Introduction
Sepsis is a clinical condition that results from an
overwhelming systemic host response to infection (1). Under certain circumstances, components
of the innate immune system that are responsible for the host's
defense against invading pathogens can cause damage to the host's
tissues and organs, resulting in multiple organ failure, a hallmark
of sepsis (2). Previously, studies
have been conducted on the extensive crosstalk between inflammation
and coagulation during sepsis (3).
Systemic inflammation leads to activation of the coagulation system
with concurrent inhibition of anticoagulant mechanisms; coagulation
in turn exacerbates the inflammatory response (4). The protein C pathway is an important
pathway in the crosstalk between coagulation and inflammation
during sepsis (5). In normal
situations, circulating protein C is cleaved and activated by
thrombin to generate activated protein C (APC) that regulates blood
coagulation. The endothelial cell layer provides an anticoagulant
surface by expressing thrombomodulin (TM) and endothelial protein C
receptor (EPCR), which support thrombin in generating APC (6,7).
However, the protein C system is impaired during sepsis; APC is
rapidly consumed during coagulation, while TM and EPCR are cleaved
from the endothelial surface by proteases, including neutrophil
elastase (8) and metalloproteases
(9,10). Thus, a rapid drop in circulating APC,
and concurrent rises in the serum levels of soluble TM (sTM) and
soluble EPCR (sEPCR), are commonly observed in sepsis.
Previous studies have introduced human recombinant
APC to replenish APC levels in order to reduce the effects of
severe sepsis (11–13). However, the clinical trials performed
discovered no evidence suggesting the effectiveness of recombinant
APC for treating patients with severe sepsis or septic shock, and
these trials have recently been discontinued (14). Comparatively, a number of in
vitro and in vivo studies have suggested a potentially
life-saving effect of heparin in the treatment of sepsis due to its
beneficial anti-inflammatory actions (15–18).
Additionally, treatment with unfractionated heparin (UFH) can also
attenuate coagulation in endotoxemic mice (19). A previous study by our group reported
that UFH inhibited inflammatory responses in endothelial cells and
protected endothelial barrier integrity in in vitro models
of lipopolysaccharide (LPS)-induced sepsis (20–22). In
the present study, the effect of UFH on the protein C system in a
mouse model of LPS-induced sepsis, as well as in human umbilical
vein endothelial cells (HUVECs) with LPS challenge, was
investigated.
Materials and methods
Mouse model of LPS-induced sepsis
Male C57BL/6J mice that were 10–12 weeks old and
weighed 20–25 g were provided by the Animal Center of China Medical
University (Shenyang, China). The mice were housed in the animal
center under standard conditions (23±2°C, 65±5% humidity, 12 h
light/dark cycle) with access to food and water ad libitum.
The mice were randomly assigned into three groups as follows:
Control, LPS and LPS + UFH, (12 mice/group). To induce sepsis, LPS
derived from Escherichia coli 055:B5 (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) was administered via an intraperitoneal
injection at a dose of 20 mg/kg body weight, while the control mice
were administered an intraperitoneal injection of 100 µl saline.
UFH with a mean molecular weight of 12,000 Da (Shanghai no. 1
Biochemical and Pharmaceutical Co., Ltd., Shanghai, China) was
administered via the tail vein (5 U/20 g of body weight) 30 min
prior to LPS administration, and the other two groups received an
equal amount of saline.
The animal care and experimental procedures were
conducted in accordance with the Guide for the Care and Use of
Laboratory Animals (23), and the
protocol was approved by the Institutional Animal Care and Use
Committee of China Medical University.
Histopathology
A total of 6 h after LPS administration, 4 mice from
each group were sacrificed. The lung, liver and kidneys were
removed, fixed in 4% paraformaldehyde and embedded in paraffin. The
paraffin blocks were cut into 5-µm-thick sections, and the sections
were stained with hematoxylin for 5 min at room temperature and
then with eosin for 3 min at room temperature. Tissue sections were
observed under an optical microscope.
ELISA
Blood was sampled from the posterior orbital venous
plexus of the mice at 3, 6 and 12 h post LPS administration. Serum
levels of APC, sTM and sEPCR were measured using the respective
commercial ELISA kits (APC, cat. no. SEA738Mu; sTM, cat. no.
SEA529Mu; protein C receptor, cat. no. SEA022Mu; USCN Life
Sciences, Inc., Wuhan, China) according to the manufacturer's
protocol.
Cell culture
Primary HUVECs were obtained from American Type
Culture Collection (Manassas, VA, USA) cultured in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 20% fetal bovine serum (HyClone; GE Healthcare
Life Sciences, Logan, UT, USA), 100 µg/ml streptomycin, 100 U/ml
penicillin and 2 mM L-glutamine. The cells were cultured in a
humidified incubator at 37°C with 5% CO2. The cells were
pretreated with 0.1, 1 or 10 U/ml UFH for 15 min, followed by
treatment with 10 µg/ml LPS for 2, 6, 12, 24 or 48 h. Untreated
cells were used as a control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
HUVECs were harvested at 2, 6, 12 and 24 h after LPS
induction, and total RNA was isolated using TRIzol reagent (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
RT was performed at 40°C for 45 min in a reaction mix (10 µl total
volume) consisting of 1 µl total RNA, 2.0 µl 5X
PrimeScript™ Buffer, 0.5 µl recombinant RNase inhibitor,
0.5 µl dNTPs, 0.5 µl PrimeScript™ RTase (Takara Bio,
Inc., Otsu, Japan), 0.5 µl random primers and 1 µl oligo (dT). qPCR
for TM and EPCR was performed using SYBR-Green Master mix (Beijing
Solarbio Science and Technology Co., Ltd., Beijing, China) with the
following primers: TM forward, 5′-CTGCCGATGTCATTTCCTTGC-3′ and
reverse, 5′-GCTGGTGTTGTTGTCTCCCGTA-3′; EPCR forward,
5′-GAGGCTGGCAAGGGAAAGT-3′ and reverse, 5′-GCAGATGTGGGAGAAGAAAG-3′;
β-actin forward, 5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ and reverse,
5′-CTGTCACCTTCACCGTTCCAGTTT−3′. The thermocycling conditions for
amplification were as follows: Initial denaturation at 95°C for 10
min, followed by 35 cycles of denaturation at 95°C for 10 sec and
annealing/extension at 60°C for 30 sec. The housekeeping gene
β-actin was used for normalization. Quantification of gene
expression was performed using the 2−ΔΔCq method
(24), and was expressed relative to
the control group.
Western blot analysis
Proteins were extracted from HUVECs at 6, 12, 24 and
48 h post LPS induction to examine the expression of TM and EPCR,
and the activation of signaling molecules was assessed 6 h after
LPS induction. Whole cell lysates, nuclear extracts and cytosolic
extracts were prepared as previously described (20). Protein concentration was determined
using the Bradford assay. Equal amounts of protein (40 µg) were
loaded into each lane, separated by 10% SDS-PAGE and transferred
onto polyvinylidene difluoride membranes (EMD Millipore, Billerica,
MA, USA). The membranes were blocked at room temperature for 1 h
with 5% non-fat milk solution in TBS, 50 mM Tris-HCl (pH 7.5) and
150 mM NaCl containing 0.1% Tween-20. The membranes were then
incubated overnight at 4°C with primary antibodies directed against
the following proteins: TM (cat. no. bs-20395R), EPCR (cat. no.
bs-9506R), mitogen-activated protein kinase 14 (p38 MAPK; cat. no.
bs-0637R), phosphorylated (p)-p38 MAPK (cat. no bs-5477R),
proto-oncogene tyrosine-protein kinase Src (Src; cat. no.
bs-10604R), p-Src (cat. no. bs-7619R), p-nuclear factor (NF)-κB
inhibitor-α (IκBα; cat. no bs-1287R; all 1:500; all BIOSS, Beijing,
China) or NF-κB p65 subunit (cat. no. BA0610; 1:400; Wuhan Boster
Biological Technology, Wuhan, China). Following incubation with
horseradish peroxidase-conjugated goat anti-rabbit (cat. no. A0208)
or goat anti-mouse antibodies (cat. no. A0216) (both 1:5,000; both
Beyotime Institute of Biotechnology, Haimen, China) for 45 min at
room temperature, chemiluminescent detection was performed using an
enhanced chemiluminescent reagent (7Sea-ECL; 7Sea Biotech,
Shanghai, China) according the manufacturer's protocol. To verify
equal loading and transfer, the membranes were stripped, and then
re-blotted overnight at 4°C with anti-β-actin (1:1,000; cat. no.
sc-47778; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
antibodies for total and cytosolic proteins, or with anti-histone
H3 (1:1,000; cat. no. bsm-33042M; BIOSS) antibodies for nuclear
proteins. For quantification of the target proteins, the intensity
of the bands was measured using ImageJ software (version 1.6.0;
National Institutes of Health, Bethesda, MD, USA). The protein
levels were presented as the ratio of the target protein to the
respective internal reference protein (β-actin or H3), and then
normalized to the control group.
Statistical analysis
The data are expressed as the mean ± standard
deviation among the individuals in each group for the in
vivo assays or of three independent experiments for the in
vitro assays. Differences between multiple groups were assessed
by one-way analysis of variance with a post hoc Bonferroni
correction test. The statistical analyses were performed using
GraphPad Prism 5.0 (GraphPad software, Inc., La Jolla, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
UFH preconditioning attenuates
LPS-induced damage of the APC system in a mouse model
Previous studies have demonstrated that UFH could
rescue sepsis-associated acute lung injury and lethality in
LPS-induced rodents (17,25). In the present study, LPS was
administered to the mice with or without UFH preconditioning, and
histopathological examinations of the lung, liver and kidneys were
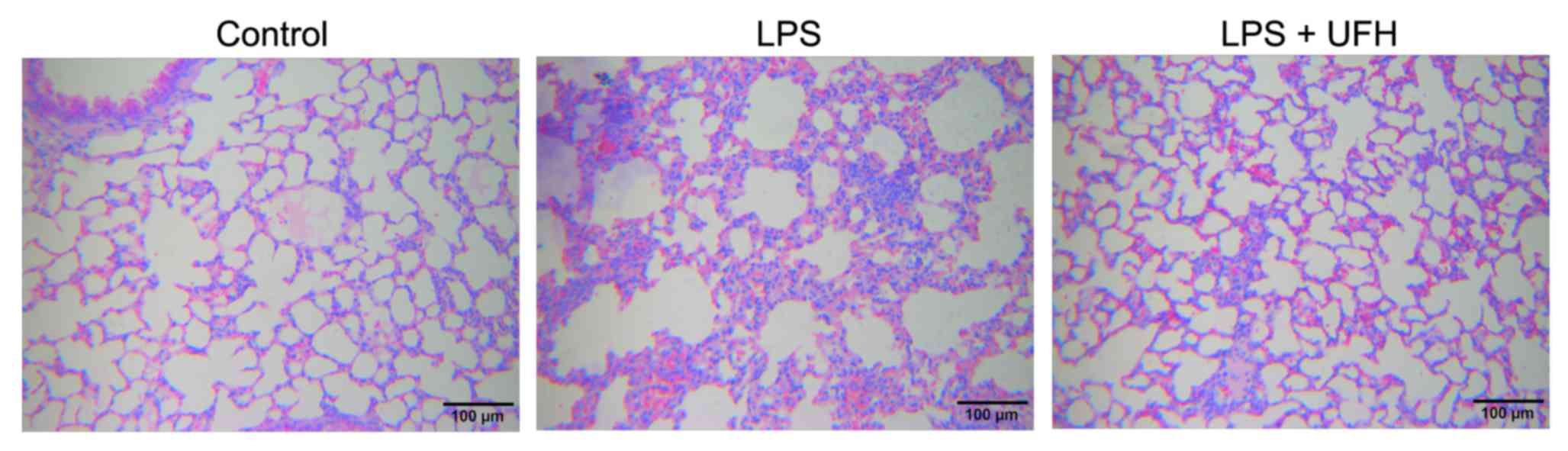
conducted 6 h after LPS induction. As shown in Fig. 1, administration of LPS led to
disruption of alveolar structure and integrity, as well as the
infiltration of leukocytes into the pulmonary tissues. In the mice
pretreated with UFH, the alveolar structure was preserved with
markedly reduced leukocyte infiltration compared to the LPS-induced
mice. No notable pathological changes were observed in the liver or
kidney 6 h post LPS induction (data not shown). These results were
consistent with those of previous studies, which demonstrated that
UFH attenuated LPS-induced acute lung injury in mice, thus
indicating the successful establishment of the animal model.
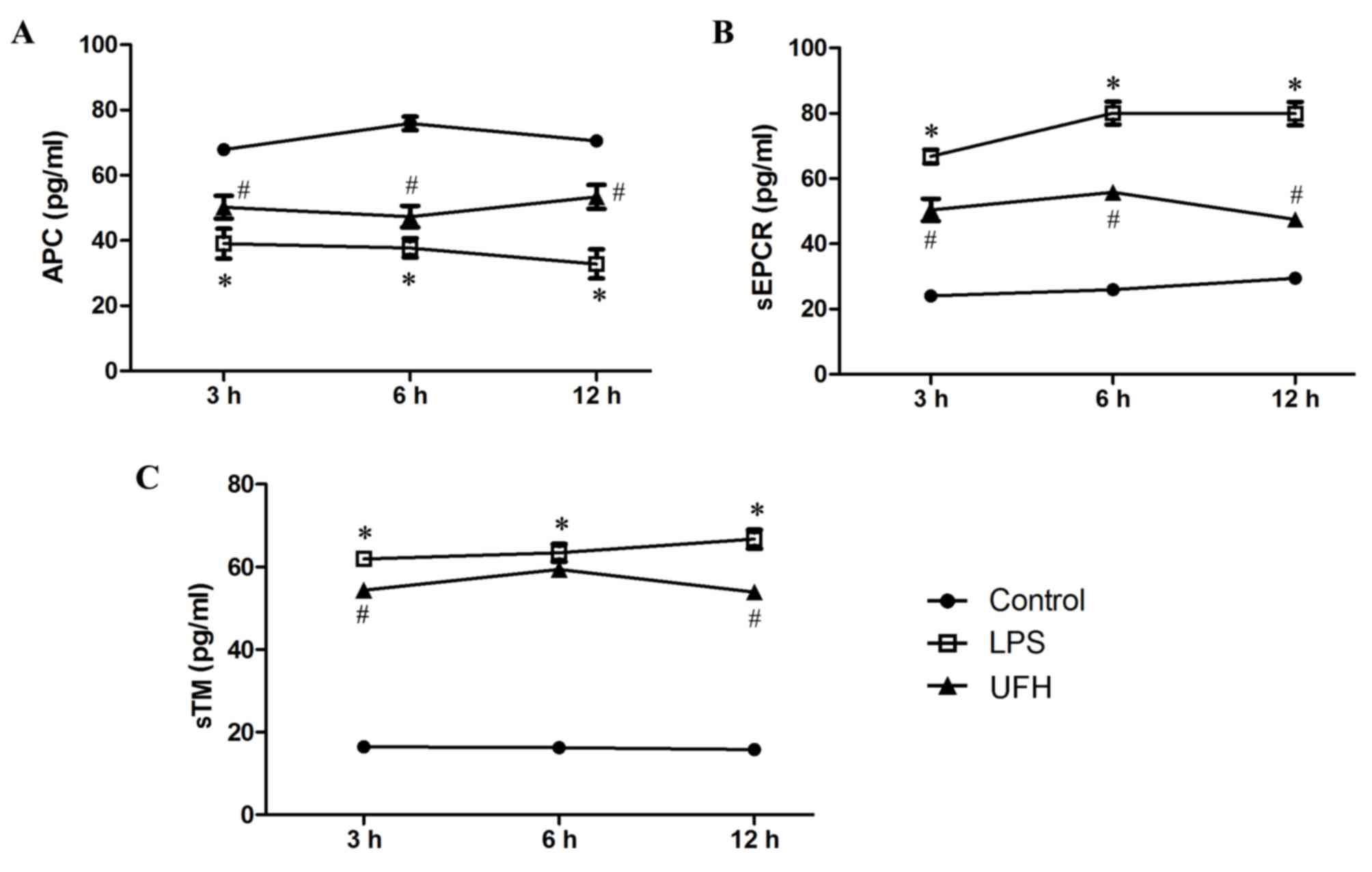
Levels of APC, sEPCR and sTM in the serum of
LPS-induced mice were measured during early sepsis. Serum APC
dropped significantly in LPS-treated mice (39.06±4.58 pg/ml)
compared with the control mice (67.88±0.51 pg/ml) at 3 h post LPS
induction, and the level slowly declined with time (37.72±2.95
pg/ml at 6 h; 32.81±4.45 pg/ml at 12 h) (Fig. 2A). Serum APC was significantly
increased at all time points in the mice that received UFH
preconditioning compared with LPS-treated mice (50.22±3.47 pg/ml at
3 h; 47.32±3.31 pg/ml at 6 h; 53.35±3.67 pg/ml at 12 h).
Furthermore, serum levels of sEPCR and sTM experienced >3-fold
increases following LPS induction (Fig.
2B and C). UFH preconditioning reduced LPS-induced elevation of
serum sEPCR by 38.41–62.48% during the early response (3, 6 and 12
h), while it attenuated LPS-induced elevation of serum sTM to a
lesser extent (8.53–25.15%). These results demonstrate that UFH
preconditioning prevented the exhaustion of circulating APC, and
inhibited the shedding of EPCR and TM, following LPS induction.
LPS-induced downregulation of EPCR and
TM expression is inhibited by UFH in HUVECs
The activation of protein C is enhanced by the
presence of EPCR and TM on the endothelial surface (6,7). As
considerable shedding of EPCR and TM was observed following LPS
induction in the mouse model, it was then investigated whether
endothelial cells could regenerate and replenish these two
molecules during sepsis, and whether UFH had an effect on the
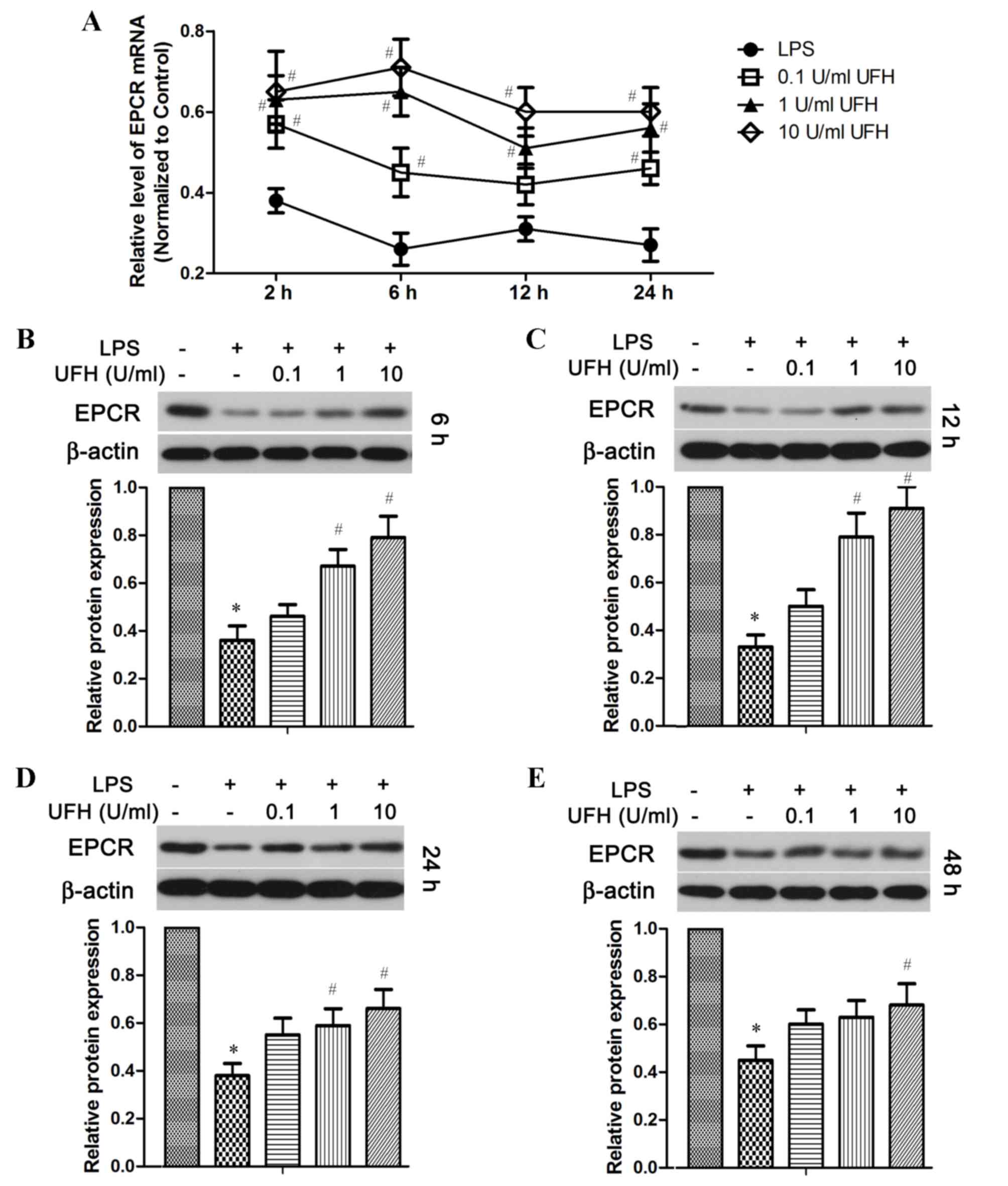
expression of these two molecules in endothelial cells (Figs. 3 and 4). RT-qPCR analysis revealed that the level
of EPCR mRNA was reduced by ~70% in LPS-treated HUVECs compared
with the control cells, whereas pretreatment with UFH significantly
reduced LPS-induced downregulation of EPCR mRNA expression in a
dose-dependent manner compared with LPS-treated cells (Fig. 3A). At the protein level, EPCR was
significantly decreased in LPS-treated HUVECs compared with the
control group shortly following LPS induction (6 h), as well as in
the long term (48 h) (Fig. 3B and
E). A prominent dose-dependent increase in EPCR protein levels
with UFH pretreatment was observed in HUVECs at 6 h and 12 h
following LPS induction (Fig. 3B and
C); however, the dose-dependency was not as strong after 24 and
48 h (Fig. 3D and E). Notably, UFH
of a high dose (10 U/ml) abated LPS-repressed EPCR transcription at
24 h as effectively as it did at 6 h (Fig. 3A). However, high-dose UFH-mediated
protection of EPCR protein was weakened at a later stage of LPS
treatment (48 h) compared with that at an earlier stage (6 and 12
h; Fig. 3 B-E).
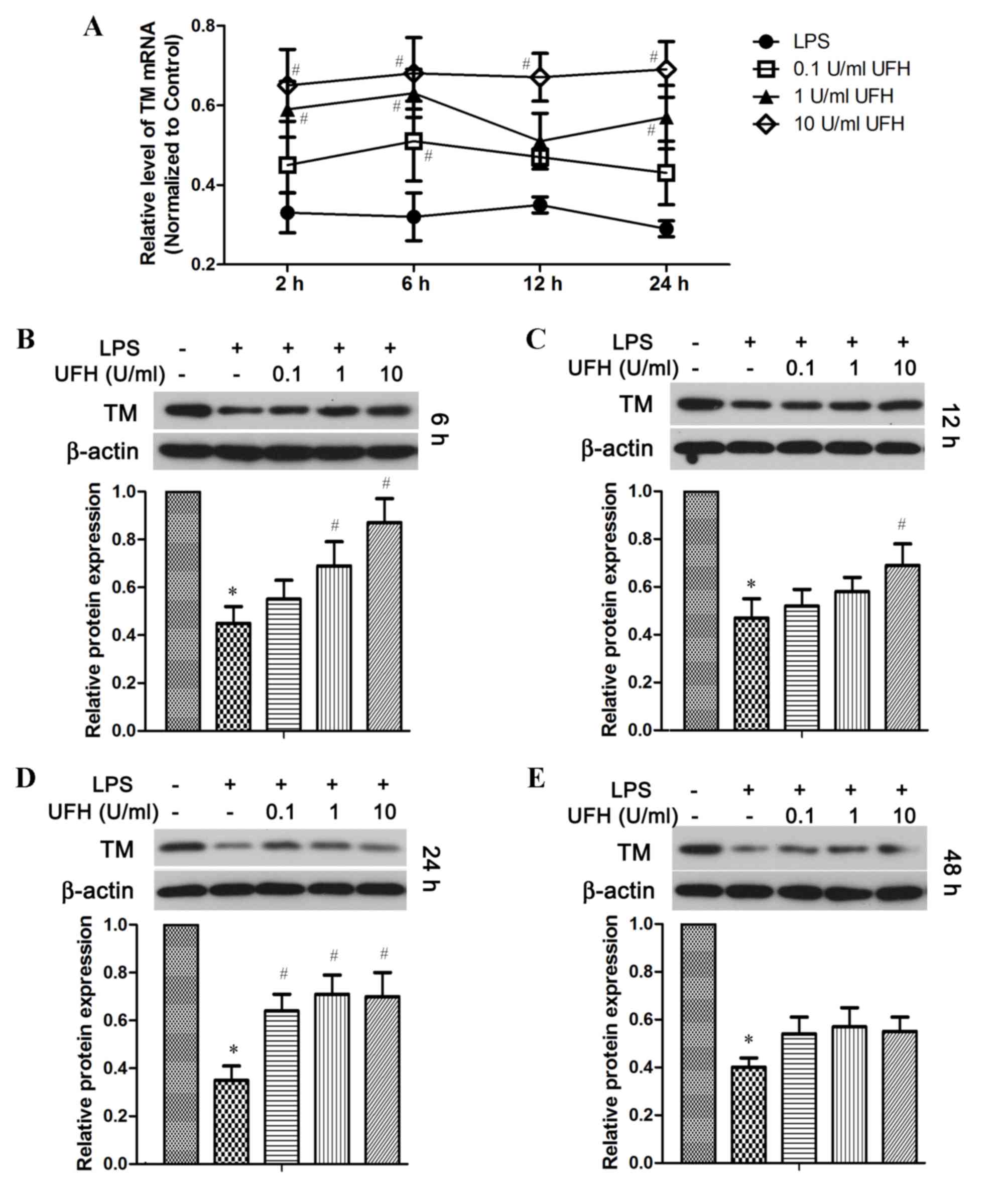
TM mRNA expression was also significantly reduced in
HUVECs following LPS stimulation, and UFH dose-dependently
increased TM mRNA expression in LPS-treated HUVECs (Fig. 4A). In addition, compared with
LPS-treated cells, UFH treatment led to dose-dependent elevation of
the TM protein at 6 h post LPS induction (Fig. 4B). UFH of all doses significantly
attenuated LPS-induced loss of TM protein at 24 h, whereas UFH did
not show significant rescue of TM protein after 48 h LPS treatment
(Fig. 4C-E).
UFH suppresses LPS-induced activation
of the p38 MAPK, Src and NF-κB signaling pathways
The possible mechanisms underlying UFH-mediated
rescue of EPCR and TM in LPS-stimulated HUVECs were explored by
examining the activation status of several associated signaling
molecules, including p38 MAPK, Src and NF-κB. As shown in Fig. 5A, LPS significantly stimulated the
phosphorylation of p38 MAPK in HUVECs compared with the control
group, while a low dose of UFH (0.1 U/ml) was able to significantly
reduce LPS-induced p38 MAPK phosphorylation. A low dose of UFH also
significantly lowered LPS-induced activation of Src in HUVECs
(Fig. 5B). In addition, UFH
dose-dependently inhibited phosphorylation of IκBα, an inhibitor of
NF-κB, and blocked the nuclear translocation of NF-κB following LPS
stimulation (Fig. 5C and D). Taken
together, these resulted demonstrated that UFH at a low dose was
sufficient to block LPS-induced activation of p38 MAPK and Src in
HUVECs, while it inhibited activation of the NF-κB pathway in a
dose-dependent manner.
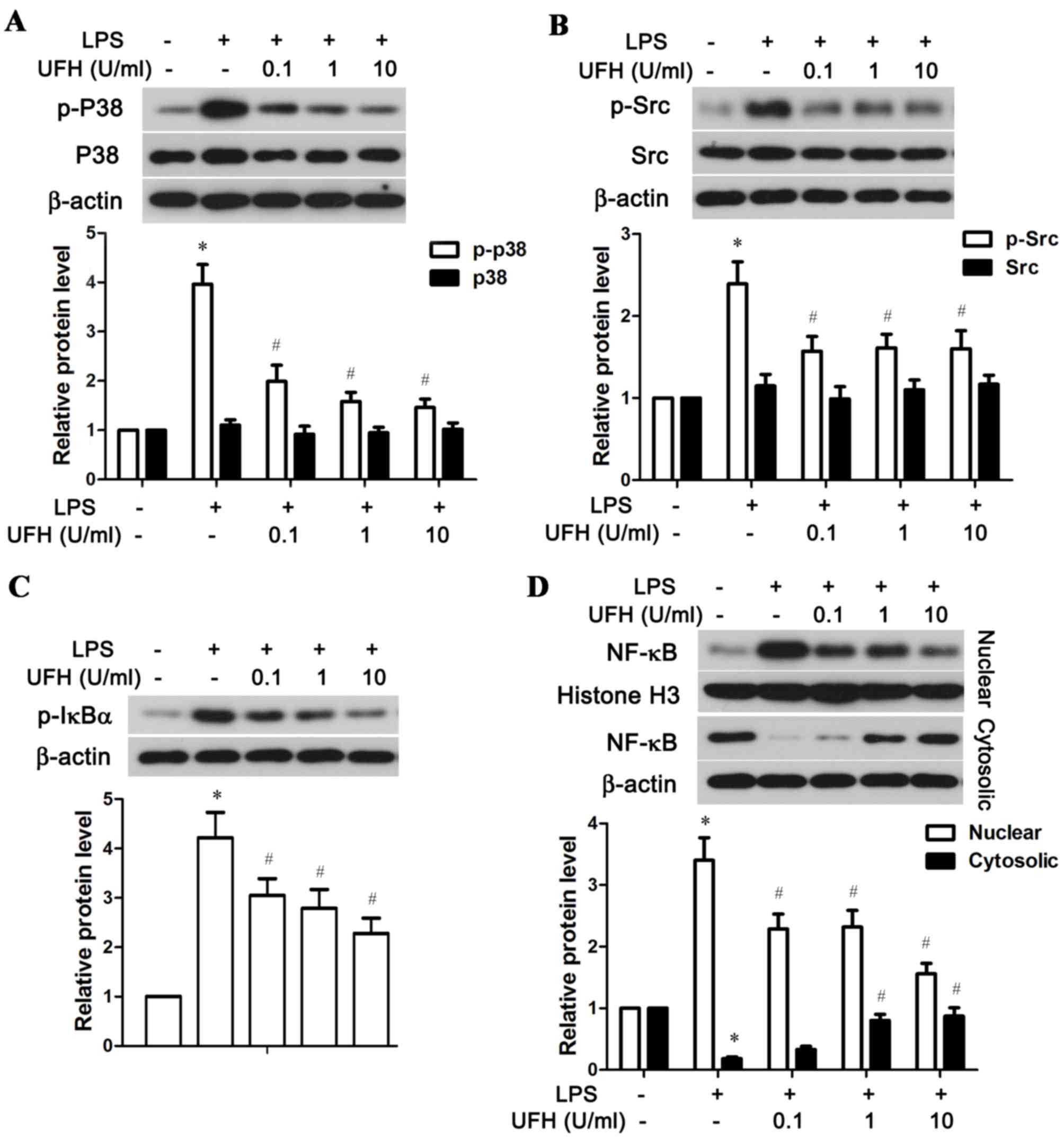 | Figure 5.UFH inhibits activation of the p38
MAPK, Src and NF-κB signaling pathways in LPS-stimulated human
umbilical vein endothelial cells. A total of 6 h after LPS
treatment, the phosphorylation status of (A) p38 MAPK, (B) Src, (C)
IκBα, and (D) nuclear and cytosolic NF-κB were detected by western
blot analysis. β-actin was used as the internal control for total
protein and cytosolic protein, and histone H3 was used as the
internal control for nuclear protein. *P<0.05 vs. the control
group; #P<0.05 vs. the LPS group. UFH, unfractionated
heparin; p38, p38 mitogen-activated protein kinase; NF-κB, nuclear
factor-κB; LPS, lipopolysaccharide; IκBα, NF-κB inhibitor α; p,
phosphorylated. |
Discussion
The impairment of the protein C anticoagulant
pathway is an important contributor to sepsis-associated
hypercoagulability. LPS and cytokine-induced alterations in various
components of the protein C pathway lead to an increased risk of
thrombosis and uncontrolled inflammation during sepsis (26). Consistent with previous studies, an
immediate decline in APC, and elevated levels of sEPCR and sTM were
observed in mice serum following LPS administration in the present
study. By contrast, preconditioning with UFH prevented APC
depletion and reduced the shedding of EPCR and TM from the
endothelial surface in LPS-treated mice. The protection of
endothelial TM and EPCR was possibly achieved by UFH-exerted direct
inhibition of neutrophil elastase (27) and metalloproteinases (28), the key sheddases for TM and EPCR. The
intact membrane-bound TM and EPCR guaranteed regular production of
APC, and thus maintained a physiological level of circulating APC.
The in vivo findings of the present study demonstrated that
UFH conveyed immediate protection upon protein C pathway components
against LPS-induced disruption, and thus disruption of the
anticoagulant system.
It has been reported that TM expression begins to
diminish in a dose- and time-dependent manner in the endothelium of
lung, liver and kidneys of rats within 2–4 h following LPS
administration (29). In addition,
decreased expression of TM and EPCR mRNA has been observed in
HUVECs following LPS stimulation (30). The present study demonstrated that
LPS-induced downregulation of TM and EPCR transcription in HUVECs
was reversed by UFH in a dose-dependent manner. UFH has been
demonstrated to induce TM expression in HUVECs in the presence or
absence of LPS (31), and it could
also prevent tumor necrosis factor α-induced downregulation of EPCR
mRNA in trophoblast cells (32).
These findings support the protective role of UFH on TM and EPCR
expression under inflammatory conditions. In addition, UHF can
suppress inflammation-mediated expression of procoagulant tissue
factors and increase the release of tissue factor pathway
inhibitors in endothelial cells, thereby restoring the
anticoagulant activity and modulating the hemostatic properties of
the endothelium (31).
Although a high dose of UFH maintained mRNA
expression of TM and EPCR in HUVECs at a comparative efficiency
during early and late phase of LPS stimulation in the present
study, it failed to preserve the protein levels of TM and EPCR at a
later stage of LPS treatment. Ishii et al (33) previously reported that an increased
release of sTM in LPS-stimulated HUVECs was correlated with the
degree of cell damage in a time-dependent manner. Thus, the
decrease of TM observed in the present study may be attributed to
TM shedding and release in damaged HUVECs due to LPS. In addition,
sEPCR can be generated in vitro through proteolytic cleavage
by metalloproteases, and this process is inducible by several
inflammatory mediators (10).
Therefore, UFH is able to protect TM and EPCR expression in
endothelial cells during early endotoxemia, while anti-inflammatory
agents and inhibitors of metalloproteases are also recommended for
optimal protection of these protein C pathway components.
A previous study demonstrated that inhibition of
endothelial NF-κB activation prevented LPS-induced downregulation
of EPCR and TM expression, reduced EPCR shedding and restored
plasma APC levels (34). In
addition, activation of the MAPK signaling pathway is implicated in
basal and induced EPCR shedding (35), and Src activation is involved in the
initiation of TM expression (36).
The present study demonstrated that UFH interfered with the
activation of the p38 MAPK, Src and NF-κB signaling pathways in
LPS-stimulated HUVECs, suggesting that it may exert a protective
effect on protein C system molecules by inhibiting these signaling
pathways.
In conclusion, the current study demonstrated that
UFH has protective effects on the protein C system in a mouse
sepsis model and in LPS-stimulated HUVECs. The mechanisms of action
of these protective effects may involve direct inhibition of
sheddases and the maintenance of endothelial EPCR and TM
expression. The preliminary clinical data suggest that heparin is
associated with reduced mortality in patients with sepsis (37,38). The
results of the present study support the potential therapeutic
value of UFH as a treatment for sepsis; however, its efficacy and
safety should be evaluated in future clinical trials.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Liaoning Province (grant no. 2013021073).
References
|
1
|
Cohen J: The immunopathogenesis of sepsis.
Nature. 420:885–891. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Annane D, Bellissant E and Cavaillon JM:
Septic shock. Lancet. 365:63–78. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levi M, van der Poll T and Büller HR:
Bidirectional relation between inflammation and coagulation.
Circulation. 109:2698–2704. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Opal SM and Esmon CT: Bench-to-bedside
review: Functional relationships between coagulation and the innate
immune response and their respective roles in the pathogenesis of
sepsis. Crit Care. 7:23–38. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schouten M, Wiersinga WJ, Levi M and van
der Poll T: Inflammation, endothelium, and coagulation in sepsis. J
Leukoc Biol. 83:536–545. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Esmon CT: The protein C pathway. Chest.
124 3 Suppl:26S–32S. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Van de Wouwer M, Collen D and Conway EM:
Thrombomodulin-protein C-EPCR system: Integrated to regulate
coagulation and inflammation. Arterioscler Thromb Vasc Biol.
24:1374–1383. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miyazaki Y, Inoue T, Kyi M, Sawada M,
Miyake S and Yoshizawa Y: Effects of a neutrophil elastase
inhibitor (ONO-5046) on acute pulmonary injury induced by tumor
necrosis factor alpha (TNFalpha) and activated neutrophils in
isolated perfused rabbit lungs. Am J Respir Crit Care Med.
157:89–94. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qu D, Wang Y, Esmon NL and Esmon CT:
Regulated endothelial protein C receptor shedding is mediated by
tumor necrosis factor-alpha converting enzyme/ADAM17. J Thromb
Haemost. 5:395–402. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu J, Qu D, Esmon NL and Esmon CT:
Metalloproteolytic release of endothelial cell protein C receptor.
J Biol Chem. 275:6038–6044. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bemard GR, Ely EW, Wright TJ, Fraiz J,
Stasek JE Jr, Russell JA, Mayers I, Rosenfeld BA, Morris PE, Yan SB
and Helterbrand JD: Safety and dose relationship of recombinant
human activated protein C for coagulopathy in severe sepsis. Crit
Care Med. 29:2051–2059. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bemard GR: Drotrecogin alfa (activated)
(recombinant human activated protein C) for the treatment of severe
sepsis. Crit Care Med. 31 1 Suppl:S85–S93. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fowler RA, Hill-Popper M, Stasinos J,
Petrou C, Sanders GD and Garber AM: Cost-effective of recombinant
human activated protein C and the influence of severity of illness
in the treatment of patients with severe sepsis. J Crit Care.
18:181–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Martí-Carvajal AJ, Solà I, Gluud C,
Lathyris D and Cardona AF: Human recombinant protein C for severe
sepsis and septic shock in adult and paediatric patients. Cochrane
Database Syst Rev. 12:CD0043882012.PubMed/NCBI
|
|
15
|
Cornet AD, Smit EG, Beishuizen A and
Groeneveld AB: The role of heparin and allied compounds in the
treatment of sepsis. Thromb Haemost. 98:579–586. 2007.PubMed/NCBI
|
|
16
|
Li Y, Sun JF, Cui X, Mani H, Danner RL, Li
X, Su JW, Fitz Y and Eichacker PQ: The effect of heparin
administration in animal models of sepsis: A prospective study in
Escherichia coli-challenged mice and a systematic review and
metaregression analysis of published studies. Crit Care Med.
39:1104–1112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao D, Ding R, Mao Y, Wang L, Zhang Z and
Ma X: Heparin rescues sepsis-associated acute lung injury and
lethality through the suppression of inflammatory responses.
Inflammation. 35:1825–1832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wildhagen KC, de Frutos García P,
Reutelingsperger CP, Schrijver R, Aresté C, Ortega-Gómez A, Deckers
NM, Hemker HC, Soehnlein O and Nicolaes GA: Nonanticoagulant
heparin prevents histone-mediated cytotoxicity in vitro and
improves survival in sepsis. Blood. 123:1098–1101. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ding R, Zhao D, Guo R, Zhang Z and Ma X:
Treatment with unfractionated heparin attenuates coagulation and
inflammation in endotoxemic mice. Thromb Res. 128:e160–e165. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li X, Zheng Z, Li X and Ma X:
Unfractionated heparin inhibits lipopolysaccharide-induced
inflammatory response through blocking P38 MAPK and NF-κB
activation on endothelial cell. Cytokine. 60:114–121. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li X, Zheng Z, Mao Y and Ma X:
Unfractionated heparin promotes LPS-induced endothelial barrier
dysfunction: A preliminary study on the roles of angiopoietin/Tie2
axis. Thromb Res. 129:e223–e228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, Li X, Zheng Z, Liu Y and Ma X:
Unfractionated heparin suppresses lipopolysaccharide-induced
monocyte chemoattractant protein-1 expression in human
microvascular endothelial cells by blocking Krüppel-like factor 5
and nuclear factor-κB pathway. Immunobiology. 219:778–785. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
National Research Council, . Guide for the
care and use of laboratory animals (8th edition). The National
Academies Press; Washington DC: 2011
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mu E, Ding R, An X, Li X, Chen S and Ma X:
Heparin attenuates lipopolysaccharide-induced acute lung injury by
inhibiting nitric oxide synthase and TGF-β/Smad signaling pathway.
Thromb Res. 129:479–485. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hayashi T and Suzuki K: Changes of
expression of the protein C pathway components in LPS-induced
endotoxemia-implication for sepsis. Cardiovasc Hematol Disord Drug
Targets. 15:2–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Redini F, Tixier JM, Petitou M, Choay J,
Robert L and Hornebeck W: Inhibition of leucocyte elastase by
heparin and its derivatives. Biochem J. 252:515–519. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kenagy RD, Nikkari ST, Welgus HG and
Clowes AW: Heparin inhibits the induction of three matrix
metalloproteinases (stromelysin, 92-kD gelatinase and collagenase)
in primate arterial smooth muscle cells. J Clin Invest.
93:1987–1993. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Terada Y, Eguchi Y, Nosaka S, Toba T,
Nakamura T and Shimizu Y: Capillary endothelial thrombomodulin
expression and fibrin deposition in rats with continuous and bolus
lipopolysaccharide administration. Lab Invest. 83:1165–1173. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao XH, Xu XX, Pan R, Li Y, Luo YB, Xia
YF, Murata K, Matsuda H and Dai Y: Saponin fraction from Astragalus
membranaceus roots protects mice against polymicrobial sepsis
induced by cecal ligation and puncture by inhibiting inflammation
and upregulating protein C pathway. J Nat Med. 63:421–429. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vignoli A, Marchetti M, Balducci D, Barbui
T and Falanga A: Differential effect of the low-molecular-weight
heparin, dalteparin, and unfractionated heparin on microvascular
endothelial cell hemostatic properties. Haematologica. 91:207–214.
2006.PubMed/NCBI
|
|
32
|
Faioni EM, Fontana G, Razzari C, Avagliano
L, Bulfamante G, Calvi E, Doi P and Marconi AM: Activation of
protein C in human trophoblasts in culture and downregulation of
trophoblast endothelial protein C receptor by TNF-α. Reprod Sci.
22:1042–1048. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ishii H, Uchiyama H and Kazama M: Soluble
thrombomodulin antigen in conditioned medium is increased by damage
of endothelial cells. Thromb Haemost. 65:618–623. 1991.PubMed/NCBI
|
|
34
|
Song D, Ye X, Xu H and Liu SF: Activation
of endothelial intrinsic NF-{kappa}B pathway impairs protein C
anticoagulation mechanism and promotes coagulation in endotoxemic
mice. Blood. 114:2521–2529. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Menschikowski M, Hagelgans A, Eisenhofer G
and Siegert G: Regulation of endothelial protein C receptor
shedding by cytokines is mediated through differential activation
of MAP kinase signaling pathways. Exp Cell Res. 315:2673–2682.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lo IC, Lin TM, Chou LH, Liu SL, Wu LW, Shi
GY, Wu HL and Jiang MJ: Ets-1 mediates platelet-derived growth
factor-BB-induced thrombomodulin expression in human vascular
smooth muscle cells. Cardiovasc Res. 81:771–779. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang C, Chi C, Guo L, Wang X, Guo L, Sun
J, Sun B, Liu S, Chang X and Li E: Heparin therapy reduces 28-day
mortality in adult severe sepsis patients: A systematic review and
meta-analysis. Crit Care. 18:5632014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zarychanski R, About-Setta AM, Kanji S,
Turgeon AF, Kumar A, Houston DS, Rimmer E, Houston BL, McIntyre L,
Fox-Robichaud AE, et al: The efficacy and safety of heparin in
patients with sepsis: A systemati review and metaanalysis. Crit
Care Med. 43:511–518. 2015. View Article : Google Scholar : PubMed/NCBI
|