indicator of apoptotic rate. It was observed that
HK-2 cells exposed to H/R had significantly higher (2.5-fold)
levels of caspase-3 activity than normoxic cells (P<0.05;
Fig. 2B). In turn, H/R-induced
caspase-3 activation was significantly inhibited by pre-treatment
with emodin (10 and 50 µM). In addition, levels of B-cell lymphoma
(Bcl)-2 and Bcl-2-associated X protein (Bax), as primary regulators
of apoptosis, were measured by western blot analysis. H/R-exposed
cells exhibited a significant upregulation in Bax and a significant
downregulation in Bcl-2, relative to normoxic cells (both
P<0.05; Fig. 2C). In turn,
pre-treatment with emodin significantly upregulated Bcl-2 and
significantly downregulated Bax in H/R-exposed HK-2 cells, leading
to restoration of the ratio between Bax and Bcl-2 (Fig. 2C).
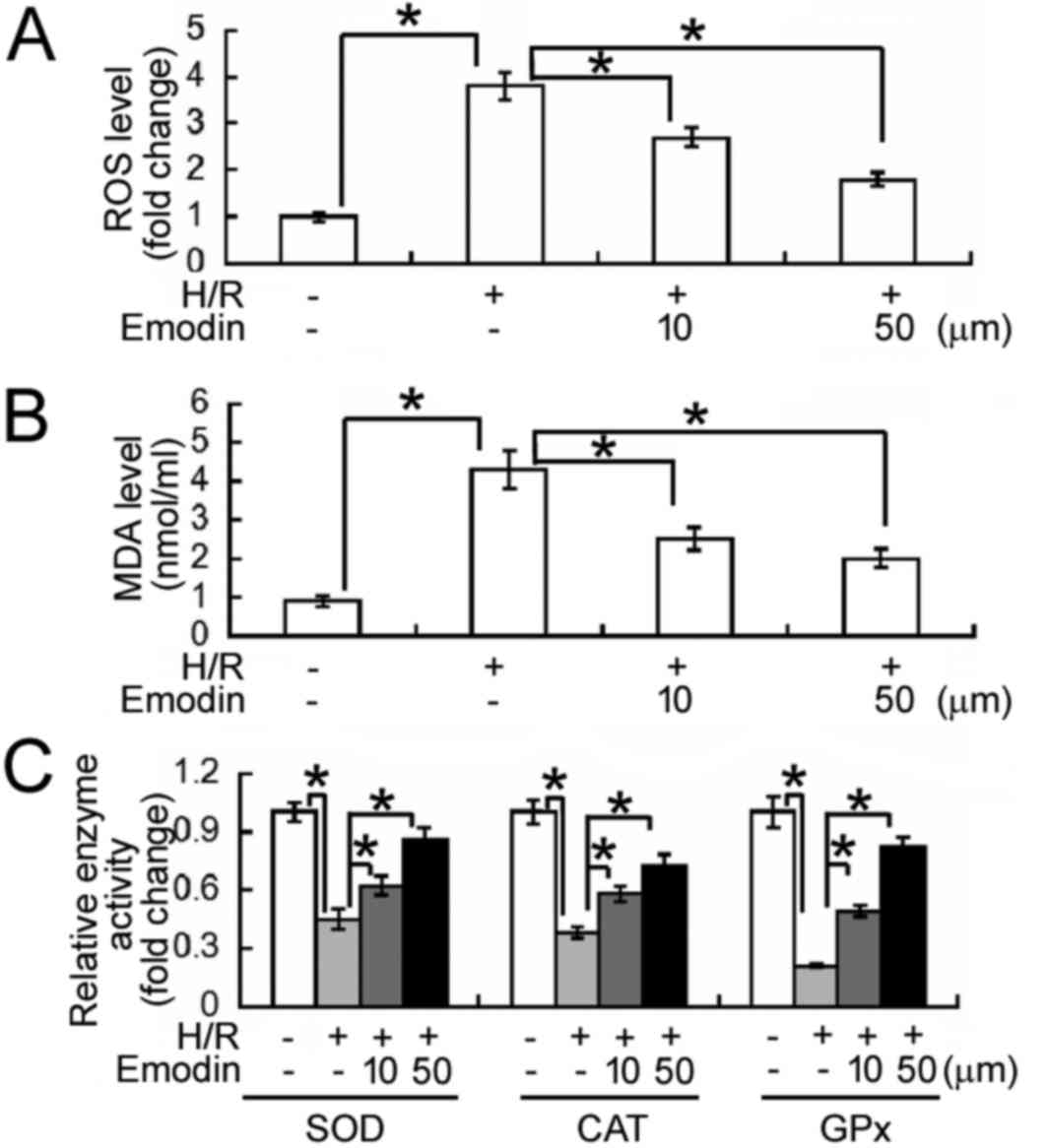
Emodin alleviates oxidative stress in
HK-2 cells following H/R
The effects of emodin on H/R-induced oxidative
stress were evaluated. It was observed that levels of ROS and MDA
were significantly increased following H/R treatment in HK-2 cells,
relative to normoxic cells (P<0.05; Fig. 3A and B, respectively). In turn,
pre-treatment with emodin (10 and 50 µM) significantly decreased
the elevated levels of ROS and MDA induced by H/R (all P<0.05).
By contrast, H/R treatment significantly decreased the activities
of SOD, CAT, and GPx in HK-2 cells; an effect significantly
reversed by emodin pre-treatment (P<0.05; Fig. 3C). These results suggest that emodin
may aid to alleviate oxidative stress under H/R conditions.
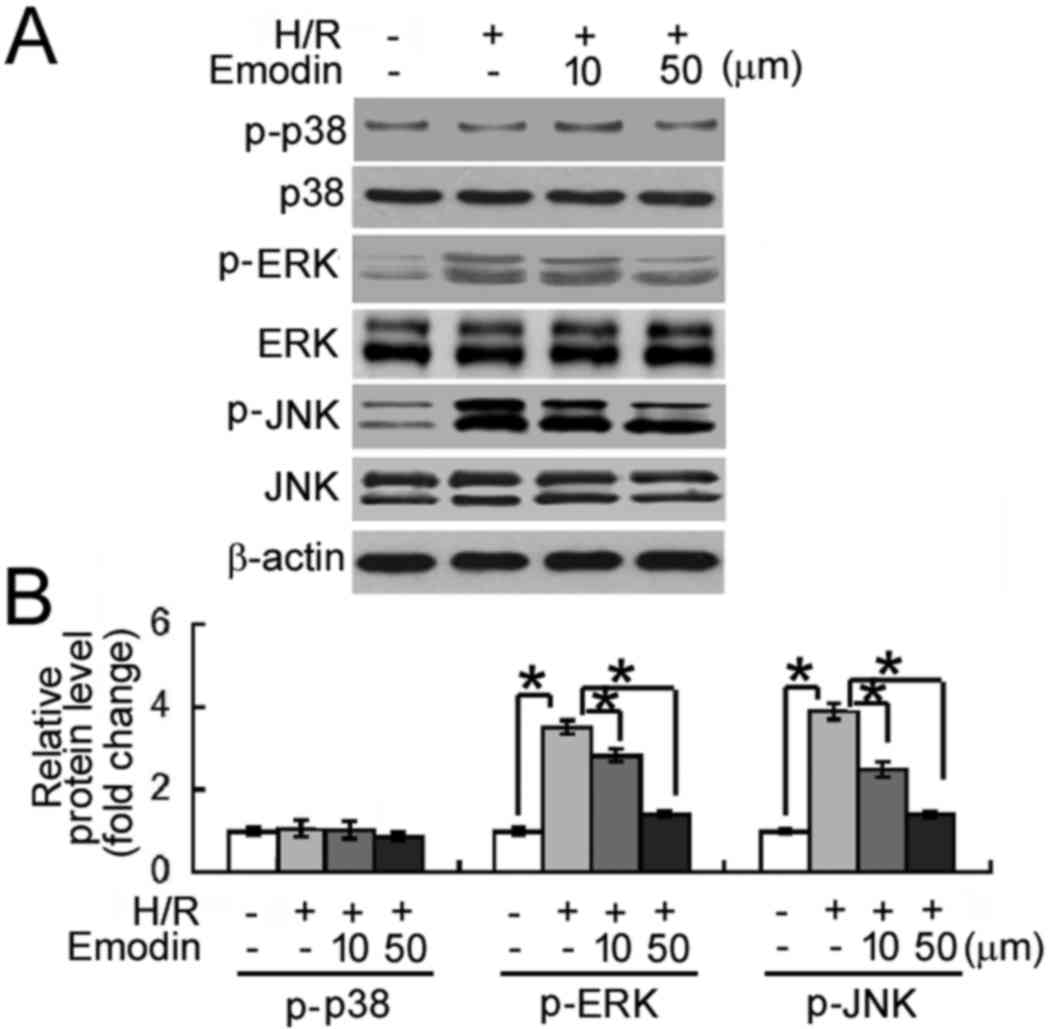
Emodin inhibits H/R-induced activation
of MAPK signaling
The potential molecular mechanisms underlying the
protective effects of emodin against H/R-induced cellular damage
were investigated. As depicted in Fig.
4, levels of phosphorylated ERK and JNK MAPKs were
significantly increased following exposure to H/R, relative to the
normoxic control group (P<0.05). By contrast, the
phosphorylation status of p38 MAPK was unaffected by H/R exposure.
Notably, relative to the H/R group, emodin pre-treatment (10 and 50
µM) significantly inhibited H/R-induced phosphorylation of ERK and
JNK MAPKs (all P<0.05).
Discussion
Oxidative stress mediated by ROS is involved in the
pathogenesis of I/R injury (10).
Numerous natural agents with antioxidant properties, including
butin (11), catalpol (12) and ginsenoside Rg1 (13), have been evaluated as potential
therapeutics in the prevention of ROS production during I/R. The
natural compound emodin is of particular interest due to its
multiple biological activities (6).
Notably, emodin has demonstrated antioxidant activities in
different pathological conditions (14,15). Xue
et al (14) documented that
emodin may protect against lung injury induced by cigarette smoke
by suppressing the formation of ROS. In addition, Nemmar et
al (15) demonstrated that
emodin attenuated pulmonary inflammation and subsequent oxidative
stress induced by diesel exhaust particles in mice. Therefore, the
present study evaluated whether emodin had similar protective
activities in renal I/R injury.
It was demonstrated that emodin pre-treatment
significantly prevented reductions in the viability of HK-2 renal
tubular cells following H/R. Furthermore, this protective activity
of emodin was concentration-dependent. It has previously been
demonstrated that renal tubular cells are sensitive to I/R injury
and undergo substantial levels of cell apoptosis (16). In the present HK-2 cell model, H/R
exposure significantly promoted cell apoptosis, as determined by
Annexin-V/PI staining and measurements of caspase-3 activity.
Notably, the H/R-induced apoptotic response in HK-2 cells was
significantly prevented by emodin pre-treatment. Bcl-2 is an
antiapoptotic protein that maintains the integrity of the outer
mitochondrial membrane, and is inhibited by other members of the
Bcl-2 family (including Bax), leading to cytochrome c
release and caspase-3 activation (17). It has previously been documented that
Bcl-2 protects tubular epithelial cells from I/R injury through the
inhibition of apoptosis (16). In
addition, it has been demonstrated that adenoviral delivery of
Bcl-2 may inhibit proximal and distal tubular apoptosis induced by
renal I/R (14). Consistent with
these previous results, the present study observed that the
protective activity of emodin in H/R-exposed HK-2 cells was
associated with increased Bcl-2 and decreased Bax expression.
Therefore, emodin may aid the protection of renal tubular cells
against H/R-induced apoptosis by restoring the ratio between Bax
and Bcl-2.
The present study also aimed to determine whether
the protective effects of emodin were associated with the
modulation of ROS generation and MAPK signaling. It was observed
that H/R-induced oxidative stress in HK-2 cells was significantly
alleviated by emodin treatment, as indicated by lower levels of ROS
and MDA. Furthermore, the activities of antioxidant enzymes (SOD,
CAT and Gpx) in H/R-exposed cells were significantly increased by
emodin. The results suggest that emodin exerts antioxidant effects
in renal tubular cells under H/R conditions. It has previously been
suggested that oxidative damage induced by ROS overproduction may
be in part mediated by alterations in MAPK activation (3). Bae et al (18) also documented that
4-hydroxy-2-hexenal, a lipid oxidation-derived toxic product,
induced ROS production and apoptosis in HK-2 cells through the
activation of ERK and JNK MAPKs, and that pharmacological
inhibition of ERK or JNK attenuated the damaging effects of
4-hydroxy-2-hexenal. Similarly, the present study found that emodin
pre-treatment suppressed the activation of ERK and JNK MAPKs in
H/R-exposed HK-2 cells. Inactivation of MAPK signaling by emodin
has also been documented in several other biological conditions
(19,20). For instance, in a rat model of
sepsis, emodin attenuated lung injury through inhibition of p38
MAPK (20). The regulation of ROS
production and MAPK activation may represent an important mechanism
for the protection against H/R-induced cellular injury induced by
emodin.
In conclusion, the present data indicates that
emodin prevents H/R-induced apoptosis in human renal tubular cells.
In addition, the protective effects of emodin were associated with
the regulation of cellular oxidative stress and MAPK activation,
along with the restoration of the Bcl-2/Bax ratio. Further studies
are now required in animal models to validate the protective
effects of emodin against renal I/R injury.
Acknowledgements
This study was supported by the Wenzhou Science and
Technology Project of China (grant no. Y20140516).
References
|
1
|
Lee D, Park S, Bae S, Jeong D, Park M,
Kang C, Yoo W, Samad MA, Ke Q, Khang G and Kang PM: Hydrogen
peroxide-activatable antioxidant prodrug as a targeted therapeutic
agent for ischemia-reperfusion injury. Sci Rep. 5:165922015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang Y, Liao H, Zhong S, Gao F, Chen Y,
Huang Z, Lu S, Sun T, Wang B, Li W, et al: Effect of N-n-butyl
haloperidol iodide on ROS/JNK/Egr-1 signaling in H9c2 cells after
hypoxia/reoxygenation. Sci Rep. 5:118092015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Son Y, Cheong YK, Kim NH, Chung HT, Kang
DG and Pae HO: Mitogen-activated protein kinases and reactive
oxygen species: How can ROS activate MAPK pathways? J Signal
Transduct. 2011.792–639. 2011.
|
|
4
|
Kunduzova OR, Bianchi P, Pizzinat N,
Escourrou G, Seguelas MH, Parini A and Cambon C: Regulation of
JNK/ERK activation, cell apoptosis, and tissue regeneration by
monoamine oxidases after renal ischemia-reperfusion. FASEB J.
16:1129–1131. 2002.PubMed/NCBI
|
|
5
|
Aichner D and Ganzera M: Analysis of
anthraquinones in rhubarb (Rheum palmatumRheum officinale) by
supercritical fluid chromatography. Talanta. 144:1239–1244. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen R, Zhang J, Hu Y, Wang S, Chen M and
Wang Y: Potential antineoplastic effects of Aloe-emodin: A
comprehensive review. Am J Chin Med. 42:275–288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu Y, Tu X, Lin G, Xia H, Huang H, Wan J,
Cheng Z, Liu M, Chen G, Zhang H, et al: Emodin-mediated protection
from acute myocardial infarction via inhibition of inflammation and
apoptosis in local ischemic myocardium. Life Sci. 81:1332–1338.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Du Y and Ko KM: Effects of emodin
treatment on mitochondrial ATP generation capacity and antioxidant
components as well as susceptibility to ischemia-reperfusion injury
in rat hearts: Single versus multiple doses and gender difference.
Life Sci. 77:2770–2782. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guan X, Qian Y, Shen Y, Zhang L, Du Y, Dai
H, Qian J and Yan Y: Autophagy protects renal tubular cells against
ischemia/reperfusion injury in a time-dependent manner. Cell
Physiol Biochem. 36:285–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou T, Chuang CC and Zuo L: Molecular
characterization of reactive oxygen species in myocardial
ischemia-reperfusion injury. Biomed Res Int. 2015:8649462015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duan J, Guan Y, Mu F, Guo C, Zhang E, Yin
Y, Wei G, Zhu Y, Cui J, Cao J, et al: Protective effect of butin
against ischemia/reperfusion-induced myocardial injury in diabetic
mice: Involvement of the AMPK/GSK-3β/Nrf2 signaling pathway. Sci
Rep. 7:414912017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cai Q, Ma T, Li C, Tian Y and Li H:
Catalpol protects pre-myelinating oligodendrocytes against
ischemia-induced oxidative injury through ERK1/2 signaling pathway.
Int J Biol Sci. 12:1415–1426. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zu G, Guo J, Che N, Zhou T and Zhang X:
Protective effects of ginsenoside Rg1 on intestinal
ischemia/reperfusion injury-induced oxidative stress and apoptosis
via activation of the Wnt/β-catenin pathway. Sci Rep. 6:384802016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xue WH, Shi XQ, Liang SH, Zhou L, Liu KF
and Zhao J: Emodin attenuates cigarette smoke induced lung injury
in a mouse model via suppression of reactive oxygen species
production. J Biochem Mol Toxicol. 29:526–532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nemmar A, Al-Salam S, Yuvaraju P, Beegam S
and Ali BH: Emodin mitigates diesel exhaust particles-induced
increase in airway resistance, inflammation and oxidative stress in
mice. Respir Physiol Neurobiol. 215:51–57. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suzuki C, Isaka Y, Shimizu S, Tsujimoto Y,
Takabatake Y, Ito T, Takahara S and Imai E: Bcl-2 protects tubular
epithelial cells from ischemia reperfusion injury by inhibiting
apoptosis. Cell Transplant. 17:223–229. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kvansakul M and Hinds MG: The Bcl-2
family: Structures, interactions and targets for drug discovery.
Apoptosis. 20:136–150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bae EH, Cho S, Joo SY, Ma SK, Kim SH, Lee
J and Kim SW: 4-Hydroxy-2-hexenal-induced apoptosis in human renal
proximal tubular epithelial cells. Nephrol Dial Transplant.
26:3866–3873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park SY, Jin ML, Ko MJ, Park G and Choi
YW: Anti-neuroinflammatory effect of emodin in LPS-stimulated
microglia: Involvement of AMPK/Nrf2 activation. Neurochem Res.
41:2981–2992. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yin JT, Wan B, Liu DD, Wan SX, Fu HY, Wan
Y, Zhang H and Chen Y: Emodin alleviates lung injury in rats with
sepsis. J Surg Res. 202:308–314. 2016. View Article : Google Scholar : PubMed/NCBI
|