Introduction
Pulmonary arterial hypertension (PAH) is a disease
with poor prognosis and high mortality rate, and is defined as a
mean pulmonary arterial pressure (mPAP) of ≥25 mmHg in the resting
state and ≥30 mmHg when moving. Its main feature is progressive
pulmonary occlusion resulting in gradually increased pulmonary
vascular resistance and pulmonary arterial pressure, accompanied by
irreversible pulmonary vascular remodeling (PVR) and the occurrence
of right-sided heart failure (1).
Therefore, PAH is harmful to patients. The survival time of
idiopathic PAH following diagnosis is only 2.5–3.4 years (2), and the condition currently has no
effective treatment.
The pathogenesis of PAH is unclear; however, it
appears to involve PVR on a genetic and immunological basis
(3,4). Cells, circulatory mediators and
molecular genetics have an involvement in the origination and
development of PAH; currently, activin receptor-like kinase 1
(ALK-1) is considered to be closely associated with idiopathic PAH,
and its role in the pathogenesis of PAH has attracted growing
attention (5–7). The ALK-1 gene is expressed in
endothelial, smooth muscle, hepatic stellate and cartilage cells,
fibroblasts, monocytes and macrophages, and is critical in the
regulation of developmental and pathological angiogenesis
angiogenesis (8,9). The overexpression of ALK-1 leads to
signaling abnormalities, thus causing changes in the growth state
of endothelial and smooth muscle cells, followed by PVR and PAH
(9–11).
The glutathione precursor N-acetylcysteine (NAC) may
be used to treat pulmonary fibrosis, inhibit the proliferation,
adhesion and migration of endothelial cells, reduce the
transforming growth factor-β (TGF-β)-induced production of
collagens, regulate gene expression and signal transduction,
improve endothelial functions, attenuate inflammation and
oxidation, and regulate the immune system, and thus exhibits great
significance in the reversal of PVR (12,13). A
previous study has revealed that NAC is able to ameliorate PAH and
right ventricular functions through immunoregulatory mechanisms
(14).
The present study used a monocrotaline (MCT)-induced
rat PAH model to observe the protein expression of ALK-1 and
mothers against decapentaplegic homolog 1 (Smad1) in the ALK-1
signal transduction pathway, with the aim of clarifying their roles
in PAH and PVR. The model was also used to investigate the
pathological changes of the pulmonary artery walls in rats
following NAC intervention, and the changes of mean right
ventricular pressure (mRVP), mPAP, weight of the right ventricle
(RV), weight of the left ventricle and septum (LV+S), and the right
ventricular hypertrophy index (RVHI). In addition, the association
of NAC treatment with changes of the ALK-1 signaling pathway was
investigated. The results may provide new therapeutic rationale for
the treatment of PAH, and provide experimental evidence for the
development of effective treatment protocols.
Materials and methods
Animals and model preparation
In total, 32 male Wistar rats (age, 8 weeks; weight,
180–200 g; License No. scxk-Lu-20130001; Shandong Lukang
Pharmaceutical Group Ltd., Jining, China) were housed in the
Tianjin Experimental Animal Center. The rats were given free access
to food and water at a constant temperature of 22±2°C and humidity
of 55±5%, with natural light for 1 week of adaptive feeding prior
to the experiment. The rats were randomly divided into four groups
(each n=8): Control (group C), model (group M), low-dose NAC (group
N1) and high-dose NAC (group N2). In the M, N1 and N2 groups, each
rat was intraperitoneally injected with 60 mg/kg MCT
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for modeling, and
the rats in group C were intraperitoneally injected with an equal
amount of normal saline (NS). From the modeling day, the rats were
orally administered NS (groups C and M), 100 mg/kg/day NAC (group
N1) or 500 mg/kg/day NAC (group N2) under the same feeding
conditions for 6 weeks. A right-heart catheter and physiological
monitoring system (PowerLab; ADInstruments, Sydney, Australia) were
used to detect the right ventricular systolic pressure and mPAP of
each group. Following that, the animals were sacrificed, the chest
was opened and the heart and lungs were excised to observe and
compare the RV and LV+S weights and to determine the RVHI. The
right lower lung tissue was then sampled, and fixed in 4%
paraformaldehyde for hematoxylin and eosin (H&E) staining. One
~1-cm section of pulmonary artery was sampled 3 mm proximal to the
lung hilum, washed with PBS and then stored at −80°C for detection
of the protein expression of ALK-1 and Smad1 using western
blotting. The present study was performed in strict accordance with
the recommendations in the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health (Bethesda, MD, USA).
The animal use protocol was reviewed and approved by the
Institutional Animal Care and Use Committee of Qingdao University
(Qingdao, China). Ethical approval was obtained from the Ethics
Committee of Qingdao University Hospital.
Detection of artery pressure and
RVHI
Following anesthetization with an intraperitoneal
injection of chloral hydrate (400 mg/kg), the hemodynamics of each
rat were detected as previously described (15,16), and
mPAP and mRVP were determined using right-heart catheterization and
the PowerLab physiological monitoring system. The animals were then
sacrificed, and the whole heart was removed. The RV and LV+S along
the interventricular septum were then isolated and weighed, and the
RVHI was calculated using the following formula:
RVHI=RV/(LV+S).
H&E staining
Following sacrifice, the rat chest was surgically
opened for excision of the heart and lungs. The right lung was
fixed for 30 min via the intratracheal perfusion of 4%
paraformaldehyde in order to flatten and smooth the lung surface.
The right hilum was horizontally sliced to sample the right lower
lung tissue, which was then fixed in 4% paraformaldehyde at 20°C
for 36–48 h, paraffin-embedded, sliced into 3-µm-thick sections and
stained with H&E at 20°C for 20 min. In each slice, 10 small
arteries (50–100 µm in diameter) were randomly selected for
measurement of their wall thickness (WT), external diameter (ED),
mean total area (TA) and internal area (IA) using Image-Pro Plus
6.0 (Media Cybernetics, Inc., Rockville, MD, USA). WT% and wall
area (WA)% were then calculated in order to evaluate PVR as
follows: WT%=(2 × WT/ED) × 100; WA%=(TA-IA)/TA × 100, and the mean
values were determined. In addition, in each slice, 10 small
pulmonary arteries (50–100 µm in diameter) were selected for the
observation of peri-pulmonary artery inflammatory infiltration
under a light microscope at a magnification of ×400. The conditions
were then scored according to the degree of infiltration of
peri-pulmonary vascular inflammatory cells as follows: i) No
infiltration, 0 points; ii) mild infiltration, 1 point; iii)
moderate infiltration, 2 points; iv) severe infiltration, 3 points
and v) very severe infiltration, 4 points.
Western blot analysis
Following sacrifice, a ~1-cm section of pulmonary
artery was sampled 3 mm proximal to the lung hilum. The surrounding
connective and fat tissues were removed, and the remaining artery
was washed with PBS and stored at −80°C. Radioimmunoprecipitation
assay lysis buffer (cat. no. CW2333S; Beijing ComWin Biotech Co.,
Ltd., Beijing, China) was added to the pulmonary artery incubated
for 20 min on the ice. The protein concentration was then detected
using the bicinchoninic acid assay method. A sample containing 20
µg protein/lane was gently mixed with 100 ml/l SDS-PAGE loading
buffer for 10 min denaturation at 95°C, and then subjected to
electrophoresis in a 10% SDS-PAGE. The isolated protein bands were
transferred onto a polyvinylidene difluoride membrane (EMD
Millipore, Billerica, MA, USA), which was placed into a bovine
serum albumin blocking buffer (cat. no. CW0054; Beijing ComWin
Biotech Co., Ltd.) at room temperature, and shaken slowly for 1 h.
Rabbit anti-mouse ALK-1 (cat. no. sc-19547), rabbit anti-mouse
Smad1 polyclonal (cat. no. sc-7965) and β-actin (cat. no. sc-47778;
1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
antibodies were then applied to uniformly cover the membrane
surface, and incubated overnight at 4°C. Following repeated washing
of the membrane, the horseradish peroxidase-labeled goat
anti-rabbit secondary antibody (1:3,000; cat. no. 31460; Pierce;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) was added in for
1 h incubation at room temperature, followed by washing with TBST,
and cECL Western Blot kit (cat. no. CW0048; Beijing ComWin Biotech
Co., Ltd.). Labworks 4.0 software (UVP, LLC, Phoenix, AZ, USA) was
used to scan the film and determine the grayscale values, with
β-actin (43 kDa) serving as the internal control, and to calculate
the expression levels of the proteins.
Statistical analysis
Measurement data are expressed as the mean ±
standard deviation. Differences among the groups were analyzed
using analysis of variance followed by Tukey's test. SPSS 17.0
software (SPSS, Inc., Chicago, IL, USA) was used to conduct the
data analysis.
Results
General conditions and survival
The rats in group M exhibited a slow increase in
body mass, slow actions, dull fur and reduced food intake compared
with those in group C. Furthermore, they demonstrated shortness of
breath and a significantly reduced body weight at the end of the
6-week experimental period (P<0.05). The rats in groups N1 and
N2 exhibited improved activity, fur condition, food intake and
breathing compared with those in group M; however, their body
weights exhibited no significant difference compared with those in
group M (P>0.05). The survival rates in groups C, M, N1 and N2
were 100, 75, 88 and 100%, respectively, and the survival rates for
groups N1 and N2 were significantly higher compared with those in
group M (P<0.05; Table I).
 | Table I.Body weights and survival rates of
rats in each group. |
Table I.
Body weights and survival rates of
rats in each group.
| Groups | n | Survival rate
(%) | Week 0 (g) | Week 6 (g) |
|---|
| C | 8 | 100 | 183.92±10.32 | 293.57±16.48 |
| M | 6 |
75a | 183.85±11.37 |
264.34±12.78a |
| N1 | 7 |
88a,b | 186.59±13.78 |
267.81±14.73a |
| N2 | 8 | 100c | 185.82±9.56 |
271.75±12.94a |
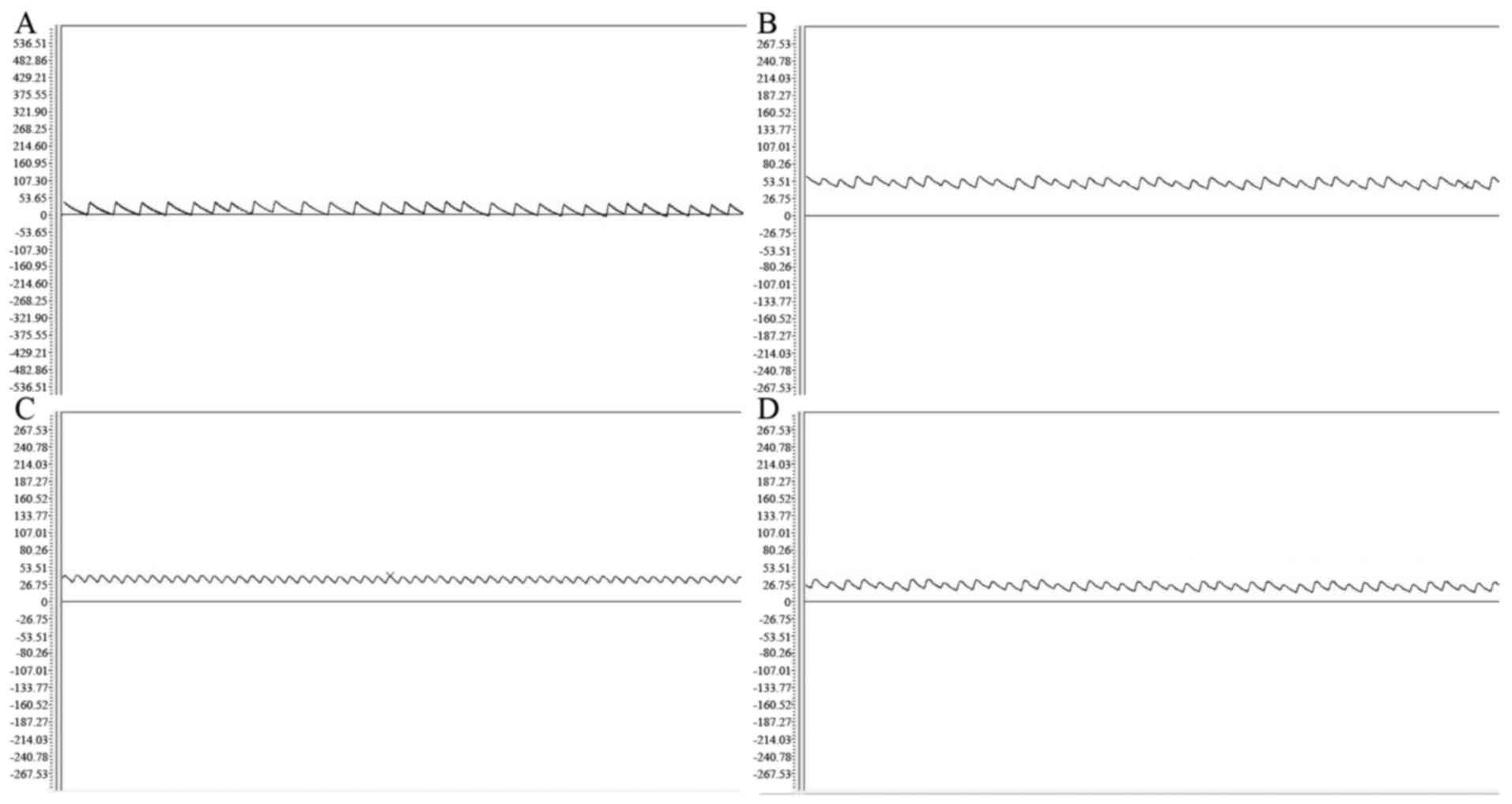
Hemodynamics and right ventricular
remodeling
At the end of the 6 weeks, mRVP, mPAP and RVHI in
group M were significantly higher compared with those in group C
(P<0.01). Furthermore, mRVP, mPAP and RVHI in groups N1 and N2
were significantly reduced compared with those in group M
(P<0.05) but were significantly higher compared with those in
group C (P<0.05). The RVHI in group N2 was significantly reduced
compared with that in group N1 (P<0.05) but mRVP and mPAP
exhibited no significant difference between groups N1 and N2
(P>0.05), as depicted in Table
II and Fig. 1.
| Table II.Hemodynamics and right ventricle
indices at 6 weeks after the monocrotaline injection. |
Table II.
Hemodynamics and right ventricle
indices at 6 weeks after the monocrotaline injection.
| Group | n | mRVP (mmHg) | mPAP (mmHg) | RVHI |
|---|
| C | 8 | 20.78±1.85 | 21.37±1.73 | 0.22±0.01 |
| M | 6 |
45.29±2.79a |
49.80±2.96a |
0.88±0.03a |
| N1 | 7 |
34.25±2.25b,c |
31.93±1.87b,c |
0.52±0.02a,c |
| N2 | 8 |
33.18±2.42b,c |
27.03±1.80b,c |
0.30±0.01b,d,e |
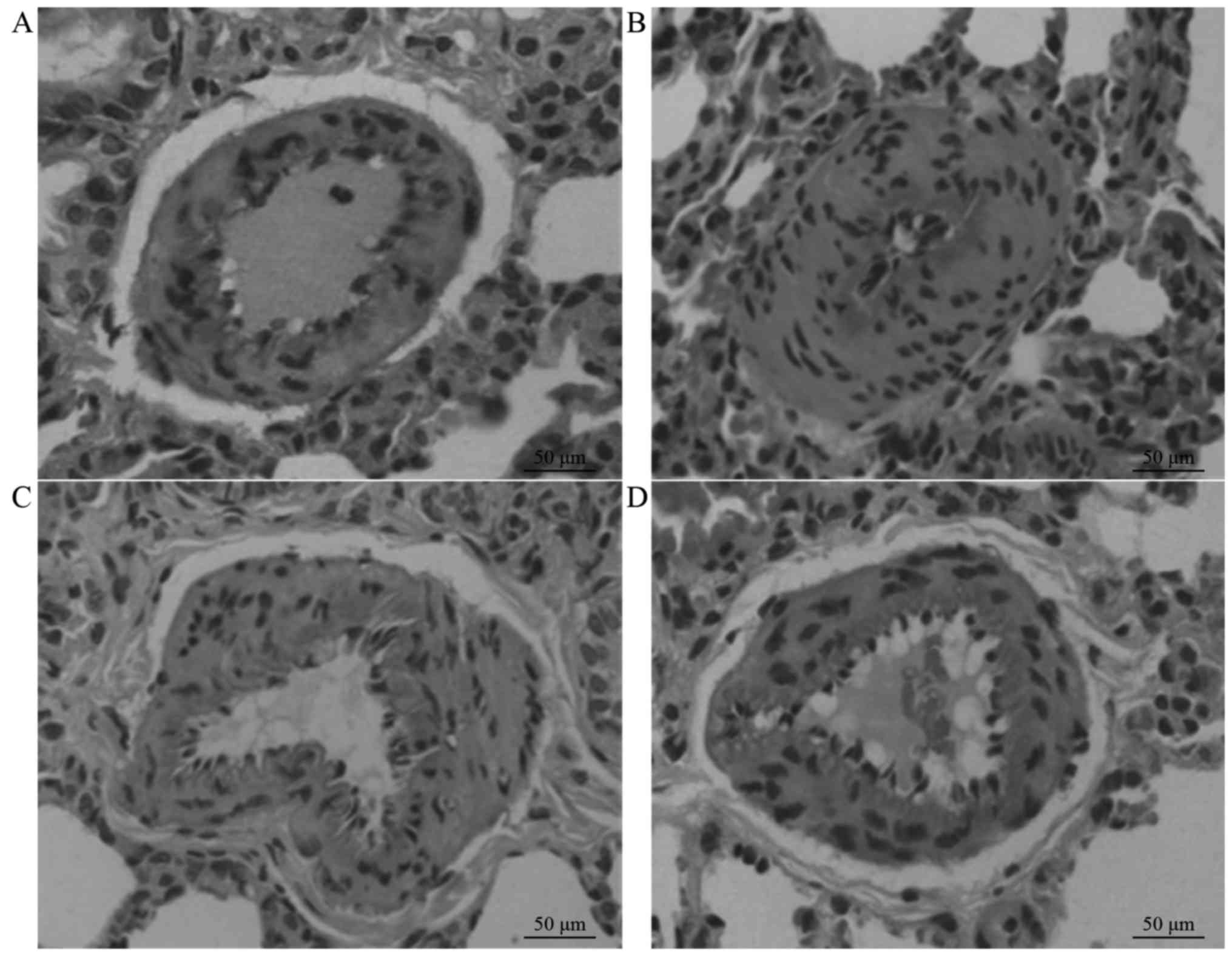
Morphological changes of pulmonary
arteries
Respiratory bronchioles (ED, 50–100 µm) or small
pulmonary arteries with integral structures accompanied by alveoli
were sampled for observation. H&E staining revealed that the
pulmonary arterial walls in group C were thin, with continuous
endothelial cells, and no cell shedding or necrosis, or luminal
stenosis. Furthermore, no inflammatory cell infiltration was
observed in and around the pulmonary arteries. Compared with group
C, the pulmonary arterial walls in group M were thickened, with
proliferation and thickening of the smooth muscle. In addition, the
lumen exhibited stenosis, and degeneration, swelling, necrosis and
shedding of the endothelial cells were observed. Furthermore,
numerous monocytes and neutrophils infiltrated the pulmonary
arterial wall and the surroundings. The WT%, WA% and inflammation
score were significantly increased in group M compared with group C
(P<0.01). Compared with group M, groups N1 and N2 demonstrated
significant attenuation of the changes in pulmonary artery WT and
stenosis, and the WT%, WA% and inflammation scores were
significantly reduced (P<0.05). Furthermore, significant
differences in these three variables were detected between groups
N1 and N2 (P<0.05). However, the WT% and WA% values for rats in
groups N1 and N2 remained higher compared with those in group C
(P<0.05), as demonstrated in Fig.
2 and Table III.
| Table III.Pulmonary artery and inflammatory
indices in each group at 6 weeks after the monocrotaline
injection. |
Table III.
Pulmonary artery and inflammatory
indices in each group at 6 weeks after the monocrotaline
injection.
| Group | n | WT% | WA% | Inflammation score
(points) |
|---|
| C | 8 | 24.67±4.21 | 38.65±3.57 | 0.74±0.03 |
| M | 6 |
60.93±6.58a |
75.84±5.83a |
3.41±0.68a |
| N1 | 7 |
48.37±5.21a,b |
61.77±5.58a,b |
2.03±0.81a,c |
| N2 | 8 |
40.65±5.12c–e |
50.63±4.26c–e |
0.82±0.02c,f |
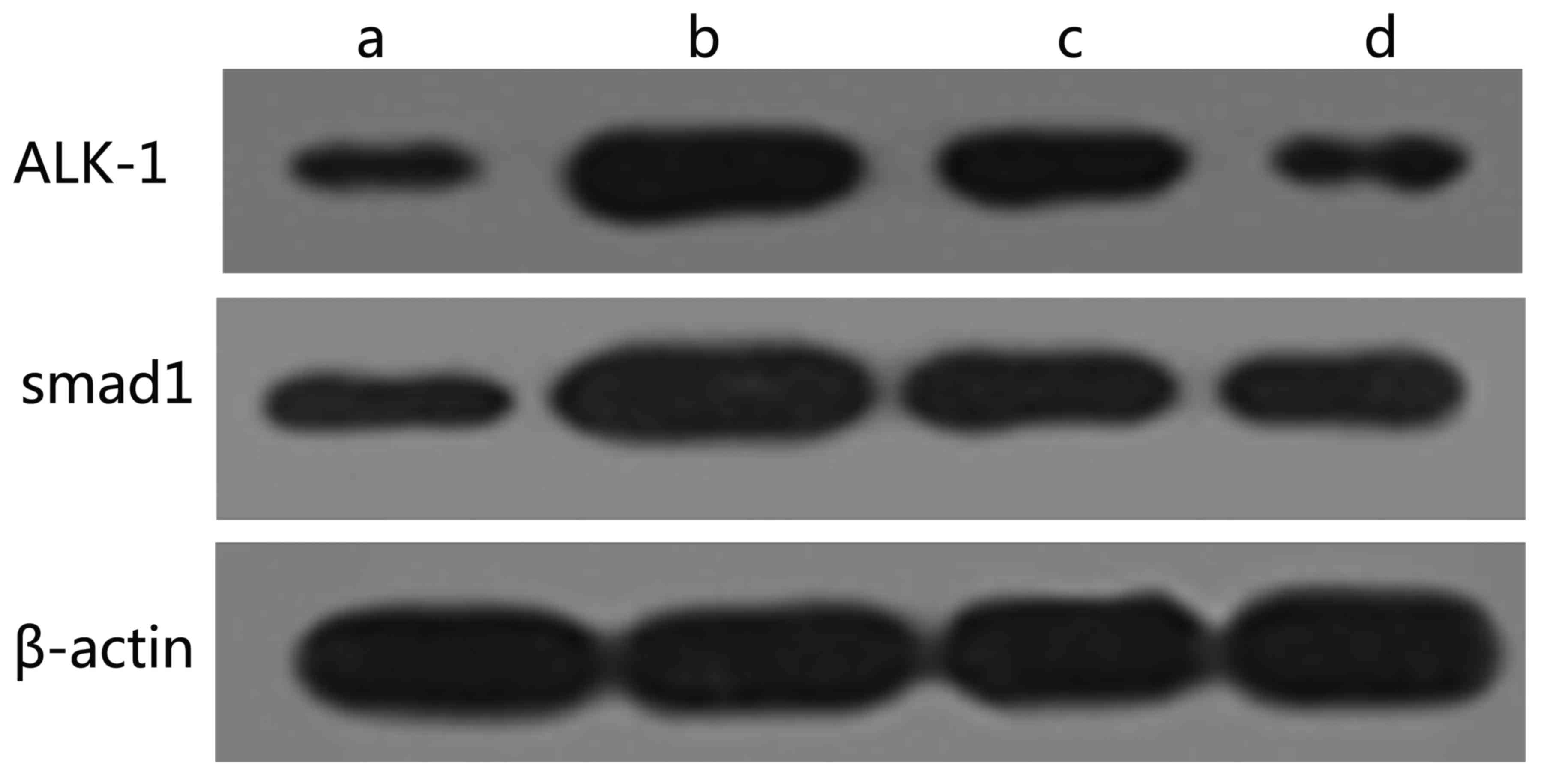
Western blotting
The western blotting results revealed that the
protein expression levels of ALK-1 and Smad1 in group M were
significantly increased compared with those in group C (P<0.01).
Additionally, the protein expression levels of ALK-1 and Smad1 in
groups N1 and N2 were significantly decreased compared with those
in group M (P<0.05), and the intergroup difference was
significant (P<0.05). However, the expression levels in groups
N1 and N2 remained higher than those in the control group
(P<0.01) as shown in Fig. 3 and
Table IV.
| Table IV.Comparison of ALK-1 and Smad1 protein
expression among different groups. |
Table IV.
Comparison of ALK-1 and Smad1 protein
expression among different groups.
| Group | ALK-1 | Smad1 |
|---|
| C | 0.1044±0.021 | 0.1735±0.034 |
| M |
0.4280±0.073a |
0.4504±0.082a |
| N1 |
0.3175±0.086a,b |
0.3524±0.078a,b |
| N2 |
0.2275±0.056a,c,d |
0.2777±0.065a,c,d |
Discussion
PAH is a progressive disease in which the pulmonary
artery pressure progressively increases, eventually leading to
progressive right-heart failure and mortality. The main
pathological feature of PAH is irreversible pulmonary arterial
remodeling. Additionally, progressive occlusion gradually increases
the pulmonary vascular resistance and pulmonary artery pressure,
which is accompanied by irreversible PVR and the occurrence of
right-heart failure (17). The
causes of PAH are complex, and the development of this disease is a
multifactorial process; however, recent studies conducted on the
pathogenesis of PAH have made considerable progress (18–20).
Genetic mutation of bone morphogenetic protein receptor 2 has been
identified in patients with familial PAH (21). Other pathophysiological changes that
have been identified for PAH include functional abnormalities of
pulmonary vascular endothelial cells, K+ channel lesions
on the cell membrane of pulmonary vascular smooth muscles, changes
to the roles of 5-hydroxytryptamine transporters, and increased
matrix synthesis in adventitia (1,22).
However, the pathogenesis of PAH remains incompletely elucidated.
As mentioned above, the ALK-1 gene is closely associated with
idiopathic PAH, and the present study used NAC for therapeutic
intervention in a rat model of PAH, with the aim of observing its
effects on the protein expression of ALK-1 and Smad1 and the
pathological changes of the pulmonary artery wall. Additionally,
changes of mRVP, mPAP, RV and LV+S weights and the RVHI were also
observed in order to explore whether treatment with NAC was
associated with changes in the ALK-1 signaling pathway.
ALK-1 is a type I receptor of the TGF-β superfamily
on the endothelial cell surface, which is able to regulate cell
proliferation, migration and the expression of extracellular matrix
proteins (23). TGF-β is distributed
in a variety of tissues and cells in the human body, serving
important roles in the regulation of cell proliferation and
differentiation, extracellular matrix synthesis and angiogenesis
(10). It has been suggested that as
the type I receptor of TGF-β, ALK-5, activates the Smad2/3 pathway
in endothelial cells and another TGF-β type I receptor, ALK-1,
activates the Smad1/5 pathway in endothelial cells, the interaction
of ALK-1 with Smad1/5 should promote the proliferation and
migration of endothelial cells (the active phase of angiogenesis),
whereas the interaction of ALK-5 with Smad2/3 should inhibit this
process, which is the regression stage of angiogenesis (24). TGF-β regulates endothelial cell
functions via interactions with ALK-1 and ALK-5, and the
overexpression of ALK-1 reduces the signaling conduction of ALK-5
in the extracellular matrix. However, the excessive expression of
ALK-5 increases the signal transduction of ALK-1 in the
extracellular matrix (25).
Therefore, it may be speculated that the balance
between ALK-1/TGF-β/ALK-5 interactions is an important signal
transduction pathway in the angiogenesis process, and that
abnormalities of any of the links in this pathway may lead to
excessive cell proliferation and disease. A previous study has
demonstrated that mutation of the gene encoding ALK-1 is associated
with PAH (6). Jerkic et al
(26), demonstrated that adult ALK-1
heterozygous mice demonstrated clear signs of PAH at an age of 9
weeks, including increased right ventricular systolic pressure,
accompanied by elevated RVHI and the muscularization of surrounding
arteries. Additionally, greater degrees of vascular occlusion and
PVR appeared at an age of 36 weeks, indicating a worsening of the
disease. These adult mice exhibited elevated levels of reactive
oxygen species in their lungs, which may promote the development of
PAH (26). In another study, Eyries
et al (27), observed that a
mutation of ACVRL1 (the gene encoding ALK-1) exists in early
carriers; PAH is observed in these patients, who exhibit a poor
prognosis.
In the present study, 6 weeks following the
subcutaneous injection of 60 mg/kg MCT, the mRVP, mPAP, RVHI, WT%
and WA% in group M were significantly increased compared with those
in the control. Furthermore, H&E staining revealed that the
thickening of the pulmonary artery wall, narrowing of the lumen,
and endothelial cell degeneration, swelling, shedding and necrosis
had occurred, and morphological changes such as the infiltration of
inflammatory cells around blood vessels were also observed. This
suggested that MCT successfully induced the rat PAH model (28). In group M, with the increased
pulmonary artery pressure and PVR, the protein expression levels of
ALK-1 and Smad1 in the pulmonary artery were also significantly
increased compared with those in the control group, indicating that
the ALK-1 signaling pathway in group M was abnormal and suggesting
that this signaling pathway may be involved in the occurrence of
PAH and PVR. Therefore, it was speculated that ALK-1 upregulation
caused increased ligand binding and complexation of Smad, and the
resultant imbalance of the ALK-1/TGF-β/ALK-5 signaling pathway led
to the activation of endothelial cells, decreased apoptosis, the
proliferation of pulmonary arteries, and occlusion and
reconstruction of the pulmonary arteries, thus causing PAH.
However, this hypothesis requires further investigation.
As a precursor of reductive glutathione, NAC
contains an active thiol group, is a commonly used expectorant drug
and is able to dissolve mucus, and attenuate oxidation or
inflammation. In addition, NAC is able to treat pulmonary fibrosis
by inhibiting the proliferation of endothelial cells, reducing the
TGF-β-induced generation of collagens and regulating gene
expression and signal transduction systems, thus exhibiting
important roles for the reversal of PVR (29). In the present study, following
treatment with NAC, the rats exhibited reductions in mRVP, mPAP,
RVHI and PVR compared with the model rats. Furthermore, the protein
expression levels of ALK-1 and Smad1 were reduced following
treatment with NAC. It may be speculated that NAC downregulated the
protein expression levels of ALK-1 and Smad1 in the ALK-1 signaling
pathway, thus inhibiting the proliferation and migration of
endothelial cells, improving PVR and reducing the pulmonary artery
pressure. However, treatment with NAC did not completely reverse
the pathological process, which indicates that other mechanisms are
involved in the formation of PAH. Therefore, further research is
required to investigate this.
The present study observed only the protein
expression of ALK-1 and Smad1 in the pulmonary artery, and other
cytokines in the ALK-1/TGF-β/ALK-5 signaling pathway were not
studied. Therefore, the association of the ALK-1/TGF-β/ALK-5
signaling pathway with PAH requires further investigation.
Furthermore, the mechanisms underlying the actions of NAC and the
feasibility of using NAC in the treatment PAH have many
uncertainties, and require further exploration. However, the
present study indicates that ALK-1 and Smad1 are involved in the
pathogenesis of PAH and PVR, and that NAC significantly inhibits
the protein expression of ALK-1 and Smad1 in the lung tissue and
pulmonary arteries in a rat model of PAH, thus attenuating PAH. In
summary, the present study provides evidence for the future
application of NAC in the treatment of PAH, and suggests its
potential as a novel method for the treatment of PAH.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WY and XS conceived and designed the study. CL and
WJ performed the experiments. XS and CL were major contributors in
writing the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The animal use protocol was reviewed and approved by
the Institutional Animal Care and Use Committee of Qingdao
University (Qingdao, China). Ethical approval was obtained from the
Ethics Committee of Qingdao University Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hecker M, Zaslona Z, Kwapiszewska G, Niess
G, Zakrzewicz A, Hergenreider E, Wilhelm J, Marsh LM, Sedding D,
Klepetko W, et al: Dysregulation of the IL-13 receptor system: A
novel pathomechanism in pulmonary arterial hypertension. Am J
Respir Crit Care Med. 182:805–818. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galiè N, Humbert M, Vachiery JL, Gibbs S,
Lang I, Torbicki A, Simonneau G, Peacock A, Noordegraaf Vonk A,
Beghetti M, et al: 2015 ESC/ERS Guidelines for the diagnosis and
treatment of pulmonary hypertension: The Joint Task Force for the
Diagnosis and Treatment of Pulmonary Hypertension of the European
Society of Cardiology (ESC) and the European Respiratory Society
(ERS) Endorsed by: Association for European Paediatric and
Congenital Cardiology (AEPC), International Society for Heart and
Lung Transplantation (ISHLT). Eur Heart J. 37:67–119. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qiao S, Fan K, Iwashita T, Ichihara M,
Yoshino M and Takahashi M: The involvement of reactive oxygen
species derived from NADPH oxidase-1 activation on the constitutive
tyrosine auto-phosphorylation of RET proteins. Free Radic Res.
48:427–434. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schermuly RT, Ghofrani HA, Wilkins MR and
Grimminger F: Mechanisms of disease: Pulmonary arterial
hypertension. Nat Rev Cardiol. 8:443–455. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cunha SI, Pardali E, Thorikay M, Anderberg
C, Hawinkels L, Goumans MJ, Seehra J, Heldin CH, ten Dijke P and
Pietras K: Genetic and pharmacological targeting of activin
receptor-like kinase 1 impairs tumor growth and angiogenesis. J Exp
Med. 207:85–100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gore B, Izikki M, Mercier O, Dewachter L,
Fadel E, Humbert M, Dartevelle P, Simonneau G, Naeije R, Lebrin F
and Eddahibi S: Key role of the endothelial TGF-β/ALK1/endoglin
signaling pathway in humans and rodents pulmonary hypertension.
PLoS One. 9:e1003102014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Girerd B, Montani D, Coulet F, Sztrymf B,
Yaici A, Jaïs X, Tregouet D, Reis A, Drouin-Garraud V, Fraisse A,
et al: Clinical outcomes of pulmonary arterial hypertension in
patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care
Med. 181:851–861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goumans MJ, Liu Z and ten Dijke P:
TGF-beta signaling in vascular biology and dysfunction. Cell Res.
19:116–127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
López-Novoa JM and Bernabeu C: The
physiological role of endoglin in the cardiovascular system. Am J
Physiol Heart Circ Physiol. 299:H959–H974. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Santibañez JF, Quintanilla M and Bernabeu
C: TGF-β/TGF-β receptor system and its role in physiological and
pathological conditions. Clin Sci (Lond). 121:233–251. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Albro MB, Nims RJ, Durney KM, Cigan AD,
Shim JJ, Vunjak-Novakovic G, Hung CT and Ateshian GA: Heterogeneous
engineered cartilage growth results from gradients of
media-supplemented active TGF-β and is ameliorated by the
alternative supplementation of latent TGF-β. Biomaterials.
77:173–185. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
López-Hernández FJ and López-Novoa JM:
Role of TGF-β in chronic kidney disease: An integration of tubular,
glomerular and vascular effects. Cell Tissue Res. 347:141–154.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tanjore H, Blackwell TS and Lawson WE:
Emerging evidence for endoplasmic reticulum stress in the
pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung
Cell Mol Physiol. 302:L721–L729. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chaumais MC, Ranchoux B, Montani D,
Dorfmüller P, Tu L, Lecerf F, Raymond N, Guignabert C, Price L,
Simonneau G, et al: N-acetylcysteine improves established
monocrotaline-induced pulmonary hypertension in rats. Respir Res.
15:652014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Idrees MM, Saleemi S, Azem MA, Aldammas S,
Alhazmi M, Khan J, Gari A, Aldabbagh M, Sakkijha H, Aldalaan A, et
al: Saudi guidelines on the diagnosis and treatment of pulmonary
hypertension: 2014 updates. Ann Thorac Med. 9 Suppl 1:S1–S15. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan P, Wu WH, Liu D, Zhang R and Jing ZC:
Determination of pulmonary vascular resistance by improved right
heart catheter in rat. Zhonghua Xin Xue Guan Bing Za Zhi.
39:901–904. 2011.(In Chinese). PubMed/NCBI
|
|
17
|
Good RB, Gilbane AJ, Trinder SL, Denton
CP, Coghlan G, Abraham DJ and Holmes AM: Endothelial to mesenchymal
transition contributes to endothelial dysfunction in pulmonary
arterial hypertension. Am J Pathol. 185:1850–1858. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Evans JD, Girerd B, Montani D, Wang XJ,
Galiè N, Austin ED, Elliott G, Asano K, Grünig E, Yan Y, et al:
BMPR2 mutations and survival in pulmonary arterial hypertension: An
individual participant data meta-analysis. Lancet Respir Med.
4:129–137. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takahashi J, Orcholski M, Yuan K and de
Jesus Perez V: PDGF-dependent β-catenin activation is associated
with abnormal pulmonary artery smooth muscle cell proliferation in
pulmonary arterial hypertension. FEBS Lett. 590:101–109. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sarrion I, Milian L, Juan G, Ramon M,
Furest I, Carda C, Gimeno Cortijo J and Roig Mata M: Role of
circulating miRNAs as biomarkers in idiopathic pulmonary arterial
hypertension: Possible relevance of miR-23a. Oxid Med Cell Longev.
2015:7928462015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cogan JD, Pauciulo MW, Batchman AP, Prince
MA, Robbins IM, Hedges LK, Stanton KC, Wheeler LA, Phillips JA III,
Loyd JE and Nichols WC: High frequency of BMPR2 exonic
deletions/duplications in familial pulmonary arterial hypertension.
Am J Respir Crit Care Med. 174:590–598. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McLaughlin VV, Archer SL, Badesch DB,
Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH,
Rosenson RS, et al: ACCF/AHA 2009 expert consensus document on
pulmonary hypertension: A report of the American College of
Cardiology Foundation Task Force on Expert Consensus Documents and
the American Heart Association: Developed in Collaboration With the
American College of Chest Physicians, American Thoracic Society,
Inc., and the Pulmonary Hypertension Association. Circulation.
119:2250–2294. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morine KJ, Qiao X, Paruchuri V, Aronovitz
MJ, Mackey EE, Buiten L, Levine J, Ughreja K, Nepali P, Blanton RM,
et al: Conditional knockout of activin like kinase-1 (ALK-1) leads
to heart failure without maladaptive remodeling. Heart Vessels.
32:628–636. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mishra A, Stueckle TA, Mercer RR, Derk R,
Rojanasakul Y, Castranova V and Wang L: Identification of TGF-β
receptor-1 as a key regulator of carbon nanotube-induced
fibrogenesis. Am J Physiol Lung Cell Mol Physiol. 309:L821–L833.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mitchell D, Pobre EG, Mulivor AW, Grinberg
AV, Castonguay R, Monnell TE, Solban N, Ucran JA, Pearsall RS,
Underwood KW, et al: ALK1-Fc inhibits multiple mediators of
angiogenesis and suppresses tumor growth. Mol Cancer Ther.
9:379–388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jerkic M, Kabir MG, Davies A, Yu LX,
McIntyre BA, Husain NW, Enomoto M, Sotov V, Husain M, Henkelman M,
et al: Pulmonary hypertension in adult Alk1 heterozygous mice due
to oxidative stress. Cardiovasc Res. 92:375–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eyries M, Coulet F, Girerd B, Montani D,
Humbert M, Lacombe P, Chinet T, Gouya L, Roume J, Axford MM, et al:
ACVRL1 germinal mosaic with two mutant alleles in hereditary
hemorrhagic telangiectasia associated with pulmonary arterial
hypertension. Clin Genet. 82:173–179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen L, Xiao J, Li Y and Ma H: Ang-(1–7)
might prevent the development of monocrotaline induced pulmonary
arterial hypertension in rats. Eur Rev Med Pharmacol Sci. 15:1–7.
2011.PubMed/NCBI
|
|
29
|
Xi Y, Tan K, Brumwell AN, Chen SC, Kim YH,
Kim TJ, Wei Y and Chapman HA: Inhibition of
epithelial-to-mesenchymal transition and pulmonary fibrosis by
methacycline. Am J Respir Cell Mol Biol. 50:51–60. 2014.PubMed/NCBI
|