Introduction
Annually, cardiovascular disease (CVD) is
responsible for >17 million mortalities around the world, and is
known as a disorder with the largest mortality rates globally
(1). Atherosclerosis, which is
characterized by lipid accumulation and long-term inflammation
within the arteries, composes the major underlying pathology of CVD
(2). It is generally considered that
unstable atherosclerotic plaques are responsible for acute
cardiovascular complications, including myocardial infarction,
stroke and sudden cardiac death (3).
Of several cells associated with plaque stability,
macrophages that are derived from the differentiation of monocyte
precursors upon migration into the sub-endothelial space (4,5) are
critical in progressive plaque formation. By the large uptake of
accumulated lipids, particularly the modified low-density
lipoprotein (LDL), macrophages turn into lipid-filled foam cells,
leading to the secretion of pro-inflammatory cytokines and the
establishment of a positive feedback loop for further monocyte
recruitment and intima migration (6,7).
Macrophage macroautophagy (hereafter referred to as
autophagy), which may affect apoptosis and efferocytosis, links
lipid metabolism and the inflammatory response to the development
of atherosclerotic lesions (8,9). In the
lipid-excessive environment of plaques, the autophagy-lysosomal
system degrades cytoplasmic lipid droplets and damaged organelles,
as well as misfolded proteins within macrophages, indicating that
autophagy has a pivotal protective role in preventing the
composition and progression of foam cells (10). However, autophagy becomes
dysfunctional as lesion development progresses (11,12).
Notably, accompanying the deficient autophagy, an evident increase
in the p62/sequestosome-1 (SQSTM1) protein level (hereafter
referred to as p62) appears in macrophage-rich areas of
atherosclerotic aortas (8,9).
p62, an autophagy receptor molecule and a selective
autophagic substrate, may associate with ubiquitin-tagged proteins
and organelles and deliver them to the lysosome for degradation
(13). p62 is also a scaffold
protein with several domains that is implicated in various signal
transduction pathways, including nuclear factor (NF)-κB, NF
erythroid 2-related factor 2, mitogen-activated protein kinase and
mechanistic target of rapamycin (mTOR) (14,15).
Increasing reports have indicated that p62 has multiple
pathophysiological functions in the inflammatory response,
metabolism regulation, inclusion body formation and tumorigenesis
(16,17). However, the regulation and
significance of p62 during atherosclerosis development have not
been characterized.
As an independent risk factor of atherosclerosis,
oxidized LDL (oxLDL) exhibits a variety of atherogenic properties,
including promoting foam cell formation and the inflammatory
response (10,18). In order to investigate the
correlation between p62 expression and oxLDL-induced foam cell
formation, the present study utilized THP-1-derived macrophages
(THP-M) as a cell model.
Materials and methods
Antibodies and reagents
The following primary antibodies were obtained for
the present study: p62/SQSTM1 (cat. no. 8025S; Cell Signaling
Technology, Inc., Danvers, MA, USA); light-chain 3 (LC3)B (cat. no.
L7543); and β-actin (cat. no. A1987) (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany). Horseradish peroxidase-conjugated secondary
antibodies were purchased from OriGene Technologies, Inc., (cat.
nos. TA-130003 and TA140003, respectively; Rockville, MD, USA) and
chloroquine (CQ) diphosphate salt was purchased from Sigma-Aldrich
(cat. no. C6628; Merck KGaA).
Cell culture
Human monocyte cell line, THP-1, was purchased from
the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). Cells were maintained in RPMI-1640 medium
supplemented with 2 mM L-glutamine and 100 U/ml
streptomycin-penicillin (Hyclone; GE Healthcare Life Sciences,
Logan, UT, USA), and 10% heat-inactivated fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C
with 5% CO2. THP-1 cells were plated in 6-well culture
plates at 1×106 cells/well and were differentiated to
macrophages using 100 ng/ml phorbol-12-myristate-13-acetate (PMA;
Sigma-Aldrich; Merck KGaA) for 24 h with serum-free RPMI-1640 at
37°C. Additionally, oxLDL (Guangzhou Yiyuan Biological Technology
Co., Ltd., Guangzhou, China) was added to a final concentration of
20 µg/ml to induce foam cell formation.
Small interfering RNA (siRNA)
knockdown of p62/SQSTM1
p62/SQSTM1 siRNA (sc-29679) and control (con) siRNA
(sc-37007) were purchased from Santa Cruz Biotechnology, Inc.,
(Dallas, TX, USA) (sequences were not released by the company). For
differentiation of THP-1 cells, 100 ng/ml PMA was used. Following
differentiation, a total of 2×105 cells/well were grown
in 6-well plates with 2 ml serum-free RPMI-1640, and transfected
with 50 nM of each siRNA for 48 h using a HiPerfect Transfection
reagent (Qiagen, Inc., Valencia, CA, USA) at 37°C. The SQSTM1 mRNA
levels were examined by reverse transcription-quantitative
polymerase chain reaction (RT-qPCR), and western blotting was
utilized to verify the efficacy of protein knockdown by siRNA.
Oil Red O staining
Following incubation with oxLDL, THP-M cells were
fixed with 4% formalin for 10 min at room temperature, and then
stained with Oil Red O solution (0.5% Oil Red O in 60% isopropanol)
for 15 min at 37°C. Foam cell formation was observed under an
inverted light microscope (magnification, ×100 and ×40), and the
accumulated lipid droplets in the cells were stained red.
Furthermore, the uptake of oxLDL was examined by extracting the
intracellular Oil Red O in isopropanol and by measuring the optical
density (OD) at 520 nm. Another replicate experiment was performed
at the same time for correcting the cell number and size in
different samples. Finally, the degree of lipid droplets was
quantified on the basis of the mean OD value for average cells,
which converts an OD value of ~1×106 cells/ml
isopropanol (19,20).
Western blotting
Cells were lysed in ice-cold RIPA lysis buffer
(POO13C; Beyotime, Shanghai, China). The protein concentration was
determined by BCA method. The total protein samples (40 µg) were
separated on 12% SDS-PAGE gels and transferred onto polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). Following
blocking with Tris-buffered saline and Tween-20 containing 5%
non-fat dry skimmed-milk for 1 h at room temperature, membranes
were incubated overnight with the previously mentioned primary
antibodies (p62/SQSTM1, 1:1,000 dilution; LC3B, 1:1,000 dilution;
and β-actin, 1:1,000 dilution) at 4°C. The appropriate horseradish
peroxidase-conjugated secondary antibodies (goat anti-mouse IgG,
1:5,000 dilution; goat anti-rabbbit IgG, 1:5,000 dilution) were
applied for 1 h at room temperature, and immunoreactivity was
subsequently visualized using a WesternBright enhanced
chemiluminescent kit (Advansta, Inc., Menlo Park, CA, USA).
Furthermore, densitometric analyses were performed using an imaging
analyzer (Tanon 5200; Tanon Science and Technology Co., Ltd.,
Shanghai, China) and Tanon Gis software (Gel Image System Ver.
4.00; Tanon Science and Technology Co., Ltd.). Experiments were
performed in triplicate.
Based on the method described above, firstly, the
conversion of LC3-I to LC3-II was determined by western blotting in
order to evaluate the activity of autophagic initiation.
Subsequently, the activity of autophagic flux was assessed by using
chloroquine (CQ), a specific lysosomal inhibitor that interferes
with vesicular acidification. A total of 2×105
cells/well were grown in 6-well plates with 2 ml serum-free
RPMI-1640 and transfected with 50 nM con siRNA or p62 siRNA for 48
h using a HiPerfect Transfection reagent. Western blotting was
subsequently performed with the anti-LC3B antibody following
exposure to 20 µg/ml oxLDL for the indicated time, with or without
CQ (30 µM) for the last 2 h.
RT-qPCR
Total cellular RNA was isolated using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions, and the integrity was confirmed by
electrophoresis on an ethidium bromide-stained 1.5% agarose gel.
cDNA was generated with an AccuPower RT PreMix (Bioneer Corp.,
Daejeon, Korea), according to the manufacturer's instructions. qPCR
was then performed using an AccuPower GreenStar qPCR PreMix
(Bioneer Corp.), according to the manufacturer's instructions, in a
7300 Real Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermocycling conditions were as follows:
95°C for 20 sec, followed by 40 cycles of 15 sec at 95°C and
annealing/extension for 45 sec at 60°C. The primers were
synthesized and purchased from Bioneer Corp., and the sequences
were as follows: SQSTM1 forward, 5′-ATCGGAGGATCCGAGTGT-3′ and
reverse, 5′-TGGCTGTGAGCTGCTCTT-3′; β-actin forward,
5′-CAACTGGGACGACATGGAGAAAAT-3′ and reverse,
5′-CCAGAGGCGTACAGGGATAGCAC-3′. The relative mRNA expression levels
of p62 were normalized to β-actin following the 2−ΔΔCq
method (21).
ELISA analysis
Interleukin-18 (IL-18) levels from culture media was
measured with an ELISA kit from R&D Systems, Inc., (cat. no.
7620; Minneapolis, MN, USA), according to the manufacturer's
instructions.
Evaluation of cell viability by MTT
assay
An MTT cell viability/cytotoxicity kit was used that
was purchased from Beyotime Institute of Biotechnology (Shanghai,
China). In total, ~10,000 cells were plated in each well of 96-well
plates. Following the treatment with oxLDL and siRNA, 20 µl MTT
reagent (5 mg/ml) was added to each well and the cells were then
cultured for 4 h at 37°C. Subsequently, the supernatant was removed
and 150 µl dimethyl sulfoxide (DMSO) was added to each well to
dissolve formazan. A well with DMSO but without cells was used as a
control, and the OD value of each well was detected at 490 nm using
a Lumo microplate reader (PHOMO; Autobio Diagnostics Co., Ltd.,
Shanghai, China). Cell viability was expressed as a percentage of
the untreated control.
Annexin V/propidium iodide (PI) double
staining and fluorescence-activated cell sorting (FACS)
analysis
Following the treatment with oxLDL and siRNA, THP-M
cells maintained in 10% FBS-RPMI-1640 were collected, re-suspended
and stained with Annexin V-fluorescein isothiocyanate/PI (Annexin
V/PI apoptosis kit; BD Biosciences, Franklin Lakes, NJ, USA),
according to the manufacturer's instructions. A total of
1×104 cells from each sample (con siRNA treated and p62
siRNA treated samples, respectively) were stained with 5 µl Annexin
V and 5 µl propidium iodide (PI) for 15 min in the dark at room
temperature, then re-suspended in 400 µl calcium binding solution,
and analyzed on a FACSCanto II flow cytometer (BD Biosciences).
Data analysis was performed with BD FACSDiva v8.0.1 software (BD
Biosciences). The percentage of Annexin V-positive/PI-negative
(considered as apoptotic cells) and Annexin V-positive/PI-positive
(considered as necrotic cells) staining was determined and compared
with the control groups.
Statistical analysis
All numerical data and error bars were provided as
the mean ± standard deviation. Statistical significance of the
differences among groups was analyzed using GraphPad Prism 6.0
software (GraphPad Software, Inc., La Jolla, CA, USA). Multi-group
comparisons of the mean data were performed using one-way analysis
of variance with the Student-Newman-Keuls test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Prolonged oxLDL treatment results in
advanced foam cell formation
THP-1-derived foam cell models were established
using oxLDL, one of the best-known atherogenic lipoproteins
(10), in order to elevate
intracellular cholesterol levels. Firstly, a time course of changes
in lipid levels was examined during the process of foam cell
formation. As demonstrated in Fig.
1, following treatment with a common dose (20 µg/ml) of oxLDL,
Oil Red O staining and oxLDL uptake assay revealed a time-dependent
increase in lipid levels within the THP-M cells. Furthermore,
incubation with oxLDL for a 48-h period induced an increase in the
size of lipid droplets in the cytoplasm, as assessed by
bright-filed microscopy, which was a typical characteristic of foam
cells. Several cells even burst when cultured with oxLDL for 72 h,
suggesting that advanced foam cells had been formed. Oil Red O
staining was significantly increased following 12, 24 (both
P<0.05), 48 and 72 h (both P<0.01) of oxLDL treatment
compared with 0 h.
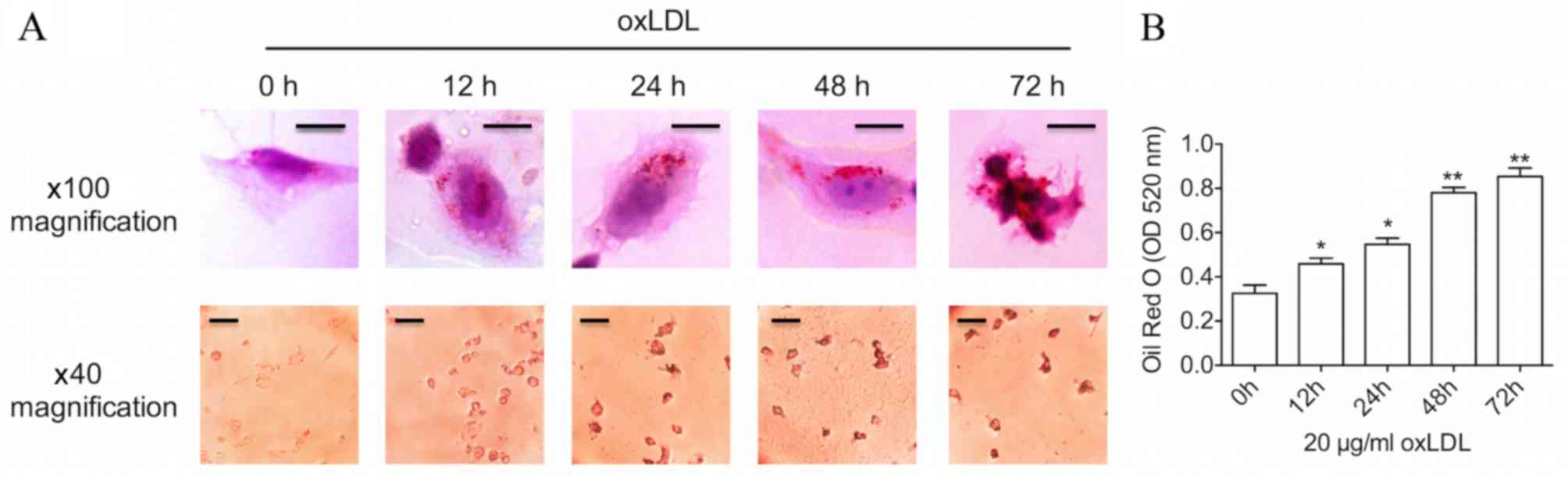 | Figure 1.Prolonged oxLDL stimulation results in
advanced foam cell formation. (A) THP-1-derived macrophages were
incubated with 20 µg/ml oxLDL for 0, 12, 24, 48 and 72 h.
Additionally, Oil Red O staining was used to label foam cells,
which was imaged by a bright-filed microscope. Upper panels:
Magnification, ×100; scale bar, 10 µm. Lower panels: Magnification,
×40; scale bar, 50 µm. (B) The graph represents OD values at 520 nm
of ~1×106 cells/ml isopropanol. Data represent the mean
± standard deviation of six independent experiments. *P<0.05 and
**P<0.01 vs. 0 h. oxLDL, oxidized low-density lipoprotein; OD,
optical density. |
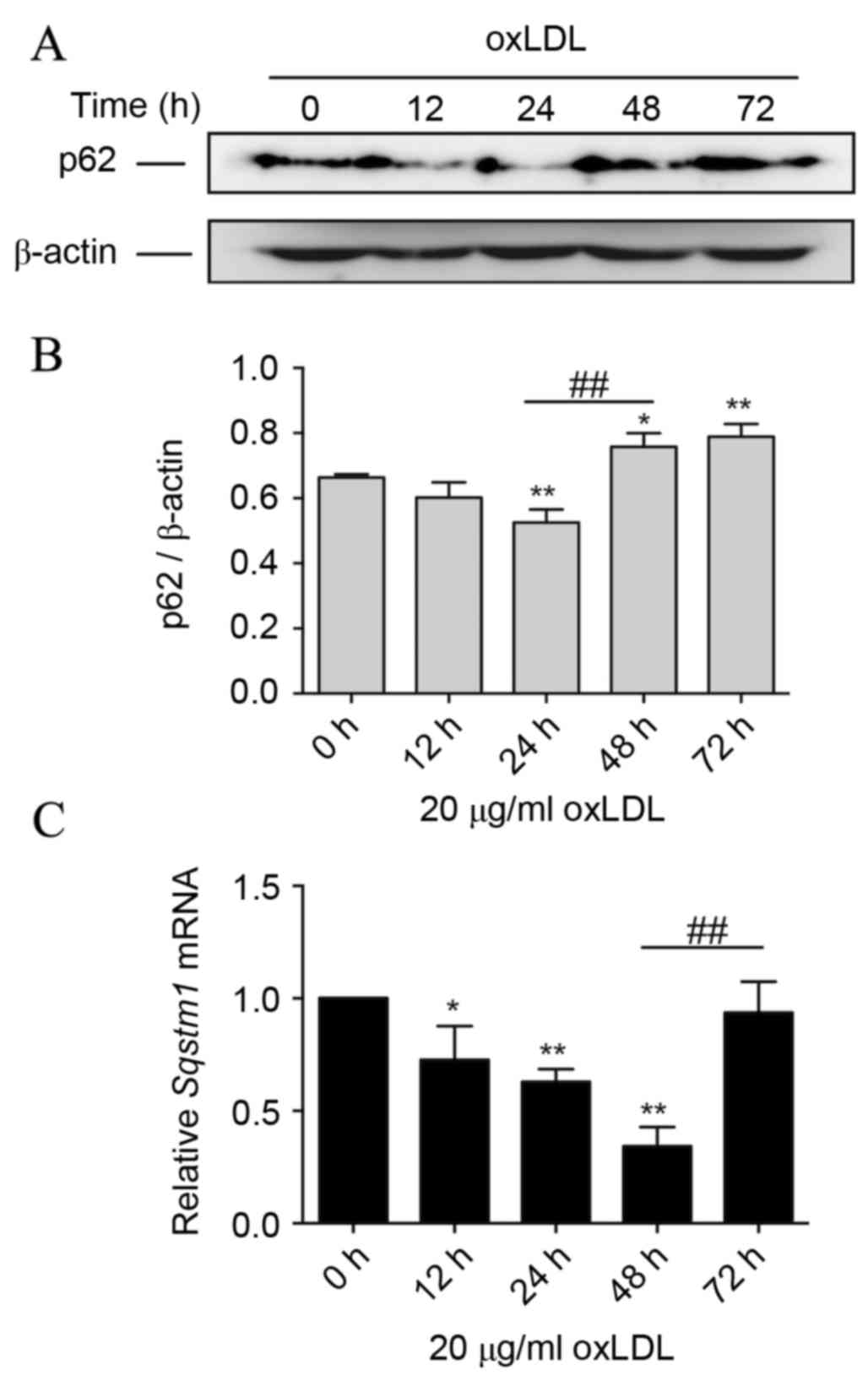
Prolonged oxLDL treatment induces p62
protein accumu-lation during foam cell formation
Data of immunoblotting revealed a significantly
reduced level of p62 protein after 24 h of oxLDL treatment compared
with 0 h (P<0.01). However, during prolonged oxLDL treatment, at
48 and 72 h, the p62 level increased significantly compared with
the level at 24 h (P<0.01), and the level at 72 h was more than
the basal level (P<0.05; Fig. 2A and
B). These data indicated that oxLDL is involved in the
regulation of the p62 protein level, and prolonged oxLDL exposure
leads to elevated expression of p62 in advanced foam cells.
Following this, the transcription level of SQSTM1 mRNA in THP-M
cells with oxLDL treatment was assessed. It was demonstrated that
during the first 48 h of oxLDL treatment, SQSTM1 mRNA was
significantly decreased compared with that at 0 h (P<0.05).
Notably, the SQSTM1 mRNA level then significantly increased
following oxLDL treatment for 72 h (P<0.01), compared with the
level at 48 h, to almost the basal level at 0 h (Fig. 2C).
Knockdown of p62 leads to a decrease
in IL-18 secretion from macrophages with prolonged oxLDL
exposure
Given the multiple pathophysiological roles of p62
reported in studies on other diseases, the present study aimed to
determine the potential role of p62 accumulation in advanced foam
cells. IL-18, one of the pro-inflammatory cytokines in
atherosclerosis, is produced from NLR family pyrin domain
containing 3 inflammasome activation and may be regulated through
the NF-κB pathway (22). In order to
examine whether increased p62 protein levels were associated with
oxLDL-induced IL-18 secretion, THP-M cells were treated with siRNA
against p62. Compared with the con siRNA group, and after oxLDL
treatment for the indicated time, p62 siRNA led to a significant
decrease in the expression of p62 protein by 41.3% at 24 h and
62.0% at 72 h (P<0.05; Fig. 3A).
On the transcriptional level, the p62 siRNA treatment group
revealed a significant reduction of SQSTM1 mRNA to 47.8% at 24 h
and 30.2% at 72 h (P<0.05; Fig.
3B).
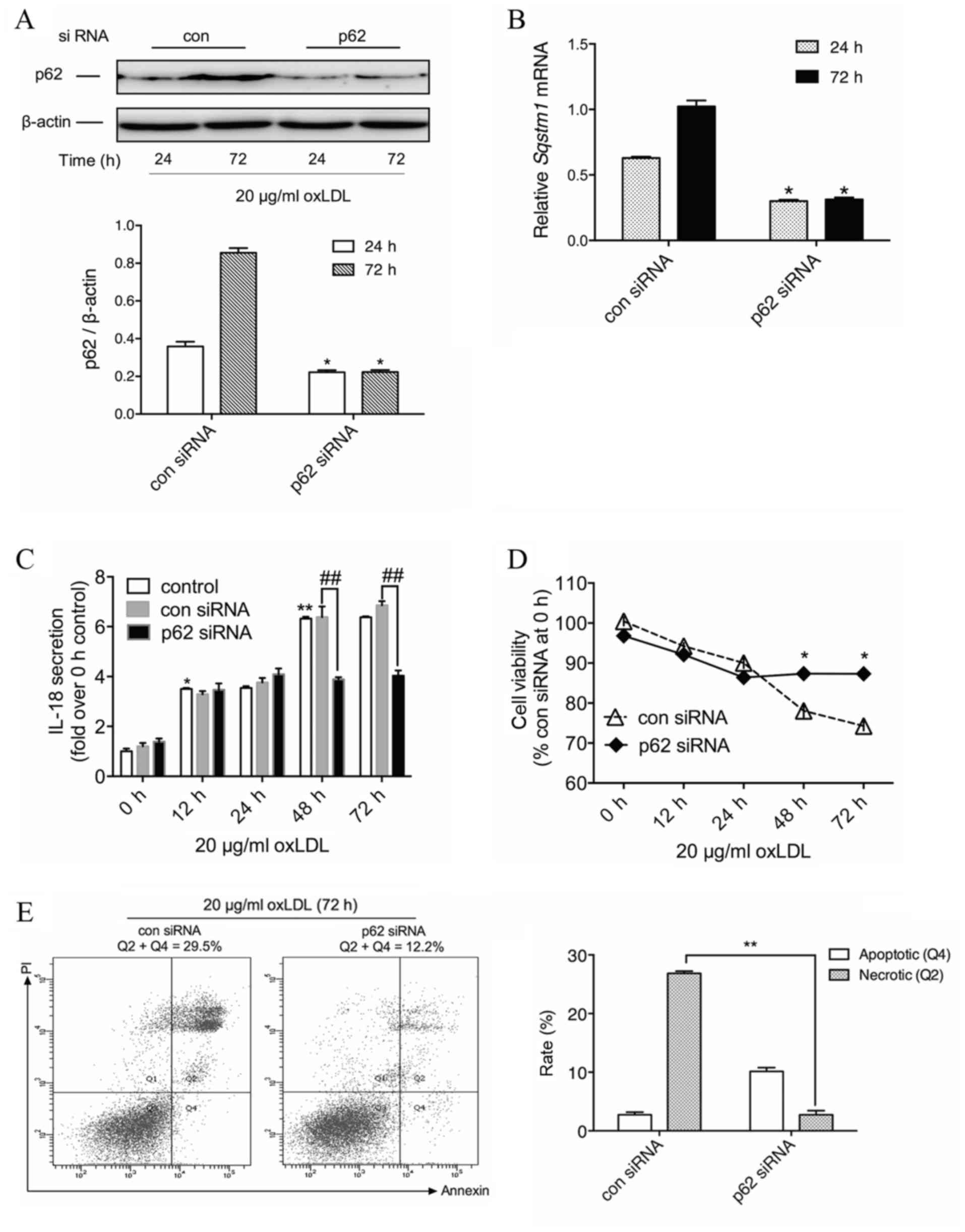 | Figure 3.Silencing p62 reduces IL-18 secretion
and promotes cell survival of macrophages with prolonged oxLDL
exposure. (A) THP-M cells treated with oxLDL (20 µg/ml) for the
indicated time following transfection with 50 nM con siRNA or p62
siRNA. Western blotting experiments were performed on total protein
extracts using anti-p62 antibody, and β-actin expression was used
as a loading control. The graph represents the values of p62 band
intensity after normalization to β-actin by densitometry.
*P<0.05 vs. 24 h group. (B) The graph represents values of the
SQSTM1 mRNA expression following normalization for β-actin mRNA by
reverse transcription-quantitative polymerase chain reaction.
*P<0.05 vs. 24 h group. (C) ELISA of secreted IL-18 in the media
of cells treated with con or p62 siRNA and 20 µg/ml oxLDL for 0,
12, 24, 48 and 72 h. *P<0.05 and **P<0.01 vs. 0 h;
##P<0.01 as indicated. (D) THP-M cells were
pretreated with con or p62 siRNA and then treated with 20 µg/ml
oxLDL for the indicated times. Cell viability was analyzed by the
MTT assay. Results are expressed as a percentage of con siRNA group
at 0 h. *P<0.05 vs. con siRNA group. (E) Determination of cell
death by flow cytometry of Annexin V-fluorescein isothiocyanate/PI
staining in THP-M cells transfected with con or p62 siRNA and then
incubated with oxLDL (20 µg/ml) for the indicated times. Data are
representatives of three independent experiments. **P<0.01 as
indicated. IL, interleukin; oxLDL, oxidized low-density
lipoprotein; THP-M, THP-1-derived macrophages; siRNA, small
interfering RNA; con, control; PI, propidium iodide. |
Time course experiments demonstrated that oxLDL
gradually induced IL-18 secretion from macrophages, and prolonged
oxLDL treatment markedly raised IL-18 levels. Although no
significant difference was observed for the first 24 h between the
con and p62 siRNA groups, 48-h and 72-h oxLDL-treated cells
revealed a significant reduction in IL-18 secretion in the presence
of p62 siRNA compared with those treated with con siRNA (P<0.01;
Fig. 3C). These results indicated
that cytoplasmic accumulation of p62 mediates oxLDL-induced IL-18
secretion in THP-M cells, which implies that p62 could serve as a
proinflammatory stimulus in advanced foam cells.
Knockdown of p62 exhibits positive
effects against prolonged oxLDL-induced cell death
Although cell death has been extensively described
in numerous oxLDL studies (5,7,10), the underlying mechanism remains
unclear. As previous reports have demonstrated, ubiquitinated
protein caspase-8 could interact with p62 and initiate a non-death
receptor-mediated pathway of apoptotic cell death (23). Therefore, the increased p62 level in
advanced foam cells could be detrimental to cell survival. In order
to examine whether p62 was functionally involved in oxLDL-induced
macrophage death, knockdown approaches were used in THP-M cells,
and the changes of cell viability were evaluated for the indicated
time.
As depicted in Fig.
3D, for the first 24 h of oxLDL treatment, there was no
significant difference between con and p62 siRNA-treated cells.
However, following prolonged incubation with oxLDL, the viability
of con siRNA-treated THP-M cells was reduced, and p62 siRNA-treated
cells revealed a significant increase in cell viability at 48 and
72 h after oxLDL treatment compared with con siRNA-treated cells
(P<0.05). Subsequently, the apoptotic and necrotic rates of
THP-M cells were measured by flow cytometry following treatment
with oxLDL for 72 h. Data demonstrated that silencing p62
significantly attenuated prolonged oxLDL-induced cell necrosis
compared with the con siRNA cells (P<0.01; Fig. 3E). This suggested a novel cytotoxic
role of p62 in foam cell progression.
Knockdown of p62 improves autophagic
activity and dampens foam cell formation
Autophagic activity, including both autophagic
initiation and flux, involves the fusion of cargo-laden
autophagosomes with lysosomes and results in cargo degradation
within the lysosome (24). The LC3
conjugation system is critical to autophagy. Cytosolic LC3 (LC3-I)
is processed and converted to the
phosphatidylethanolamine-conjugated form (LC3-II), which is located
on the autophagosomal membrane and thus commonly used as an
autophagosomal indicator (25).
Notably, compared with con siRNA-treated cells, p62 siRNA-treated
cells revealed a significant switch of LC3-I to LC3-II (P<0.01;
Fig. 4A and B), indicating that
autophagic initiation was activated by p62 inhibition. Following
this, the activity of autophagic flux was assessed using CQ. LC3-II
in p62 siRNA-treated cells was further increased in the presence of
CQ at 0 and 72 h compared with cells treated with only p62 siRNA
(P<0.05), which represented the completed autophagic process
(Fig. 4A and C). A previous study
revealed that autophagy regulates cholesterol efflux from
macrophage foam cells (26).
Therefore, it was hypothesized that silencing p62 could prevent
oxLDL-induced lipid deposition in macrophages. Results from Oil Red
O staining revealed that THP-M cells exposed to 72 h of oxLDL
exhibited a significantly reduced number and size of lipid droplets
in the presence of p62 siRNA compared with those treated with oxLDL
and con siRNA (P<0.05; Fig. 4D),
indicating that reducing the p62 level could effectively dampen
foam cell formation.
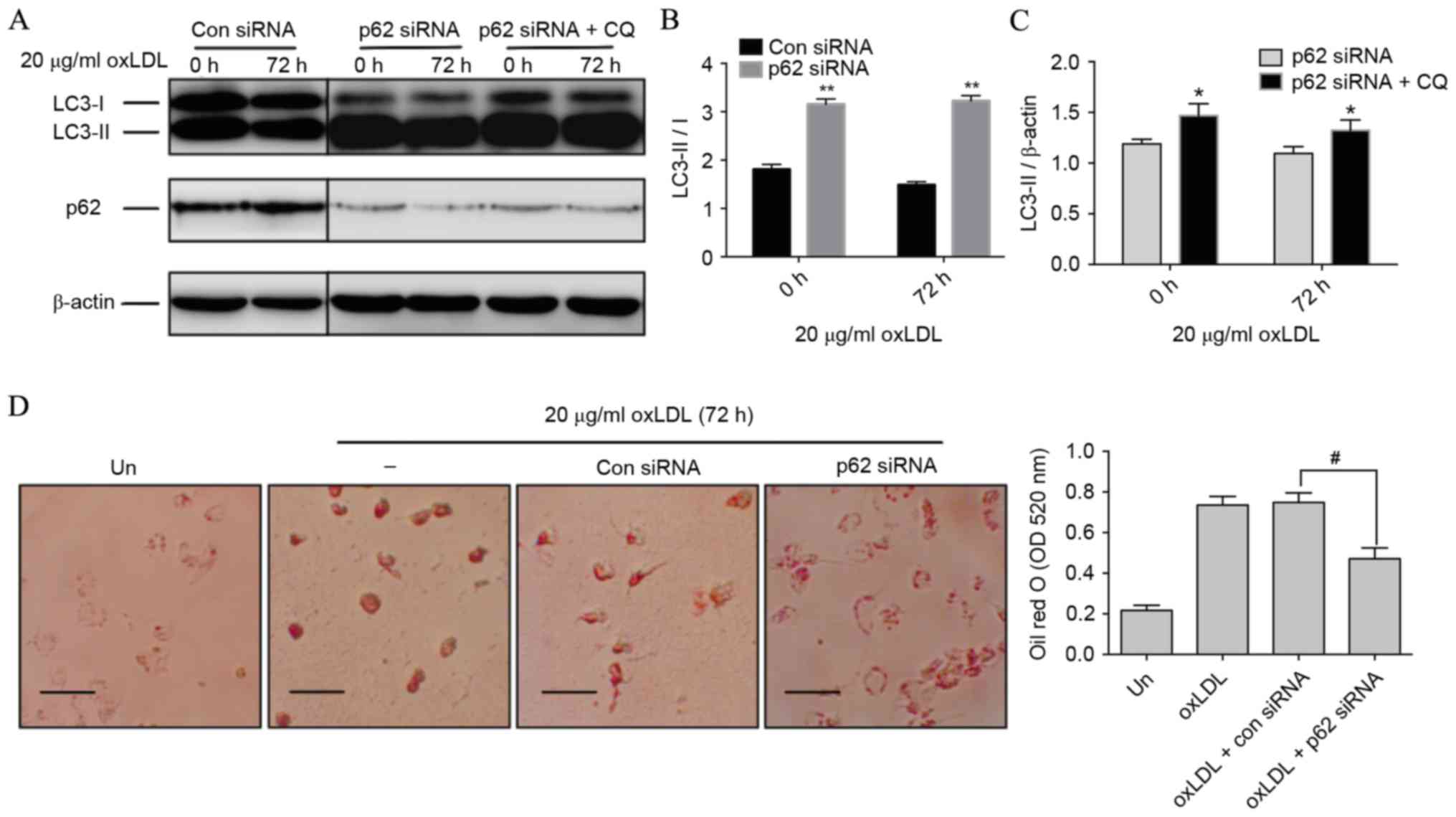 | Figure 4.Silencing p62 improves autophagic
activity and dampens foam cell formation. (A) THP-M cells were
transfected with con or p62 siRNA. Western blot analysis was
performed with the anti-LC3B antibody following exposure to 20
µg/ml oxLDL for the indicated time, with or without CQ (30 µM) for
the last 2 h. β-actin expression was used as a protein loading
control. (B) Densitometric quantification of LC3-II/I from the
western blot analysis of cells treated with con or p62 siRNA. (C)
Densitometric quantifications of LC3-II from the western blot
analysis of cells treated with p62 siRNA with or without CQ. (D)
THP-M cells were treated with con or p62 siRNA for 48 h before
stimulation with 20 µg/ml oxLDL for 72 h. Foam cells were then
assayed by Oil red O staining and imaged by a bright-filed
microscope (magnification, ×40). Scale bar, 50 µm. The graph
represents OD values at 520 nm of ~1×106 cells/ml
isopropanol. Data represent the mean ± standard deviation of three
independent experiments. **P<0.01 vs. con siRNA; *P<0.05 vs.
p62 siRNA; #P<0.05 as indicated. THP-M, THP-1-derived
macrophages; siRNA, small interfering RNA; con, control; ‘−’, oxLDL
treated cells without any siRNA; Un, untreated cells; LC3,
light-chain 3; oxLDL, oxidized low-density lipoprotein; CQ,
chloroquine; OD, optical density. |
Discussion
As a cytosolic protein, the expression of p62 may be
regulated in two ways: Generation and degradation. It was worth
noting that, although treatment with oxLDL for a 48-h period caused
an increase in p62 protein, the transcriptional level of SQSTM1 was
significantly downregulated in the present study. This difference
suggested that degradation pathway autophagic dysfunction may be
involved in the regulation of p62 expression within oxLDL-treated
macrophages.
A previous study has indicated that either excessive
lipid concentrations or chronic lipid exposure possesses the
property of inhibiting autophagy by blocking the fusion of
autophagic/lysosomal compartments or impairing lysosomal
acidification and hydrolase activity (27), which was mostly consistent with the
observations of the present study. By silencing the expression of
p62 in the present study, decreased IL-18 secretion and reduced
overall cell death was observed in prolonged oxLDL-treated cells.
Accordingly, these results indicated a proinflammatory and
cytotoxic role of p62 in foam cell progression. Notably, silencing
p62 significantly decreased the necrosis rate in prolonged
oxLDL-treated macrophages. However, the specific mechanism of this
remains to be discussed in the future.
One surprising observation of the present study was
that, in the RNA interference experiments, p62 siRNA-treated THP-M
cells revealed an evident increase in LC3 conversion, and the
autophagic flux remained normal even after a 72-h incubation with
oxLDL. These results indicated that p62 may negatively regulate
autophagic activity, and these findings are further supported by a
previous study that demonstrated that p62-silencing induced LC3-II
expression and autophagy activation by inhibiting mTOR in numerous
p62-overexpressed carcinoma cells (28).
Furthermore, results from a previous study reported
that mTOR-silencing markedly suppressed foam cell formation with a
decrease in lipid deposition by upregulating autophagy (29). This report extends our thought and
may help explain another observation of the present study, which
demonstrated an evident decrease in lipid accumulation in prolonged
oxLDL-treated THP-M cells in the presence of p62 siRNA. It has been
reported that the autophagic substrate p62 promotes mTOR complex 1
(mTORC1) activity by interacting with its key component, Raptor
(30,31). mTORC1 also has a negative role in the
regulation of autophagy, which contributes to cholesterol efflux
(25). Therefore it is hypothesized
that silencing p62 activates mTORC1, thus enhancing autophagic
activity and leading to augmented reverse cholesterol transport and
restrained foam cell formation. In addition, the discovery of
silencing p62 inducing autophagy activation provides a way to
understand how p62 influences efferocytosis. A previous study
reported that apoptotic cells that die with defective autophagy are
poorly engulfed by efferocytes (32). Therefore, it is presumed that
silencing p62 boosts autophagy, which is beneficial to effective
efferocytosis against secondary necrosis.
In light of the above points, the present study
demonstrated an important role for p62 in the nexus of autophagy
activation, lipid metabolism, inflammatory response and cell death
during foam cell progression. Such an interrelationship may trap
macrophages into a detrimental reinforcing loop, in which prolonged
oxLDL exposure promotes p62 accumulation, as well as lipid
deposition. Next, accumulated p62 further inhibits autophagy, which
in turn leads to more p62 accumulation and accelerates foam cell
formation, with growing inflammation and cell death.
The observations of the present study highlight a
previously unexplored interrelationship between p62 regulation and
lipid metabolism in macrophages, and provide novel insight into how
the plaque switches to an unstable state, allowing us to further
understand the significance of accumulated p62 in atherosclerotic
plaques. Besides being a marker of defective autophagy in
macrophages, p62 may also be a detrimental factor that triggers
increased inflammation and cell death. In addition, knowledge
gained from the present study of p62 in foam cells may be useful
for devising mechanism-based therapeutic strategies. Therefore,
more cell lines, lipid types and even in vivo studies are
required in the future to establish the actual relationship between
the regulation of p62 in macrophage foam cells and
atherosclerosis.
Acknowledgements
The present study was supported by the China
Postdoctoral Science Foundation funded project of Heilongjiang
Province (grant no. LBH-Q12033).
References
|
1
|
World Health Organization (WHO), . A
global brief on hypertension: Silent killer, global public health
crisis (World Health Day 2013). http://apps.who.int/iris/bitstream/10665/79059/1/WHO_DCO_WHD_2013.2_eng.pdfFeb
11–2015
|
|
2
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Finn AV, Nakano M, Narula J, Kolodgie FD
and Virmani R: Concept of vulnerable/unstable plaque. Arterioscler
Thromb Vasc Biol. 30:1282–1292. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Glass CK and Witztum JL: Atherosclerosis:
The road ahead. Cell. 104:503–516. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mestas J and Ley K: Monocyte-endothelial
cell interactions in the development of atherosclerosis. Trends
Cardiovasc Med. 18:228–232. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kleemann R, Zadelaar S and Kooistra T:
Cytokines and atherosclerosis: A comprehensive review of studies in
mice. Cardiovasc Res. 79:360–376. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stewart CR, Stuart LM, Wilkinson K, van
Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA,
et al: CD36 ligands promote sterile inflammation through assembly
of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol.
11:155–161. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liao X, Sluimer JC, Wang Y, Subramanian M,
Brown K, Pattison JS, Robbins J, Martinez J and Tabas I: Macrophage
autophagy plays a protective role in advanced atherosclerosis. Cell
Metab. 15:543–553. 2012. View Article : Google Scholar
|
|
9
|
Razani B, Feng C, Coleman T, Emanuel R,
Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB and Semenkovich CF:
Autophagy links inflammasomes to atherosclerotic progression. Cell
Metab. 15:534–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moore KJ and Tabas I: Macrophages in the
pathogenesis of atherosclerosis. Cell. 145:341–355. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sergin I and Razani B: Self-eating in the
plaque: What macrophage autophagy reveals about atherosclerosis.
Trends Endocrinol Metab. 25:225–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maiuri MC, Grassia G, Platt AM, Carnuccio
R, Ialenti A and Maffia P: Macrophage autophagy in atherosclerosis.
Mediators Inflamm. 2013:5847152013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Komatsu M and Ichimura Y: Physiological
significance of selective degradation of p62 by autophagy. FEBS
Lett. 584:1374–1378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Manley S, Williams JA and Ding WX: Role of
p62/SQSTM1 in liver physiology and pathogenesis. Exp Biol Med
(Maywood). 238:525–538. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bitto A, Lerner CA, Nacarelli T, Crowe E,
Torres C and Sell C: P62/SQSTM1 at the interface of aging,
autophagy and disease. Age (Dordr). 36:96262014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee HM, Shin DM, Yuk JM, Shi G, Choi DK,
Lee SH, Huang SM, Kim JM, Kim CD, Lee JH and Jo EK: Autophagy
negatively regulates keratinocyte inflammatory responses via
scaffolding protein p62/SQSTM1. J Immunol. 186:1248–1258. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Burdelski C, Reiswich V, Hube-Magg C,
Kluth M, Minner S, Koop C, Graefen M, Heinzer H, Tsourlakis MC and
Wittmer C: Cytoplasmic accumulation of sequestosome 1 (p62) is a
predictor of biochemical recurrence, rapid tumor cell
proliferation, and genomic instability in prostate Cancer. Clin
Cancer Res. 21:3471–3479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schrijvers DM, De Meyer GR, Kockx MM,
Herman AG and Martinet W: Phagocytosis of apoptotic cells by
macrophages is impaired in atherosclerosis. Arterioscler Thromb
Vasc Biol. 25:1256–1261. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Murase Y, Kobayashi J, Nohara A, Asano A,
Yamaaki N, Suzuki K, Sato H and Mabuchi H: Raloxifene promotes
adipocyte differentiation of 3T3-L1 cells. Eur J Phamacol. 538:1–4.
2006. View Article : Google Scholar
|
|
20
|
Choi SH, Gonen A, Diehl CJ, Kim J, Almazan
F, Witztum JL and Miller YI: SYK regulates macrophage MHC-II
expression via activation of autophagy in response to oxidized LDL.
Autophagy. 11:785–795. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Badimon L: Interleukin-18: A potent
pro-inflammatory cytokine in atherosclerosis. Cardiovasc Res.
96:172–175. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang YB, Gong JL, Xing TY, Zheng SP and
Ding W: Autophagy protein p62/SQSTM1 is involved in HAMLET-induced
cell death by modulating apoptosis in U87MG cells. Cell Death Dis.
4:e5502013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim KH and Lee MS: Autophagy-a key player
in cellular and body metabolism. Nat Rev Endocrinol. 10:322–337.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ouimet M, Franklin V, Mak E, Liao X, Tabas
I and Marcel YL: Autophagy regulates cholesterol efflux from
macrophage foam cells via lysosomal acid lipase. Cell Metab.
13:655–667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koga H, Kaushik S and Cuervo AM: Altered
lipid content inhibits autophagic vesicular fusion. FASEB J.
24:3052–3065. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nihira K, Miki Y, Ono K, Suzuki T and
Sasano H: An inhibition of p62/SQSTM1 caused autophagic cell death
of several human carcinoma cells. Cancer Sci. 105:568–575. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Li L, Niu X, Dang X, Li P, Qu L,
Bi X, Gao Y, Hu Y and Li M: mTOR enhances foam cell formation by
suppressing the autophagy pathway. DNA Cell Biol. 33:198–204. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ichimura Y, Kumanomidou T, Sou YS,
Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K and
Komatsu M: Structural basis for sorting mechanism of p62 in
selective autophagy. J Biol Chem. 283:22847–22857. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moscat J and Diaz-Meco MT: p62 at the
crossroads of autophagy, apoptosis and cancer. Cell. 137:1001–1004.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng
P, Hogan RN, Gilpin C and Levine B: Autophagy gene-dependent
clearance of apoptotic cells during embryonic development. Cell.
128:931–946. 2007. View Article : Google Scholar : PubMed/NCBI
|