Introduction
Early mortality in patients with acute massive
pulmonary embolism (AMPE) is as high as 30–55% (1,2).
Mortality can be even higher when accompanied by shock and heart
failure (3,4). The reasons for ventricular dysfunction
and failure in patients with PE remain unclear (5,6).
However, these patients are more likely to have myocardial damage
and increased cardiac troponin I (CTnI) levels during heart failure
(7,8). Furthermore, the plasma troponin levels
and extent of myocardial damage are significantly negatively
correlated with the clinical prognosis in these patients (9). Therefore, investigating the causes and
factors associated with myocardial damage in PE patients will aid
in determining the clinical prognosis.
Since nitric oxide (NO) acts as a vasodilator
(10–12), nitric oxide inhalation (NOI) is important for the
treatment of massive pulmonary thrombosis (PT) (13,14). NOI
mitigates lung injury by reducing pulmonary hypertension and
neutrophil migration, promotes endothelial integrity in the lung,
improves pulmonary ventilation/perfusion, increases vascular
density, and repairs vascular endothelial cells (15,16).
NO is an important signaling molecule in the
cardiovascular system and is considered to be a ubiquitous
cardioprotective mediator (17).
However, further research is required to determine whether NOI has
a protective effect against myocardial damage in AMPE.
For further study of the mechanisms of AMPE, we
prepared a rabbit model of AMPE and studied the release curve of
CTnI, the relationship between CTnI and mean pulmonary arterial
pressure (mPAP), and the impact of NOI on CTnI and mPAP.
Materials and methods
Animals and grouping
A total of 30 healthy Japanese white rabbits (not
restricted by sex) aged 6 to 9 months (weight: 2–2.5 kg) were
provided by the Laboratory Animal Center, Medical Department of
Hebei University. They were randomly divided into an experimental
group (EXP; n=10) that did not receive any treatments, an AMPE
control group (CON; n=10) that received urokinase (intravenous
drip, 20,000 U/kg/2 h), and a treatment group (TRE; n=10) that
received conventional thrombolysis plus NOI. This study was carried
out in strict accordance with the recommendations in the Guide for
the Care and Use of Laboratory Animals of the National Institutes
of Health. The animal use protocol has been reviewed and approved
by the Institutional Animal Care and Use Committee (IACUC) of Hebei
University.
Preparation of the MPE animal model
Blood sampling and preparation of
artificial thrombus
Venous blood (2 ml) was sampled from the ear vein of
each rabbit using an indwelling needle and was allowed to settle in
a sterile culture dish at room temperature for 15 min. This was
then placed in a constant-temperature water bath (70°C) for 30 min
to promote blood clotting. Thrombi were then disinfected and cut
into 1×7×1 mm emboli.
Under sterile conditions, anesthetic (20% urethane
solution, 1.0 g/kg) was slowly injected into the ear vein of each
rabbit. Rabbits were then prepared for a 2-cm conventional
transverse incision in the anterior chest for tracheal intubation
to protect the airway for spontaneous breathing and mechanical
ventilation. After the right jugular vein and left external carotid
artery were separated, while simultaneously exposing the left
femoral vein, a homemade pulmonary artery catheter and a
microvascular catheter (5-Fr TIG; Tyler Company, Japan) were
inserted into the right jugular vein and left carotid artery,
respectively. An oscilloscope was used for monitoring during
placement in order to determine the positions of the catheter tips
as indicated by high pressure waveforms, which reflected the length
of pulmonary catheter insertion. During the experiment,
physiological saline (0.3 ml/min) was continuously instilled
through the pulmonary artery catheters using a microperistaltic
pump. The two catheters were connected to a multi-channel
physiological parameter analyzer (MP150; Biopac Systems, Inc.,
Goleta, CA, USA) through a pressure sensor. The mean arterial
pressure and mPAP were controlled synchronously. The left femoral
vein was also punctured for placement of a microcatheter for
in-time rehydration and blood sampling. The prepared emboli (0.5 ml
per injection) were then repeatedly injected (time interval=3 min)
into the right jugular vein via the pulmonary artery catheter using
a 5-ml syringe, followed by an infusion of physiological saline (2
ml), until the mPAP increased to 45 mmHg, the mean arterial blood
pressure decreased to approximately 20–40% of baseline, and
hypotension was achieved and maintained (approximately 55–60 mmHg
blood pressure). AMPE modeling was determined to be successful when
symptoms such as shortness of breath or dyspnea occurred and lasted
for approximately 40 min, at which time the injection of emboli was
terminated. During modeling, if severe dyspnea developed such that
the process could not continue, short-term mechanical ventilation
(SERVO-i infant ventilator, Rontgenvagen2, SE-17154Solna; Siemens
Maquet Critical Care AB, Slona, Sweden) was performed (pressure: 20
cmH2O, respiratory rate: 28 breaths/min).
Perfusion and observation of the
results
When the experiment was completed after 48 h of
successful modeling, the rabbits were perfused with ABS solution,
with BIOPACK pressure monitoring (18–25 mmHg). The specific
procedures were as follows:
Anesthesia
The rabbit ear vein runs along the trailing edge.
The fur covering the skin over the vein was pulled or cut off, and
the skin was moistened with water. The vein was slightly rubbed or
flicked using fingers to increase blood flow, then compressed at
the ear root until the vein became engorged. After injection, the
needle was pulled out and a cotton ball was used to apply pressure
at the incision to stop bleeding.
Fixation
Each rabbit was placed in the prone position on a
laboratory table, with the limbs and jaws tightly fixed.
Preoperative preparation
Fur was removed from the chest and abdomen, which
were then coated with a layer of hair removal cream (using a
medical cotton swab) for approximately 10 sec, and gently wiped
using wet gauze until the fur was completely removed. Meanwhile, an
ABS solution (80 ml) was prepared for future use.
Surgery A
A 6-cm incision was made along the linea alba to
expose the abdominal organs. The abdominal aorta was then gradually
exposed, freed, and threaded for further use. The prepared heparin
saline was then connected to the infusion device and needle. The
needle was cut and the abdominal aorta was cannulated, followed by
knotting and fixation. The inferior vena cava was cut short, and
the infusion device was opened to rinse the blood vessels.
Surgery B
When the liver appeared pale after washing,
heparin-rinsing was stopped and 50 ml of acetone was slowly
instilled until the lung tissue exhibited a color change and the
inferior vena cava emitted a strong odor. The thoracic cavity and
manubrium were then opened in order to ligate the superior vena
cava. An inferior vena cava catheter was inserted above the
diaphragm and connected to a BIOPACK multi-function pressure
detector and the prepared ABS perfusion fluid. This was used to
inject the perfusion liquid until the pressure rose to a point
where it could not be reduced (18–25 mmHg), at which time the
injection was stopped.
The specimen was then removed for micro-computed
tomography (MicroCT; A00001514J; PerkinElmer, Inc., Yokohama,
Japan). The lung and heart tissues were taken. After fixation with
formaldehyde (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and
embedding with paraffin (Sigma-Aldrich; Merck KGaA), the tissue
slices with thickness of 5 µm were prepared. The routine H&E
staining was performed (Sigma-Aldrich; Merck KGaA). The
Histomorphological changes were observed under DVM6 optical
microscope (Leica Science Lab, Leica Camera AG Berlin, Germany).
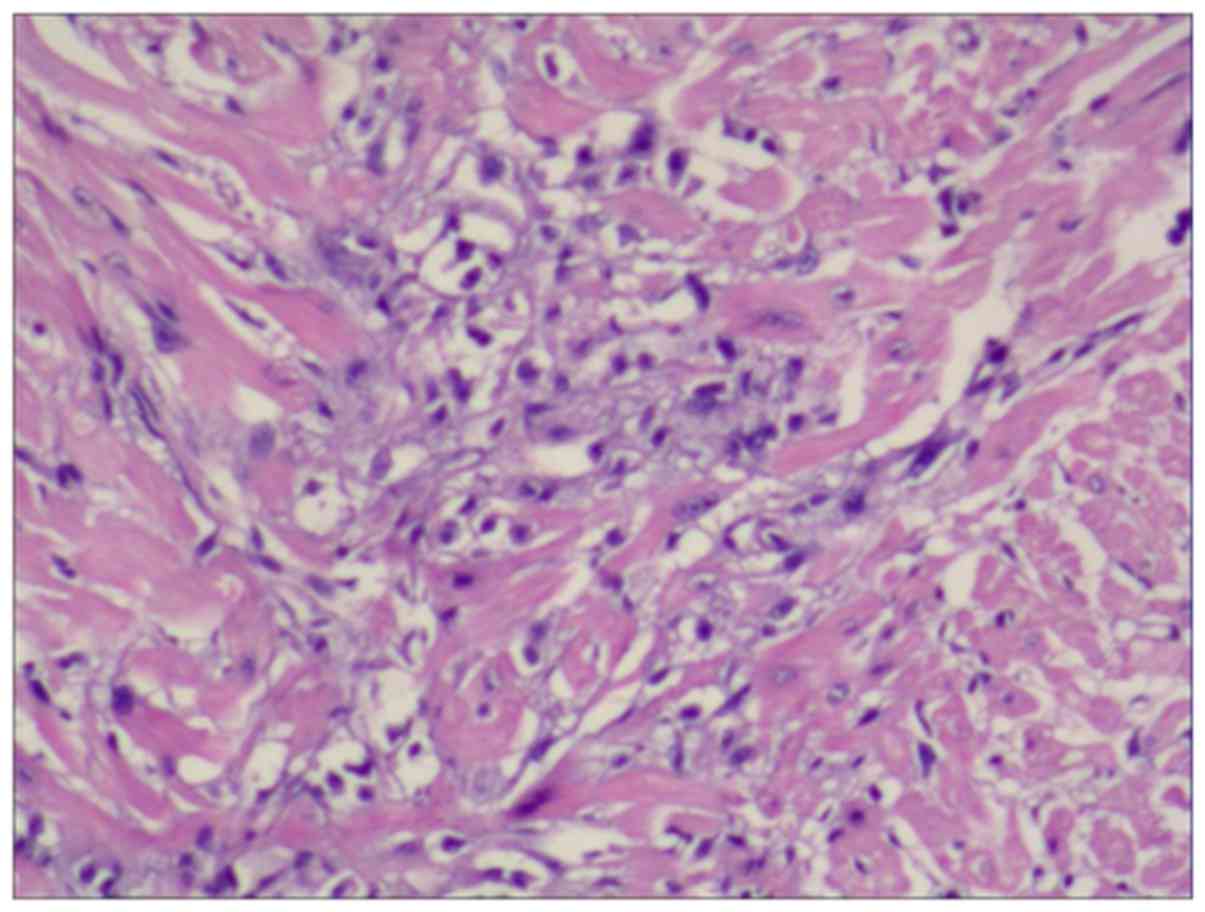
The sections with flaky aggregation of necrosis were defined as
positive (Fig. 1), and the rest were
negative.
Groups CON and TRE underwent thrombolysis or
thrombolysis+NOI 4 h after modeling. Simultaneously, mechanical
ventilation was also applied with the concentration of inhaled
oxygen at 50%.
NOI
Method of NOI
After successful modeling, defined as when the mPAP
of the rabbit reached 40%, and the rabbit developed shortness of
breath or dyspnea for 4 h, mechanical ventilation was initiated. NO
comprised 800 ppm of decompressed NO, N2, and air. When
the concentration of NOI reached 10–20 ppm, a ventilator pipe was
connected to the NOI pipe for invasive mechanical ventilation; the
ventilator was set to pressure control mode (pressure: 20
cmH2O, respiratory rate: 28 breaths/min, oxygen
concentration for mechanical ventilation: 50%). NOI was
administered via a delivery system (Datex-Ohmeda, Madison, WI,
USA). The level of methemoglobin was also monitored during the
experiment and kept at <0.3 g/l.
Blood collection and determination of
CTnI
Blood was sampled before treatment and every 4 h
after treatment. The mean arterial pressure and mPAP were also
monitored simultaneously.
The concentration of CTnI was determined using a
microparticle chemiluminescence method and an automatic immunoassay
analyzer. The kit was provided by Beckman Coulter, Inc. (Brea, CA,
USA).
Statistical analysis
The data were expressed as means ± standard
deviation and were processed using SPSS for Windows, Version 21.0
(IBM SPSS., Armonk, NY, USA). Intergroup comparisons of Indices
were performed using single-factor analysis of variance with
Tukey's post hoc test and paired comparisons were performed with
the q test. The pathological parameters were expressed by the
positive rate, and were analyzed using Fisher's test. P<0.05 was
considered to indicate a statistically significant difference.
Results
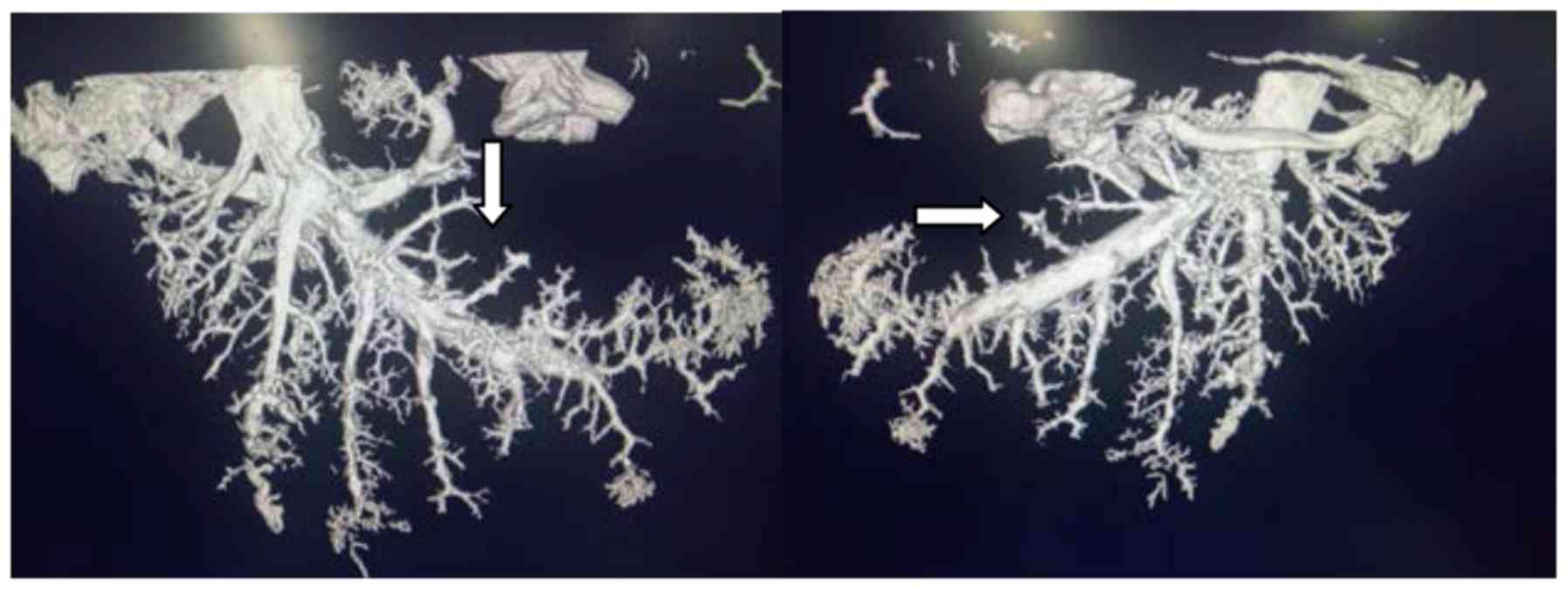
All 30 rabbits were successfully adapted to the MPE
model using this method (confirmed with MicroCT), with a 100%
success rate (Fig. 2).
Pulmonary histopathological changes in
the AMPE model
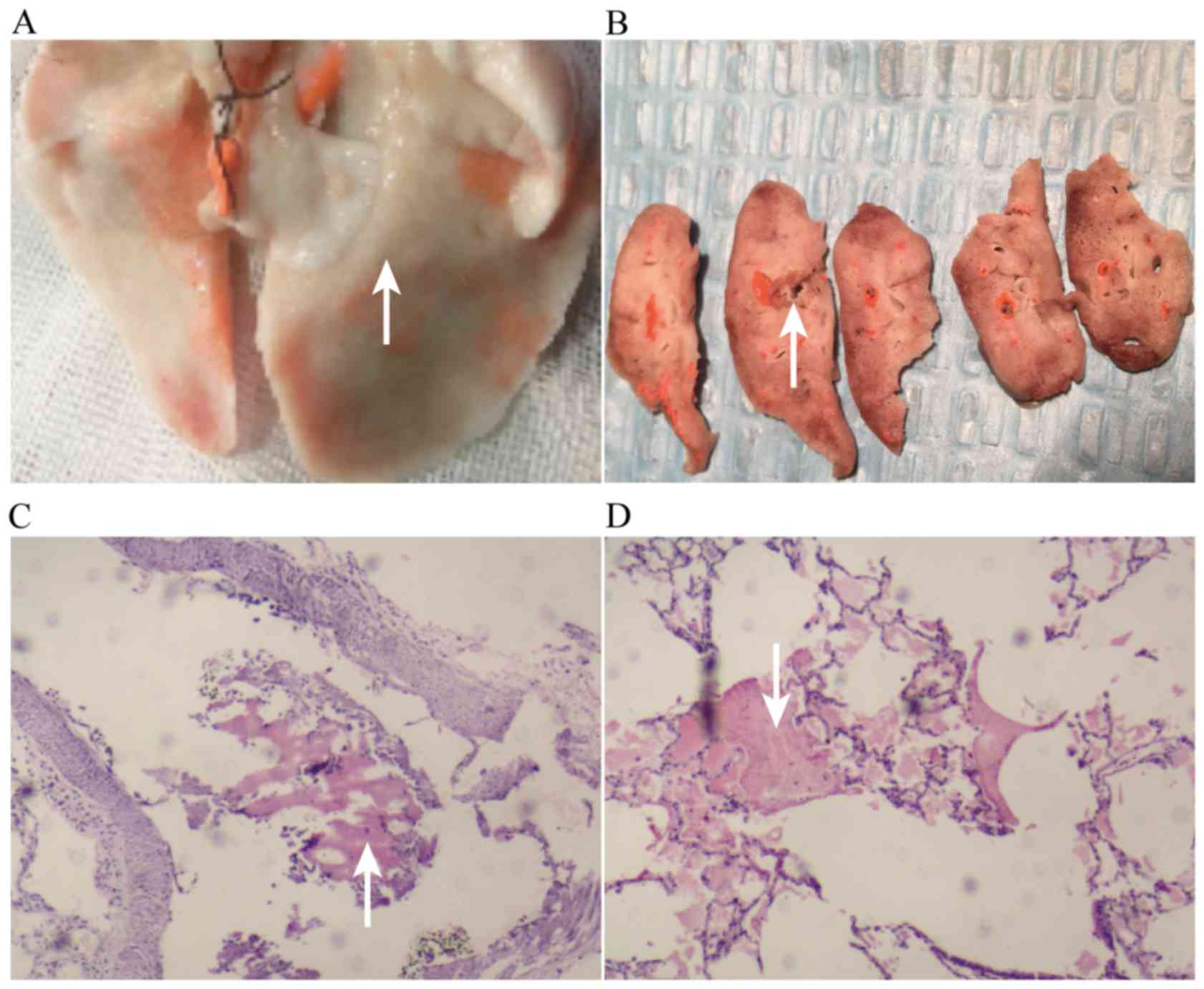
Histopathology revealed that the embolized pulmonary
region was pale and ischemic; the non-embolized region is shown in
Fig. 3A. The pale region is shown in
Fig. 3B, in which large vascular
emboli can be seen, and small vascular emboli are shown in Fig. 3C (microscopy); the bleeding
conditions around the embolized lung tissue are shown in Fig. 3D.
Cardiac histopathology
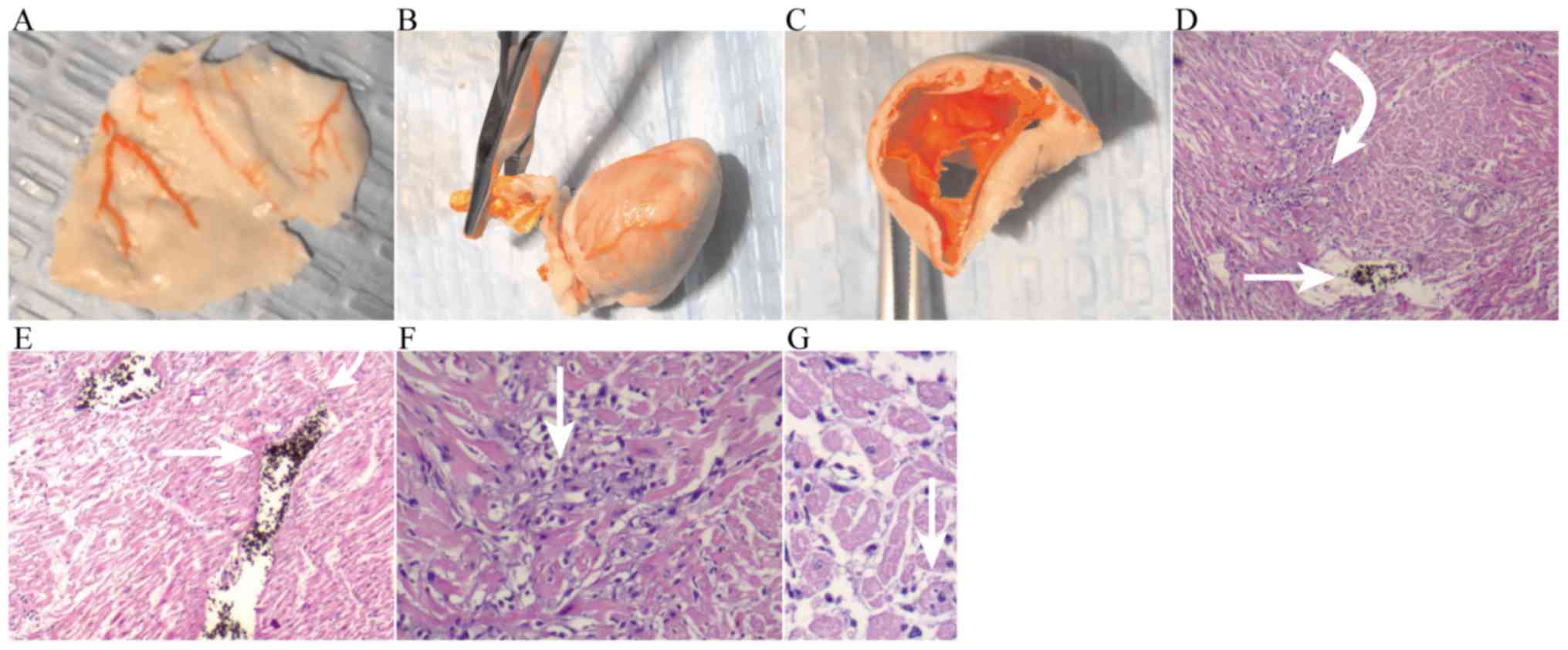
The right ventricle was enlarged, vasodilated, and
congested (Fig. 4A), and the right
ventricular wall became thinner (Fig. 4B
and C); microscopically, the left ventricle exhibited
myocardial necrosis (Fig. 4D).
In 30 pathological slices in the EXP group, there
were 27 slices of obvious myocardial necrosis. After thrombolysis,
myocardial necrosis was still obviously increased, with no
significant difference with the EXP group. The emboli could be seen
intravascularly (Fig. 4D).
Thrombolysis+NOI significantly reduced myocardial necrosis. In 30
pathological slices, there were 8 slices of obvious myocardial
necrosis, with positive rate of 27%. Only scattered necrotic areas
could be seen, together with intravascular emboli (Fig. 4E). High-power microscopy revealed
more obvious changes in group TRE; myocardial necrosis could be
clearly seen after thrombolysis (Fig.
4F). Thrombolysis+NOI scattered necrosis was seen (Fig. 4G and Table
I).
 | Table I.Slices with obvious myocardial
necrosis in different groups. |
Table I.
Slices with obvious myocardial
necrosis in different groups.
| Group (n) | Positive, n (%) | Negative, n (%) | P-values |
|---|
| EXP (30) | 27 (90) | 3 (10) | 0.007 (EXP vs.
CON) |
| CON (30) | 18 (60) | 12 (40) | 0.038 (CON vs.
TRE) |
| TRE (30) | 10 (33) | 20 (67) | <0.001 (EXP vs.
TRE) |
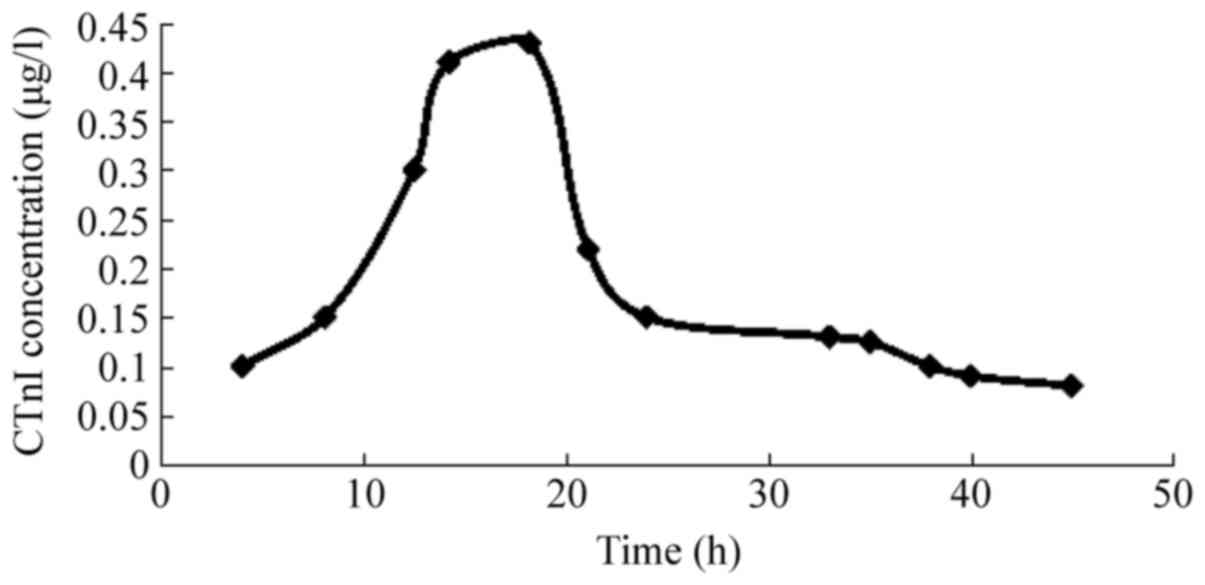
Concentration curve of CtnI
The curve of CTnI became positive 4 h after
modeling, peaked at 18.8±4.5 h, and remained positive for 38±5.2 h;
the peak CTnI concentration was 0.42±0.12 µg/l (Fig. 5).
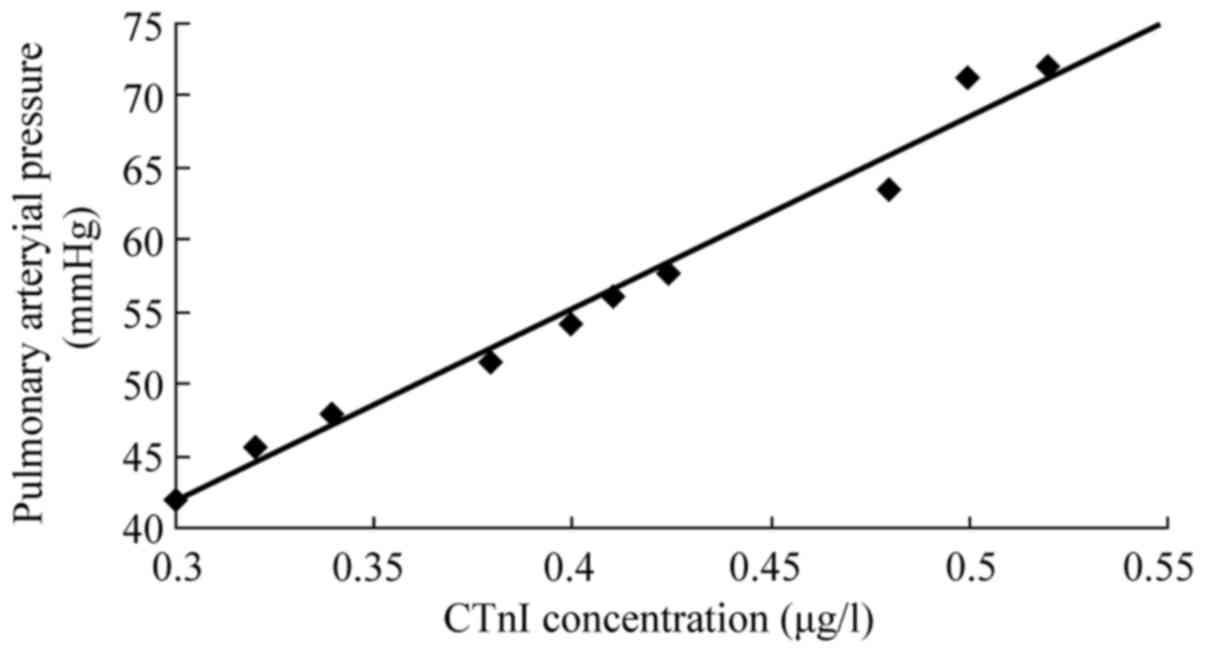
The peak CTnI concentration and mPAP were
significantly correlated in rabbits with AMPE (r=0.98, P<0.05;
Fig. 6).
CTnI peak time and sustained positive
duration
The CTnI peak time and sustained positive duration
were significantly longer and the plasma peak concentration in
group EXP was significantly higher when compared with groups CON
and TRE (P<0.047, P<0.03); corresponding durations in group
CON were significantly longer when compared to those in group TRE
(P<0.048, P<0.036).
The CTnI peak concentration in group EXP was
significantly higher than that in group CON (P<0.039), and that
of group CON was also significantly higher than that of group TRE
(P<0.014; Table II).
| Table II.Comparison of CTnI changes among the
three groups. |
Table II.
Comparison of CTnI changes among the
three groups.
|
| CTnI≥0.1
µg/1a |
|---|
|
|
|
|---|
| Group (n) | Positive rate [4 h, n
(%)] | CTnI peaking time
(h) | Positive duration
(h) | Plasma peak
concentration (µg/1) |
|---|
| EXP 10 | 10/10 (100) | 18.81±4.51 | 38.62±5.22 | 0.42±0.12 |
| CON 10 | 10/10 (100) |
15.13±3.21b |
34.10±3.53b |
0.31±0.10b |
| TRE 10 | 10/10 (100) |
12.42±2.43c |
31.04±2.22c |
0.21±0.06c |
| EXP vs. CON |
| <0.05 | <0.03 | <0.01 |
| TRE vs. CON |
| <0.05 | <0.04 | <0.04 |
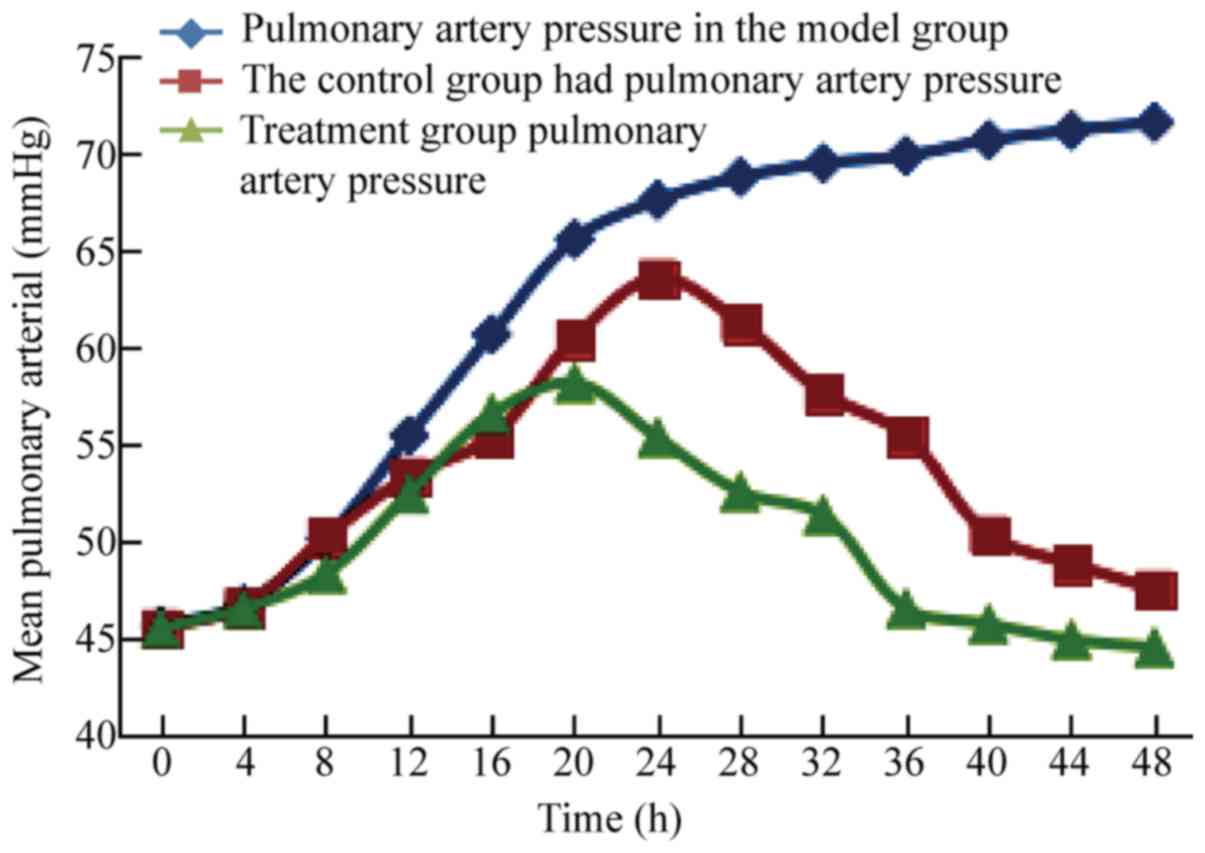
mPAP
The post-modeling mPAP in group EXP slowly increased
with prolongation of modeling time. The post-modeling mPAP in group
CON gradually increased after thrombolysis, but gradually decreased
after peaking. The post-modeling mPAP in group TRE increased slowly
after treatment, then decreased, and was significantly different at
24 h (63.5±5.9, 58.1±5.5, P<0.04), 28 h (61.2±5.7, 55.3±5.6,
P<0.03), 32 h (57.6±5.4, 52.5±5.3, P<0.05), and 34 h
(55.4±4.3, 51.3±4.2, P<0.04) in group TRE when compared with
group CON (Fig. 7).
Discussion
PE is common and is characterized by various
clinical manifestations. Patients with AMPE have increased
mortality and poor prognosis, which may be associated with
myocardial damage. Studies have confirmed that CTnI is increased in
PT, with peak concentration showing a significant correlation with
mortality (15). Therefore, further
investigate the status of impaired myocardium in PT, it is
necessary to establish a PT-induced myocardial injury model. Our
previous studies confirmed that directly injecting emboli into the
pulmonary artery can rapidly increase pulmonary artery pressure,
thus rapidly leading to myocardial necrosis; therefore, this method
has a higher success rate for creating an AMPE model. Studies have
shown that all rabbits with AMPE have accompanying myocardial
damage, which is also associated with the formation of
intravascular emboli. This has been confirmed by our pathological
results. Therefore, the levels of CTnI in the rabbit model are
significantly higher than normal (0.1 µg/l). Our studies also
confirmed that CTnI becomes elevated 4 h after establishing the MPE
model, peaks at 18.8±4.5 h, and remains elevated for 38.6±5.2 h.
This differs from the release curve in patients with myocardial
infarction, who exhibit an elevated CTnI 3 h after onset of
symptoms, with the level remaining elevated for 10 to 14 days.
Characteristic features include the late appearance of CTnI peak
concentration and short duration of elevation, with no
fluctuations. These features indicate that the mechanism of
PT-induced myocardial damage is different from that in myocardial
infarction-induced release of CTnI into the blood (16,17).
In our study, the CTnI released into the blood in
rabbits with MPE showed no significant correlation with the mean
arterial pressure in the systemic circulation, indicating that
myocardial ischemia and hypoxia-induced myocardial damage, which
are caused by the reduction of average arterial pressure, are not
the main cause of PT. This is consistent with previous studies
showing that when MPE occurs, blood pressure decreases, but does
not reduce coronary blood flow; therefore, there is no myocardial
damage (18).
In our study, the CTnI peak concentration in AMPE
was significantly correlated with the mPAP at the same time points,
indicating that pulmonary hypertension plays a major role in the
process of myocardial damage in rabbits with MPE (19,20). It
has been reported that when pulmonary arterial embolization occurs,
platelet dysfunction also occurs, which then activates platelets
and significantly increases thromboxane (TXA2) levels, thus leading
to heart failure and circulatory shock. These vasoactive substances
may partially account for myocardial damage in PT. However, the
CTnI peak concentration is significantly correlated with the mPAP
at the same time points, indicating that the increase in mPAP plays
a major role in determining the degree of myocardial damage in AMPE
(21,22). Therefore, in PT, mechanical
obstruction and vasoactive factors can lead to pulmonary
hypertension. This increases the right ventricular post-load, which
results in an increase in right ventricular work, causing
myocardial ischemia and hypoxia; therefore, this plays an important
role in the extent of myocardial damage and, in turn, patient
prognosis. This is consistent with our findings. Our study showed
that the pulmonary arteries in rabbits with MPE were widened and
the right ventricle was enlarged, but the right ventricular wall
became thinner. Although the mechanism of PT-induced myocardial
damage is not very clear (18), it
has been reported that the left and right ventricles are both
involved (21). This is consistent
with our findings. Combined with our studies, it can be deduced
that the left and right ventricles are both involved; the right
ventricle may mainly exhibit mechanical traction damage caused by
the pulmonary hypertension-induced right ventricular post-load
increase and thinning of the ventricular wall. However, the damage
in the left ventricle may be mainly related to PT-induced platelet
dysfunction, which activates platelets and significantly increases
TXA2 levels. This causes cardiac vascular damage, intravascular
thrombosis, myocardial ischemia/hypoxia, and myocardial necrosis.
The above pathological changes have also been confirmed by our
results.
After successfully creating the animal model, the
mPAP increased slowly and remained at a high level within 48 h of
modeling. This phenomenon is another indication that the
development of pulmonary hypertension is not simply due to
mechanical obstruction. As disease progresses, vasoactive factors
play certain roles in the development of pulmonary hypertension.
Hence, simple thrombolysis cannot immediately decrease the mPAP.
With time, the mPAP increases to a peak, followed by a significant
decrease. This may be caused by the shorter thrombolysis time;
furthermore, when the emboli dissolve, the roles of vasoactive
factors also gradually decrease (23).
Thrombolysis+NOI exhibited a similar change in the
mPAP curve as a result of simple thrombolysis. However, with time,
the mPAP increased slowly. This was mainly due to the role of NOI
(24). NO is an endogenous factor
that promotes vascular smooth muscle relaxation (25) with an obvious and rapid effect. Thus,
the changes in the mPAP curves in groups TRE and CON are very
similar. However, the mPAP changes at 24, 28, 32, and 34 h showed
statistical significance between the two groups, indicating that
NOI can significantly reduce pulmonary hypertension in AMPE, and
that thrombolysis and NOI synergistically reduce pulmonary
hypertension in AMPE.
A comparison of cardiac damage between groups TRE
and CON shows that these two treatment methods can both shorten the
CTnI peak time and duration of elevation, as well as reduce the
CTnI peak concentration. This indicates that thrombolysis and NOI
individually and synergistically have protective effects against
MPE-induced myocardial damage; the mechanism of the latter is
related to the fact that NO can significantly reduce pulmonary
hypertension, restore platelet function, and reduce TXA2 levels
(26).
In conclusion, pulmonary hypertension is not an
independent factor, but is still a major factor leading to
myocardial damage in AMPE. NOI can significantly reduce pulmonary
hypertension and protect against myocardial damage, and exhibits a
synergistic effect with thrombolysis.
Acknowledgements
The authors would like to thank Professor Baoyuan
Chen of the Department of Respiratory, Tianjin Medical University
General Hospital (Tianjin China) for helping in writing this
manuscript.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZZ and KP participated in the design of this study,
and ZZ, LC and YW performed the experiments and collected the data.
LC performed the statistical analysis. ZZ and YW collected
important background information. ZZ, KP and LC drafted the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was carried out in strict accordance with
the recommendations in the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health. The animal use
protocol has been reviewed and approved by the Institutional Animal
Care and Use Committee (IACUC) of Hebei University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Darze ES, Casqueiro JB, Ciuffo LA, Santos
JM, Magalhães IR and Latado AL: Pulmonary embolism mortality in
Brazil from 1989 to 2010: Gender and regional disparities. Arq Bras
Cardiol. 106:4–12. 2016.PubMed/NCBI
|
|
2
|
Yavuz S, Toktas F, Goncu T, Eris C, Gucu
A, Ay D, Erdolu B, Tenekecioglu E, Karaagac K, Vural H and
Ozyazicioglu A: Surgical embolectomy for acute massive pulmonary
embolism. Int J Clin Exp Med. 7:5362–5375. 2014.PubMed/NCBI
|
|
3
|
Dalen JE and Alpert JS: Natural history of
pulmonary embolism. Prog Cardiovasc Dis. 17:259–270. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Soloff A and Rodman T: Acute pulmonary
embolism. II. Clinical. Am Heart J. 74:829–847. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jones AE, Watts JA, Debelak JP, Thornton
LR, Younger JG and Kline JA: Inhibition of prostaglandin synthesis
during polystyrene microsphere-induced pulmonary embolism in the
rat. Am J Physiol Lung Cell Mol Physiol. 284:L1072–L1081. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cho JH, Sridharan Kutti G, Kim SH, Kaw R,
Abburi T, Irfan A and Kocheril AG: Right ventricular dysfunction as
an echocardiographic prognostic factor in hemodynamically stable
patients with acute pulmonary embolism: A meta-analysis. BMC
Cardiovasc Disord. 14:642014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meyer T, Binder L, Hruska N, Luthe H and
Buchwald AB: Cardiac troponin I elevation in acute pulmonary
embolism is associated with right ventricular dysfunction. J Am
Coll Cardiol. 36:1632–1636. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
French Intensive Care Society, .
International congress-Réanimation 2016. Ann Intensive Care. 6
Suppl 1:S502016. View Article : Google Scholar
|
|
9
|
Kreit JW: The impact of right ventricular
dysfunction on the prognosis and therapy of normotensive patients
with pulmonary embolism. Chest. 125:1539–1545. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kline JA, Hernandez J, Garrett JS and
Jones AE: Pilot study of a protocol to administer inhaled nitric
oxide to treat severe acute submassive pulmonary embolism. Emerg
Med J. 31:459–462. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Trummer G, Berchtold-Herz M, Martin J and
Beyersdorf F: Successful treatment of pulmonary hypertension with
inhaled nitric oxide after pulmonary embolectomy. Ann Thorac Surg.
73:1299–1301. 2012. View Article : Google Scholar
|
|
12
|
Waldow T, Witt W, Janke A, Ulmer A, Buzin
A and Matschke K: Cell-cell junctions and vascular endothelial
growth factor in rat lung as affected by ischemia/reperfusion and
preconditioning with inhaled nitric oxide. J Surg Res. 157:30–42.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qi Y, Qian L, Sun B, Liu L, Wu P and Sun
L: Inhaled NO contributes to lung repair in piglets with acute
respiratory distress syndrome via increasing circulating
endothelial progenitor cells. PLoS One. 7:e338592012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Strijdom H, Chamane N and Lochner A:
Nitric oxide in the cardiovascular system: A simple molecule with
complex actions. Cardiovasc J Afr. 20:303–310. 2009.PubMed/NCBI
|
|
15
|
Elias A, Mallett S, Daoud-Elias M, Poggi
JN and Clarke M: Prognostic models in acute pulmonary embolism: A
systematic review and meta-analysis. BMJ Open. 6:e0103242016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Müller-Bardorff M, Weidtmann B, Giannitsis
E, Kurowski V and Katus HA: Release kinetics of cardiac troponin T
in survivors of confirmed severe pulmonary embolism. Clin Chem.
48:673–675. 2002.PubMed/NCBI
|
|
17
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al:
Heart disease and stroke statistics-2014 update: A report from the
American Heart Association. Circulation. 129:e28–e292. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zagorski J, Gellar MA, Obraztsova M, Kline
JA and Watts JA: Inhibition of CINC-1 decreases right ventricular
damage caused by experimental pulmonary embolism in rats. J
Immunol. 179:7820–7826. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lannan KL, Phipps RP and White RJ:
Thrombosis, platelets, microparticles and PAH: More than clot. Drug
Discov Today. 19:1230–1235. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
von Brühl ML, Stark K, Steinhart A,
Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A,
Coletti R, Köllnberger M, et al: Monocytes, neutrophils, and
platelets cooperate to initiate and propagate venous thrombosis in
mice in vivo. J Exp Med. 209:819–835. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sullivan DM, Watts JA and Kline JA:
Biventricular cardiac dysfunction after acute massive pulmonary
embolism in the rat. J Appl Physiol (1985). 90:1648–1656. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Montani D, Günther S, Dorfmüller P, Perros
F, Girerd B, Garcia G, Jaïs X, Savale L, Artaud-Macari E, Price LC,
et al: Pulmonary arterial hypertension. Orphanet J Rare Dis.
8:972013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu Q, Huang K, Zhai Z, Yang Y, Wang J and
Wang C: Initial thrombolysis treatment compared with
anticoagulation for acute intermediate-risk pulmonary embolism: A
meta-analysis. J Thorac Dis. 7:810–821. 2015.PubMed/NCBI
|
|
24
|
Tanus-Santos JE and Moreno H Jr: The use
of inhaled nitric oxide during gas embolism. Chest. 115:1220–1221.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Y, Feng Y, Zhou XG, Pan JJ and Zhou
XY: Inhaled nitric oxide in preterm infants: An updated
meta-analysis. J Res Med Sci. 21:412016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nong Z, Hoylaerts M, Van Pelt N, Collen D
and Janssens S: Nitric oxide inhalation inhibits platelet
aggregation and platelet-mediated pulmonary thrombosis in rats.
Circ Res. 81:865–869. 1997. View Article : Google Scholar : PubMed/NCBI
|