Introduction
Lung cancer is a multistep process involving
alterations in the expression of oncogenes and tumor suppressor
genes via numerous factors, including alcohol, smoking, pathogenic
infections and genetic factors (1–3). Lung
cancer is the most frequently diagnosed cancer and its incidence
has been increasing in recent years (2,3). It
remains a leading cause of cancer-associated mortality worldwide
for men and women (4,5), but the pathogenesis of this disease
remains unknown (6).
PDZ-binding-kinase/T-LAK cell-originated protein
kinase (PBK/TOPK) is a 322-amino acid serine/threonine kinase
(7). In normal tissues, is difficult
to detect PBK/TOPK protein, other than in the germ cells of the
testis and numerous fetal tissues (8). The overexpression of PBK/TOPK has been
observed in activated T-LAK cells, which interacted with fruit
plate-large human homologues via the C-terminal PDZ binding motif
of TOPK (9). PBK/TOPK is widely
expressed in a variety of malignancies, including leukemia,
lymphoma and breast cancer, and malignant peripheral nerve sheath
tumors (10–12). PBK/TOPK overexpression contributes to
tumor proliferation and growth in Ewing sarcoma, colorectal cancer
and breast cancer (13,14). In lung cancer, patients exhibit poor
clinical outcome with high expression of PBK/TOPK (15). Recently, PBK/TOPK overexpression was
reported to be closely associated with tumor malignancy potential
and poor outcome of patients with gastric cancer (16). In addition, high expression of
PBK/TOPK may serve as a favorable prognostic marker for patients
with oral cancer (17). In molecular
studies, PBK/TOPK expression was associated with cell mitotic
regulation, inflammation and apoptosis (18).
MicroRNAs (miRNAs/miRs) are non-coding RNAs that
inhibit gene expression by inhibiting the translation of target
mRNAs or by degrading them. miRNAs have been demonstrated to
interact directly with the 3′-untranslated region (3′-UTR) of
target mRNAs (19). miRNAs regulate
numerous cell functions, including proliferation, differentiation,
metastasis and apoptosis (19).
Studies have indicated that altered miRNA expression levels are
associated with cancer (20,21). miRNAs may serve as oncogenes or tumor
suppressors, depending on the organs and tumors in which they are
expressed (22–24). miR-216b has been studied in a variety
of cancer types. For instance, miR-216b was reported to be closely
associated with the prognosis of cervical cancer patients and may
inhibit cervical cancer cell proliferation via regulating forkhead
box protein M1 (FOXM1) expression (25). miR-216b prevented the migration and
invasion of glioma cells by suppressing FOXM1 expression (26). Liu et al (27) reported that miR-216b was involved in
cisplatin resistance in ovarian cancer by the regulation of poly
adenosine 5′-diphosphate-ribose polymerase 1. Furthermore, miR-216b
may inhibit hepatocellular carcinoma cell proliferation, migration
and invasion by regulating insulin-like growth factor 2
mRNA-binding protein 2 (28).
However, the role and mechanism of miR-216b-3p in lung
adenocarcinoma requires further investigation.
The present study aimed to explore the effects of
miR-216b-3p on lung adenocarcinoma, and the association between
miR-216b-3p and PBK/TOPK. Herein, the molecular functions and
underlying mechanism of miR-216b-3p were investigated.
Materials and methods
Reagents
Human lung adenocarcinoma cell lines (A549, GLC-82
and H358 cells) and the human normal lung epithelial cell line
BEAS-2B were acquired from Scien Cell Research Laboratories, Inc.
(San Diego, CA, USA) and cultured in the laboratory. Fetal bovine
serum (FBS), penicillin/streptomycin, and trypsin-EDTA were
obtained from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Dulbecco's modified Eagle medium (DMEM) was obtained from
Corning, Inc. (Corning, NY, USA).
Cell culture
A549, GLC-82, H358 and BEAS-2B cells were cultured
in DMEM containing 10% FBS, 100 U/ml penicillin and 100 U/ml
streptomycin (pH 7.2) in a 5% CO2 atmosphere at
37°C.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated from A549, GLC-82, H358 and
BEAS-2B cells using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
first strand cDNA synthesis was performed with 1 µg RNA in the
reaction mixture of 10 µl with 50 pmol random hexamers and 50 units
of M-MLV RTase. Conditions were as following: 16°C for 30 min, 42°C
for 30 min, 85°C for 5 min, and hold at 4°C. Subsequently, qPCR was
performed to analyze the synthesized cDNA using the QuantiTect SYBR
Green PCR kit (Qiagen GmbH, Hilden, Germany). The master mix (20
µl) contained 2 µl 10× reverse transcription buffer, 1 µl
deoxynucleotides (100 mM; with thymidine triphosphate), 0.25 µl 2
µM forward primer, 0.25 µl 2 µM reverse primer, 5 µl 1 ng/µl cDNA
and 11.5 µl nuclease-free water. The amplification conditions were
as follows: 38 cycles of denaturation at 95°C for 10 sec, followed
by 60°C for 60 sec to allow annealing and extension. U6 and GAPDH
served as the internal controls for miR-216b-3p and PBK expression
respectively. qPCR was conducted with the following primers:
miR-216b-3p forward, 5′-CAGGCACACACTTACCCGTA-3′ and reverse,
5′-GCAGGGTCCGAGGTATTC-3′; U6 forward, 5′-CTCGCTTCGGCAGCACATATACT-3′
and reverse, 5′-ACGCTTCACGAATTTGCGTGTC-3′; GAPDH forward,
5′-GAAGGTGAAGGTCGGAGTC-3′ and reverse, 5′-GAAGATGGTGATGGGATTTC-3′;
and PBK forward, 5′-CCAAACATTGTTGGTTATCGTGC-3′ and reverse,
5′-GGCTGGCTTTATATCGTTCTTCT-3′. Relative gene expression was
calculated by using the 2−ΔΔCq method (29). The test was performed three times in
triplicate.
Western blot analysis
Harvested A549, GLC-82, H358 and BEAS-2B cells were
briefly washed with cold PBS. On ice, they were lysed in
radioimmunoprecipitation complete lysis buffer [50 mM Tris, pH 7.2;
1% sodium deoxycholate; 150 mM NaCl; 0.1% SDS; 10 mM NaF; 1%
Triton-X 100; 1 mM Na3VO4; protease inhibitor cocktail (1:800)].
Lysates were sonicated 3 times at 4°C, each time 10 sec (frequency,
20 kHz) and centrifuged at 13,000 × g for 10 min at 4°C. Serum
albumin was used as the standard and protein concentration was
determined with bicinchoninic acid as previously described
(30). Total protein (30 µg/lane)
was separated using 12% SDS-PAGE and transferred to polyvinylidene
difluoride membranes. Subsequently, membranes were incubated with
PBS containing 0.05% Tween-20 and 5% non-fat dry milk at room
temperature for 1.5 h to block non-specific binding and were
incubated with PBK/TOPK (cat no. 4942), p53 (Cat no. 2527), p21
(cat no. 2947), p-p38 (cat no. 4511) and β-actin (cat no. 4970)
primary antibodies (all 1:1,000; Cell Signaling Technology,
Danvers, MA, USA), then treated with anti-rabbit Immunoglobulin G
horseradish peroxidase-coupled secondary antibody (cat no. 7074;
1:1,000; Cell Signaling Technology) as described (31). Immunoreactive bands were observed
with enhanced chemiluminescence using the SignalFire™
Plus ECL Reagent (cat no. 12630; Cell Signaling Technology) and
imaged by ChemiDoc XRS+ System (Bio-Rad Laboratories). The band
density was quantified with Gel-Pro Analyzer densitometry software
(version 6.3; Media Cybernetics, Inc., Rockville, MD, USA).
Transient transfection
Cells were transiently transfected with 50 nM
miR-216b-3p mimic (Pre-miR™ miR-216b-3p; sense,
5′-AAAUCUCUGCAGGCAAAUGUGA-3′ and antisense,
5′-ACAUUUGCCUCCAGAGAUUUUU-3′; Thermo Fisher Scientific, Inc.), 50
nM negative control (negative control miRNA; sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
ACGUGACACGUUCGGAGAATT-3′; Thermo Fisher Scientific, Inc.), 2 µg
control plasmids, 2 µg PBK/TOPK plasmids (PBK CRISPR/Cas9 KO
Plasmid (h); cat no. sc-404069; Santa Cruz Biotechnology, Inc.,
Dallas, CA, USA) or 50 nM miR-216b-3p mimic + 2 µg PBK/TOPK
plasmids using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
Cells were seeded at a density of 5×104 cells per well
in a 6-well plate. At 48 h post-transfection, the cells were
subjected to subsequent analysis.
Luciferase reporter assay
Bioinformatics software (targetscan.org/vert_71) was used to predict the
targets of miR-216b-3p; PBK was identified as a potential target of
miR-216b-3p. To determine whether miR-216b-3p directly targets the
3′-UTR of PBK, the psiCHECK-2 reporter plasmid (Sangon Biotech Co.,
Ltd., Shanghai, China) vectors named PBK-3′UTR-WT and PBK-3′UTR-MUT
with wild type and mutated 3′UTR of PBK mRNA, respectively, were
constructed as previously described (32). GLC-82 cells (5×104
cells/well) were seeded in a 24-well plate and then co-transfected
with PBK-3′UTR-WT or PBK-3′UTR-MUT and miR-216b-3p or its negative
control (miR-NC) vector using Lipofectamine® 2000
transfection reagent according to the manufacturer's protocols. A
total of 48 h after transfection, the luciferase activity was
detected using the Dual-Luciferase Reporter Assay kit (Promega
Corporation, Madison, WI, USA) according to the manufacturer's
protocol and normalized to Renilla luciferase activity.
Flow cytometry for apoptosis
analysis
For apoptosis analysis, 1×105 cells/well
were seeded in 6-well plates and incubated in DMEM containing 10%
FBS at 37°C for 48 h. After 48 h, the harvested floating and
adherent cells were washed twice with cold PBS. Cells were stained
with Annexin V-fluorescein isothiocyanate and propidium iodide
(cat. no. 6592; Cell Signaling Technology) in 500 µl binding buffer
at room temperature for 15 min, and then analyzed with a BD
FACSCelesta™ flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA) using WinMDI (Version 2.5; Purdue University
Cytometry Laboratories, West Lafayette, IN, USA) within 1 h
(28).
Cell proliferation assay
Cell proliferation was examined by a MTT assay. A
total of 48 h after transfection, GLC-82 cells (4×103
cells/well) were seeded into 96-well culture plates and incubated
in 5% CO2 at 37°C. Then, 20 µl MTT (5 mg/ml) was added
to each well and the cells were incubated for another 4 h at 37°C.
Following removal of MTT, 100 µl dimethyl sulfoxide was added to
each well, and the plate was gently agitated for 10 min at room
temperature. The absorbance was measured with a microplate reader
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) at 490 nm. All
experiments were repeated ≥3 times (33).
Statistical analysis
Experiments were performed for at least three times.
Data are expressed as the mean ± standard deviation. SPSS 17.0
statistical software (SPSS, Inc., Chicago, IL, United States) was
performed for all statistical analyses. Statistical analysis was
performed using a Student's t-test or one-way analysis of variance
followed by Tukey's test where appropriate. P<0.05 was
considered to indicate a statistically significant difference.
Results
Low expression of miR-216b-3p in lung
adenocarcinoma cell lines
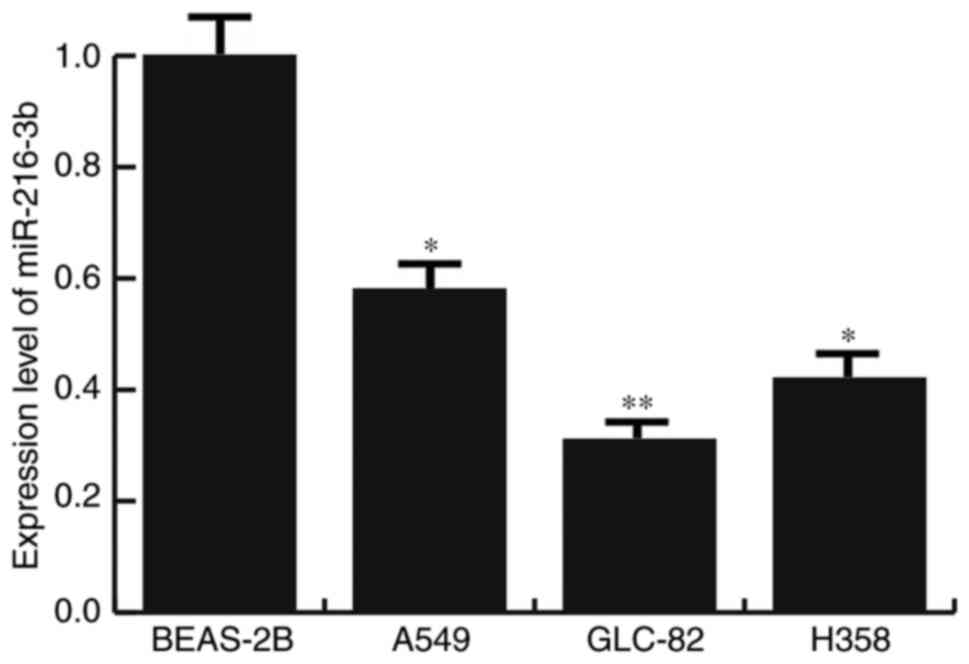
The expression levels of miR-216b-3p in human lung
adenocarcinoma cell lines (A549, GLC-82 and H358 cells) and human
normal lung epithelial cell line BEAS-2B were analyzed. The results
of the present study indicated that the expression levels of
miR-216b-3p in all lung adenocarcinoma cell lines (A549, GLC-82 and
H358 cells) were significantly lower compared with BEAS-2B cells.
The lowest expression level of miR-216b-3p was in GLC-82 cells
(Fig. 1). Therefore, GLC-82 cells
were selected for subsequent in vitro studies in the present
study.
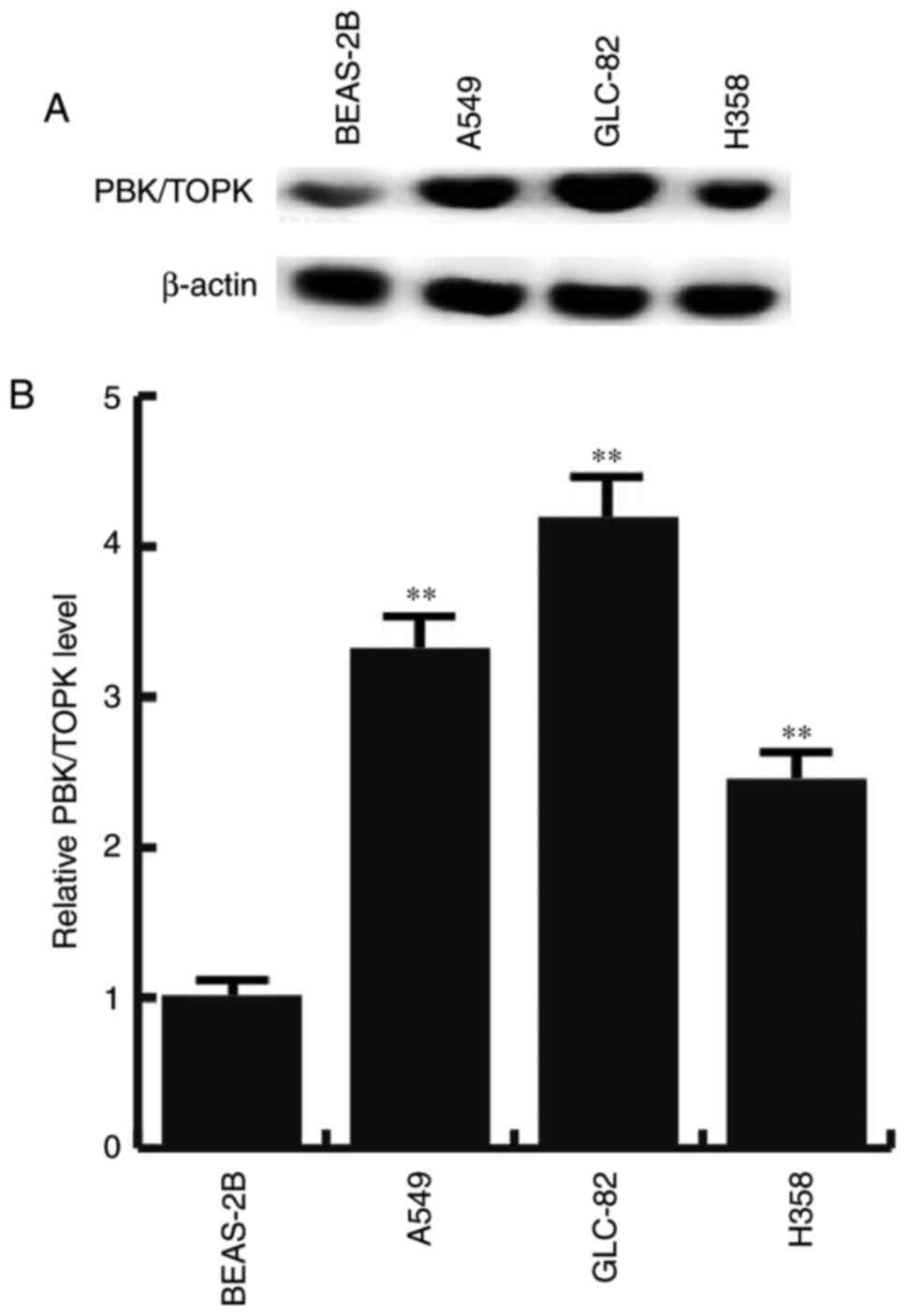
Overexpression of PBK/TOPK in lung
adenocarcinoma cell lines
Western blotting and RT-qPCR were performed to test
whether PBK/TOPK was overexpressed in lung adenocarcinoma cell
lines. PBK/TOPK was expressed at low levels within BEAS-2B cells.
However, the protein and mRNA expression levels of PBK/TOPK were
increased in all lung adenocarcinoma cell lines (A549, GLC-82 and
H358 cells), compared with BEAS-2B cells, with the largest increase
in GLC-82 (Fig. 2). These results
suggested that PBK/TOPK may be a target of activation in lung
adenocarcinoma.
PBK is a direct target gene of
miR-216b-3p
The aforementioned results demonstrated that
miR-216b-3p and PBK/TOPK expression may be dysregulated in lung
adenocarcinoma cell lines. Therefore, miR-216b-3p and PBK/TOPK may
have a direct association. TargetScan was employed to predict the
target gene of miR-216b-3p (Fig.
3A). PBK was identified as a direct target gene of miR-216b-3p
and a double luciferase reporter assay revealed the same result
(Fig. 3B).
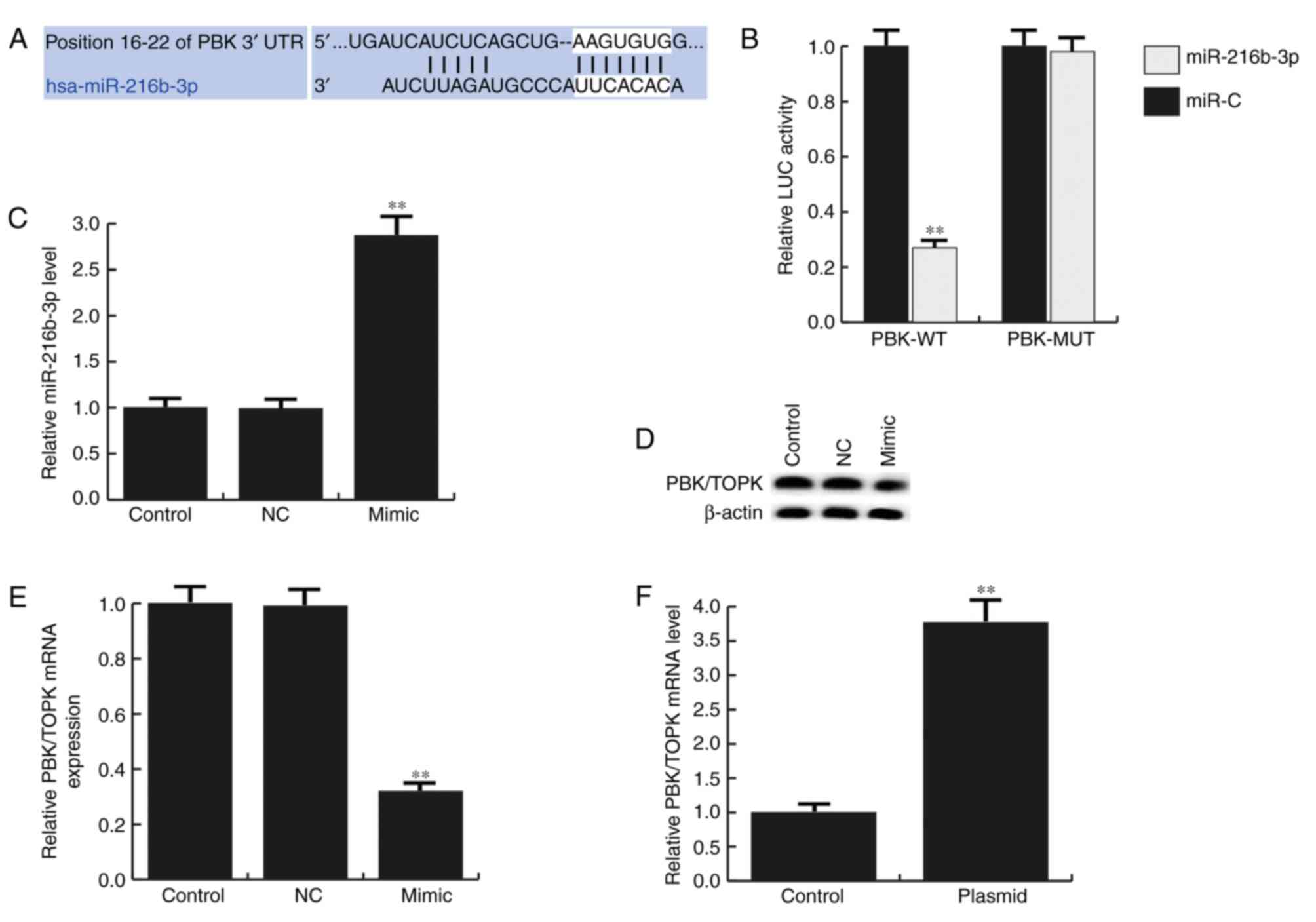 | Figure 3.PBK is a target gene of miR-216b-3p.
(A) TargetScan was applied to predict the interaction between
miR-216b-3p and 3′UTR of PBK. (B) Luciferase activity was detected
via a dual luciferase assay. (C) Relative miR-216b-3p expression
levels in GLC-82 cells. (D) Effects of miR-216b-3p on PBK/TOPK
protein expression in GLC-82 cells were determined by western
blotting. (E) Effects of miR-216b-3p on PBK/TOPK mRNA expression in
GLC-82 cells were determined by reverse transcription-quantitative
polymerase chain reaction. (F) Relative PBK/TOPK mRNA expression
levels in GLC-82 cells. Data are presented as the mean ± standard
deviation. **P<0.01 vs. control. Control, control group, cells
without any treatment; NC, negative control group, cells
transfected with the negative control vector of miR-216b-3p mimics;
mimic, cells transfected with miR-216b-3p mimics; plasmid, cells
transfected with PBK/TOPK plasmids; LUC, luciferase; miR, microRNA;
PBK/TOPK, PDZ-binding-kinase/T-LAK cell-originated protein kinase;
MUT, mutated 3′-untranslated region; WT, wild type 3′-untranslated
region. |
To investigate the function of miR-216b-3p in lung
adenocarcinoma, synthesized miR-216b-3p mimics, miR-NC, control
plasmids, PBK/TOPK plasmids or miR-216b-3p mimic + PBK/TOPK
plasmids were transfected into GLC-82 cells (Fig. 3C-F). At 48 h post-transfection, the
transfection efficiency was determined by RT-qPCR (Fig. 3C and F). The results indicated that
miR-216b-3p mimics significantly enhanced miR-216-3p expression in
GLC-82 cells and PBK/TOPK plasmids significantly increased the mRNA
level of PBK/TOPK. To further reveal whether miR-216b-3p may
regulate PBK/TOPK in GLC-82 cells, at 48 h after GLC-82 cell
transfection with miR-216b-3p mimics or miR-NC, the protein and
mRNA expression levels of PBK/TOPK were detected by western
blotting and RT-qPCR respectively. The results of the present study
indicated that miR-216b-3p mimics may inhibit PBK/TOPK expression
in GLC-82 cells (Fig. 3D and E).
Collectively, the data suggested that PBK may be a direct target
gene of miR-216b-3p and may be negatively regulated by
miR-216b-3p.
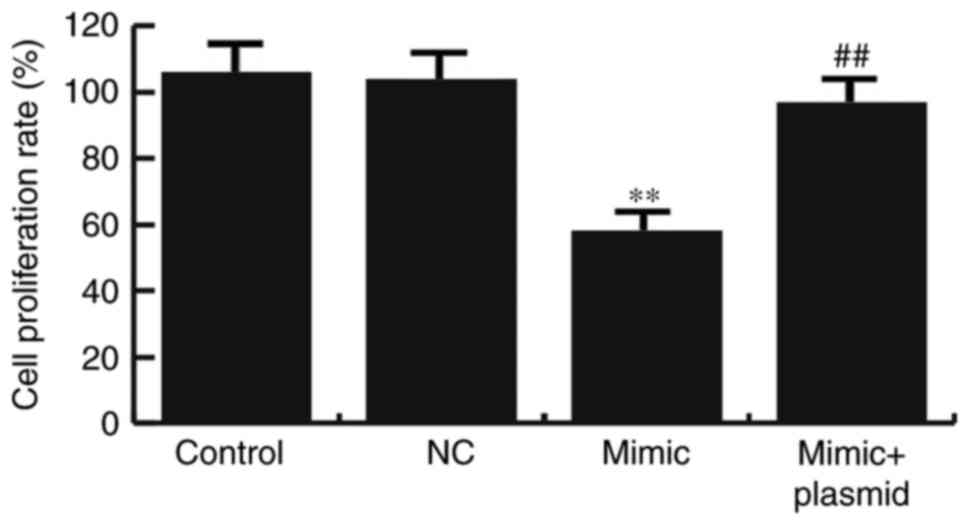
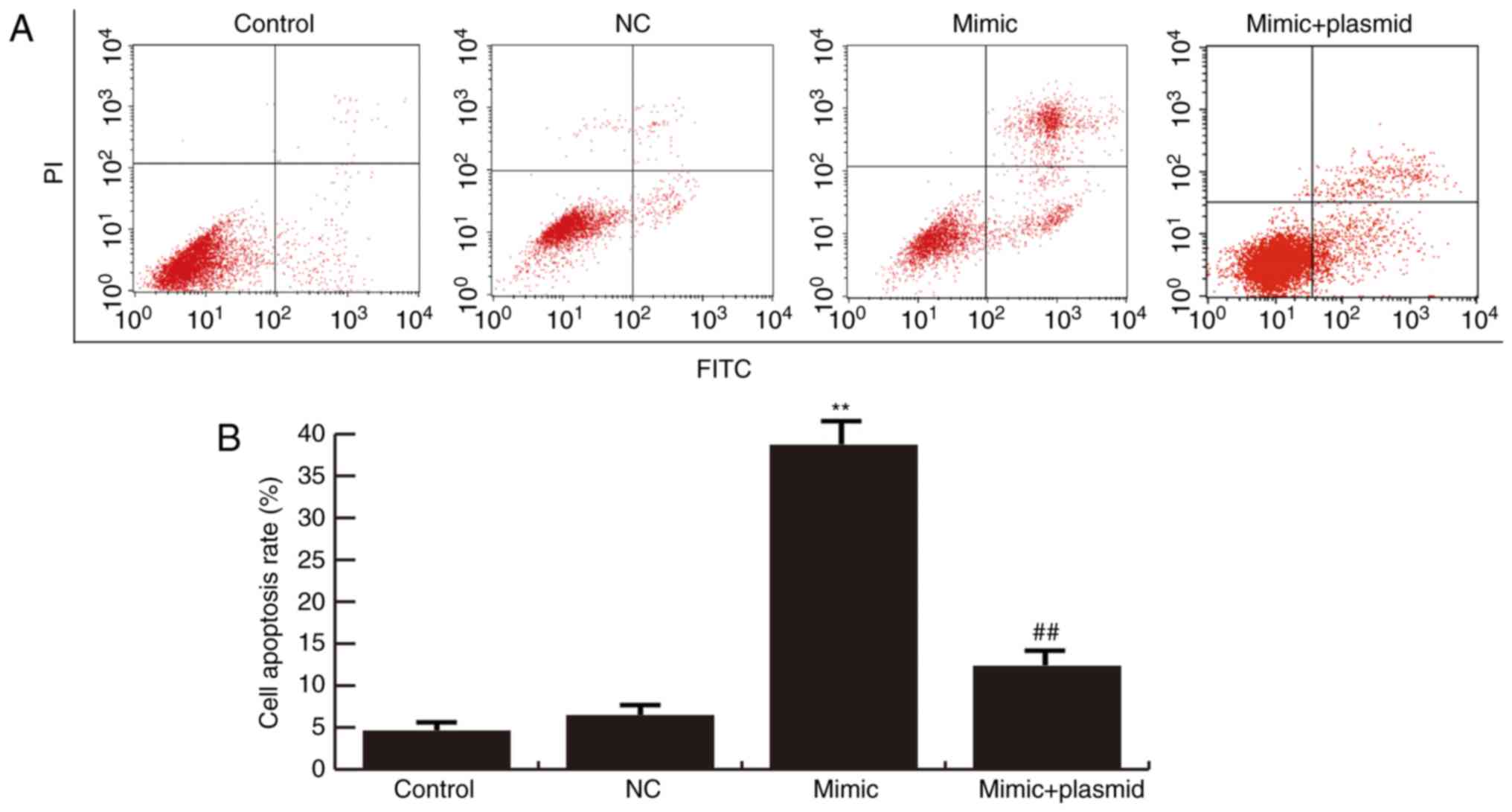
miR-216b-3p overexpression inhibits
GLC-82 cell proliferation and induces cell apoptosis via regulating
PBK/TOPK expression
Cells were transiently transfected with miR-216b-3p
mimics, miR-NC or miR-216b-3p mimics + PBK/TOPK plasmids. An MTT
assay was conducted to detect the proliferation of GLC-82 cells;
flow cytometry was used to analyze apoptosis. The results
demonstrated that miR-216b-3p overexpression significantly inhibits
GLC-82 cell proliferation (Fig. 4)
and induces cell apoptosis (Fig. 5)
compared with the control. These effects were eliminated by
PBK/TOPK overexpression (Figs. 4 and
5).
miR-216b-3p overexpression affects
p53, p21 and phosphorylated (p) -p38 expression
To further investigate the molecular mechanism of
miR-216b-3p, the expression of p53, p21 and p-p38 was detected by
western blot analysis. The results demonstrated that the
overexpression of miR-216b-3p increased the expression of p53 and
p21, and decreased the expression of p-p38. These effects also
appeared to be eliminated by PBK/TOPK overexpression (Fig. 6).
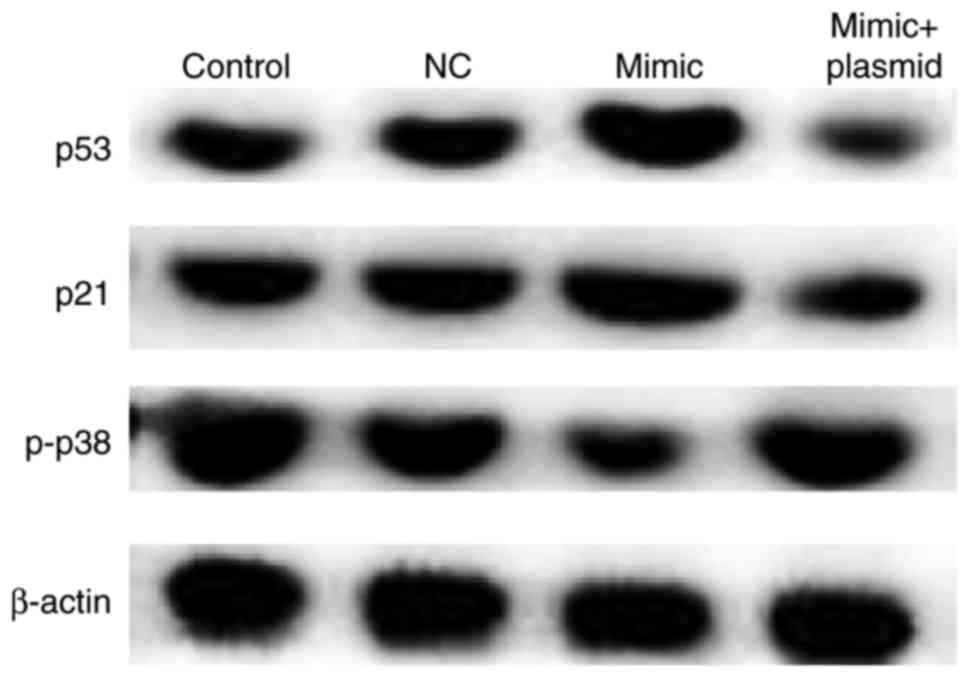 | Figure 6.Effects of miR-216b-3p on the
expression of p53, p21 and p-p38 in GLC-82 cells. After 48 h
post-transfection, western blotting was performed to determine the
effect of miR-216b-3p the expression of p53, p21 and p-p38 in
GLC-82 cells. Control, control group, cells without any treatment;
NC, negative control group, cells transfected with the negative
control vector of miR-216b-3p mimics; mimic, cells transfected with
miR-216b-3p mimics; mimic + plasmid, cells co-transfected with
miR-216b-3p mimics and PDZ-binding-kinase/T-LAK cell-originated
protein kinase plasmids. miR, microRNA; p-, phosphorylated. |
Discussion
According a previous report, miRNAs serve a
regulatory role in cell growth, proliferation, apoptosis,
differentiation, migration and metabolism (34). In the present study, the expression
of miR-216b-3p was detected in a variety of lung adenocarcinoma
cell lines and the results demonstrated that within all lung
adenocarcinoma cell lines (A549, GLC-82 and H358 cells),
miR-216b-3p expression levels was significantly lower compared with
BEAS-2B cells. This indicated that miR-216b-3p was associated with
lung adenocarcinoma. GLC-82 cells, which expressed the lowest level
of miR-216b-3b, were selected for further analysis.
PBK/TOPK gene expression is increased in various
types of cancer, including bladder cancer, brain tumors, breast
cancer, liver cancer, lung cancer and sarcoma (35,36). In
the present study, the expression levels of PBK/TOPK were observed
to be significantly increased within lung adenocarcinoma cells.
These results suggested that the expression of miR-216b-3p and
PBK/TOPK was dysregulated in lung cancer. Therefore, miR-216b-3p
may be involved in tumor development by regulating PBK/TOPK
expression. TargetScan was used to predict the target gene of
miR-216b-3p, which suggested that miR-216b-3p was targeted to PBK
and a double luciferase reporter gene system validated this result.
Furthermore, the overexpression of miR-216b-3p in GLC-82 cells
significantly inhibited cell proliferation and induced cell
apoptosis; when co-transfected with PBK/TOPK, the inhibitory effect
exerted by miR-216b-3p was reversed. These results indicated that
miR-216b-3p may be considered to be a tumor suppressor by
decreasing the expression of PBK/TOPK.
As a member of the mitogen-activated protein kinase
kinase family, PBK/TOPK protein is highly expressed in various
types of cancer. p53 is considered to be one of the most common
tumor suppressors; wild type p53 is involved in apoptosis,
cell-cycle arrest and DNA repair (37). The loss of p53 function caused by
mutations, however, is associated with the majority of human
cancers (38). Mutant p53 is able to
not only promote invasion, migration and proliferation, but also
enhance genomic instability and chemoresistance (39). It was reported that a p53 mutation is
closely associated with the occurrence and progression of lung
cancer (40). Additionally, Hu et
al (7) revealed that PBK/TOPK
may inhibit p53 function and promote the development and
progression of tumor cells. The expression levels of p53, p21 and
p-p38 were also determined in the present study, and the data
demonstrated that the overexpression of miR-216b-3p may inhibit
PBK/TOPK. Thus, increases in p53 and p21, and decreases in p-p38
expression, may exert an antitumor effect.
In summary, the present study provided novel
evidence that decreased miR-216b-3p expression may contribute to
lung adenocarcinoma development by increasing the expression of
PBK/TOPK. Furthermore, PBK/TOPK expression may be beneficial to the
promotion of cell proliferation and viability. Therefore,
miR-216b-3p and PBK/TOPK may be considered as potential clinical
indicators and therapeutic targets for the treatment of lung
adenocarcinoma.
Acknowledgements
The authors would like to thank Dr Shuo Wu, Deputy
Chief Physician, Department of Respiratory Medicine, XiJing
Hospital, for assistance with the experiments.
References
|
1
|
de Groot P and Munden RF: Lung cancer
epidemiology, risk factors, and prevention. Radiol Clin North Am.
50:863–876. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Krawczyk P, Nicoś M, Ramlau R, Powrózek T,
Wojas-Krawczyk K, Sura S, Jarosz B, Szumiło J, Warda E,
Mazurkiewicz T, et al: The incidence of EGFR-activating mutations
in bone metastases of lung adenocarcinoma. Pathol Oncol Res.
20:107–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang X, de Groh M, Liu S, Liang H and
Morrison H: Rising incidence of adenocarcinoma of the lung in
Canada. Lung Cancer. 78:16–22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Travis WD, Brambilla E, Noguchi M,
Nicholson AG, Geisinger K, Yatabe Y, Powell CA, Beer D, Riely G,
Garg K, et al: International Association for the Study of Lung
Cancer/American Thoracic Society/European Respiratory Society:
International multidisciplinary classification of lung
adenocarcinoma: Executive summary. Proc Am Thorac Soc. 8:381–385.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lei B, Qi W, Zhao Y, Li Y, Liu S, Xu X,
Zhi C, Wan L and Shen H: PBK/TOPK expression correlates with mutant
p53 and affects patients' prognosis and cell proliferation and
viability in lung adenocarcinoma. Hum Pathol. 46:217–224. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hu F, Gartenhaus RB, Eichberg D, Liu Z,
Fang HB and Rapoport AP: PBK/TOPK interacts with the DBD domain of
tumor suppressor p53 and modulates expression of transcriptional
targets including p21. Oncogene. 29:5464–5474. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park JH, Lin ML, Nishidate T, Nakamura Y
and Katagiri T: PDZ-binding kinase/T-LAK cell-originated protein
kinase, a putative cancer/testis antigen with an oncogenic activity
in breast cancer. Cancer Res. 66:9186–9195. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gaudet S, Branton D and Lue RA:
Characterization of PDZ-binding kinase, a mitotic kinase. Proc Natl
Acad Sci USA. 97:5167–5172. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Simons-Evelyn M, Bailey-Dell K, Toretsky
JA, Ross DD, Fenton R, Kalvakolanu D and Rapoport AP: PBK/TOPK is a
novel mitotic kinase which is upregulated in Burkitt's lymphoma and
other highly proliferative malignant cells. Blood Cells Mol Dis.
27:825–829. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ayllón V and O'connor R: PBK/TOPK promotes
tumour cell proliferation through p38 MAPK activity and regulation
of the DNA damage response. Oncogene. 26:3451–3461. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stricker TP, Henriksen KJ, Tonsgard JH,
Montag AG, Krausz TN and Pytel P: Expression profiling of 519
kinase genes in matched malignant peripheral nerve sheath
tumor/plexiform neurofibroma samples is discriminatory and
identifies mitotic regulators BUB1B, PBK and NEK2 as overexpressed
with transformation. Mod Pathol. 26:930–943. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim DJ, Li Y, Reddy K, Lee MH, Kim MO, Cho
YY, Lee SY, Kim JE, Bode AM and Dong Z: Novel TOPK inhibitor
HI-TOPK-032 effectively suppresses colon cancer growth. Cancer Res.
72:3060–3068. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Herrero-Martín D, Osuna D, Ordóñez JL,
Sevillano V, Martins AS, Mackintosh C, Campos M, Madoz-Gúrpide J,
Otero-Motta AP, Caballero G, et al: Stable interference of EWS-FLI1
in an Ewing sarcoma cell line impairs IGF-1/IGF-1R signaling and
reveals TOPK as a new target. Br J Cancer. 101:80–90. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lei B, Liu S, Qi W, Zhao Y, Li Y, Lin N,
Xu X, Zhi C, Mei J, Yan Z, et al: PBK/TOPK expression in
non-small-cell lung cancer: Its correlation and prognostic
significance with Ki67 and p53 expression. Histopathology.
63:696–703. 2013.PubMed/NCBI
|
|
16
|
Ohashi T, Komatsu S, Ichikawa D, Miyamae
M, Okajima W, Imamura T, Kiuchi J, Kosuga T, Konishi H, Shiozaki A,
et al: Overexpression of PBK/TOPK relates to tumour malignant
potential and poor outcome of gastric carcinoma. Br J Cancer.
116:218–226. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang CF, Chen SL, Sung WW, Hsieh MJ, Hsu
HT, Chen LH, Chen MK, Ko JL, Chen CJ and Chou MC: PBK/TOPK
expression predicts prognosis in oral cancer. Int J Mol Sci.
17:2016. View Article : Google Scholar
|
|
18
|
Hu F, Gartenhaus RB, Zhao XF, Fang HB,
Minkove S, Poss DE and Rapoport AP: c-Myc and E2F1 drive PBK/TOPK
expression in high-grade malignant lymphomas. Leuk Res. 37:447–454.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Inui M, Martello G and Piccolo S: MicroRNA
control of signal transduction. Nat Rev Mol Cell Biol. 11:252–263.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Png KJ, Halberg N, Yoshida M and Tavazoie
SF: A microRNA regulon that mediates endothelial recruitment and
metastasis by cancer cells. Nature. 481:190–194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Croce CM: Causes and consequences of
microRNA dysregulation in cancer. Nat Rev Genet. 10:704–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ameres SL and Zamore PD: Diversifying
microRNA sequence and function. Nat Rev Mol Cell Biol. 14:475–488.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
He S, Liao B, Deng Y, Su C, Tuo J, Liu J,
Yao S and Xu L: miR-216b inhibits cell proliferation by targeting
FOXM1 in cervical cancer cells and is associated with better
prognosis. BMC Cancer. 17:6732017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang T, Ma G, Zhang Y, Huo H and Zhao Y:
miR-216b inhibits glioma cell migration and invasion through
suppression of FoxM1. Oncol Rep. 38:1751–1759. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Y, Niu Z, Lin X and Tian Y: miR-216b
increases cisplatin sensitivity in ovarian cancer cells by
targeting PARP1. Cancer Gene Ther. 24:208–214. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu FY, Zhou SJ, Deng YL, Zhang ZY, Zhang
EL, Wu ZB, Huang ZY and Chen XP: miR-216b is involved in
pathogenesis and progression of hepatocellular carcinoma through
HBx-miR-216b-IGF2BP2 signaling pathway. Cell Death Dis.
6:e16702015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meng SF, Mao WP, Wang F, Liu XQ and Shao
LL: The relationship between Cd-induced autophagy and lysosomal
activation in WRL-68 cells. J Appl Toxicol. 35:1398–1405. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feng YL, Yin YX, Ding J, Yuan H, Yang L,
Xu JJ and Hu LQ: Alpha-1-antitrypsin suppresses oxidative stress in
preeclampsia by inhibiting the p38MAPK signaling pathway: An in
vivo and in vitro study. PLoS One. 12:e01737112017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zou J, Kuang W, Hu J and Rao H: miR-216b
promotes cell growth and enhances chemosensitivity of colorectal
cancer by suppressing PDZ-binding kinase. Biochem Biophys Res
Commun. 488:247–252. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao S, Li X, Ding X, Qi W and Yang Q:
Cepharanthine Induces autophagy, apoptosis and cell cycle arrest in
breast cancer cells. Cell Physiol Biochem. 41:1633–1648. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rottiers V, Najafi-Shoushtari SH, Kristo
F, Gurumurthy S, Zhong L, Li Y, Cohen DE, Gerszten RE, Bardeesy N,
Mostoslavsky R and Näär AM: MicroRNAs in metabolism and metabolic
diseases. Cold Spring Harb Symp Quant Biol. 76:225–233. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Singh PK, Srivastava AK, Dalela D, Rath
SK, Goel MM and Bhatt ML: Expression of PDZ-binding kinase/T-LAK
celloriginated protein kinase (PBK/TOPK) in human urinary bladder
transitional cell carcinoma. Immunobiology. 219:469–474. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Joel M, Mughal AA, Grieg Z, Murrell W,
Palmero S, Mikkelsen B, Fjerdingstad HB, Sandberg CJ, Behnan J,
Glover JC, et al: Targeting PBK/TOPK decreases growth and survival
of glioma initiating cells in vitro and attenuates tumor growth in
vivo. Mol Cancer. 14:1212015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu X, Wilcken R, Joerger AC, Chuckowree
IS, Amin J, Spencer J and Fersht AR: Small molecule induced
reactivation of mutant p53 in cancer cells. Nucleic Acids Res.
41:6034–6044. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Muller PA and Vousden KH: p53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jackson JG and Lozano G: The mutant p53
mouse as a pre-clinical model. Oncogene. 32:4325–4330. 2013.
View Article : Google Scholar : PubMed/NCBI
|