Introduction
Cardiovascular disease associated with obesity,
including coronary heart disease, is caused by dysfunctions of the
heart and blood vessels (1,2). Dysregulated myocardial structures and
functions are associated with myocardial hypertrophy and
cardiovascular disease (3,4). Hypoxia and reoxygenation-induced
injuries, such as coronary syndrome, are commonly observed in the
clinic (5). Early studies have
demonstrated the effects of hypoxia and reoxygenation induced
oxidative stress and tissue damage in heart disease (5,6).
Additionally, cardiac reoxygenation injury promotes apoptosis,
alters enzyme activity, induces mitochondrial dysfunction and is
often accompanied by myocardial injury (1,7).
Cardiomyocyte apoptosis is associated with caspase-3 and −9
activity (8) and is accompanied by
the release of mitochondrial cytochrome c (Cyto c) (9,10). The
activity of a number of mitochondrial enzymes, including cytochrome
oxidase, catalase and manganese superoxide dismutase (SOD2), is
decreased under hypoxic conditions (11). Impaired cytochrome oxidase and
anti-oxidant enzyme activity results in the overproduction of
reactive oxygen species (ROS) (12).
At present, oxidative stress is considered to be a key intermediary
step of the hypoxia/reoxygenation (H/R)-induced apoptosis
process.
H/R-induced vascular remodeling involves a number of
signaling pathways, including the phosphoinositol 3-kinase
(PI3K)/protein kinase B (Akt) and mitogen-activated protein kinase
(MAPK) pathways (13–16). MAPK pathways are activated by
oxidative stress and have a number of important downstream
molecules, including extracellular signal-regulated kinase (ERK)
and PI3K (17,18). The PI3K/Akt pathway regulates
multiple biological processes and mediates apoptosis to regulate
cellular metabolism and cell growth (19,20).
Furthermore, Akt and its downstream effector mammalian target of
rapamycin (mTOR) have been reported to enhance cardiac protection
under oxidative stress (21).
Signaling factors in the Hedgehog family include
sonic hedgehog (Shh), Indian hedgehog, desert hedgehog and three
glioma-associated (GLI) proteins; GLI1, GLI2 and GLI3 (22,23). The
Shh and PI3K/Akt pathways have been reported to regulate cell
migration, proliferation and apoptosis in a number of cell lines
(24). It has previously been
reported that the Shh signaling pathway is activated in ischemia to
regulate a number of biological processes, exerting anti-apoptosis
and anti-oxidative stress effects (25) while also promoting the proliferation
of neural progenitors and muscle regeneration under hypoxic
conditions (26,27). However, the precise molecular
mechanism of the Shh signaling pathway in H/R-induced apoptosis
remains unclear. The aim of the present study was to clarify the
effects of Shh signaling on H/R-induced apoptosis and investigate
the potential downstream targets of Shh. An H9C2 myocardial cell
model was used for in vitro investigation.
Materials and methods
Cell culture
The rat cardiomyoblast H9C2 cell line was purchased
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and maintained
in DMEM (Sigma-Aldrich; Merck KGaA) supplemented with 10% fetal
bovine serum (FBS; Hyclone; GE Healthcare Life Sciences, Logan, UT,
USA) at 37°C in a humidified atmosphere containing 5%
CO2. Cells were grown to 80–90% confluence and then
exposed to hypoxic conditions as follows: Incubation in an
atmosphere containing 0.1% O2 and 5% CO2 in
1% FBS serum-starvation medium for 4 h. After hypoxia, the cells
were reoxygenated in an atmosphere containing 95% O2 and
5% CO2. In the present study, cells were treated with
H/R only (group I), H/R + 20 mM purmorphamine (group II;
Sigma-Aldrich; Merck KGaA), H/R + 10 µΜ cyclopamine (group III;
Sigma-Aldrich; Merck KGaA), or H/R + 20 mM purmorphamine + 5 µM AKT
inhibitor AZD 5363 (group IV; Cayman Chemical Company, Ann Arbor,
MI, USA). H9C2 cells were pre-treated with 20 mM purmorphamine or
10 µΜ cyclopamine or 20 mM purmorphamine + 5 µM AKT inhibitor AZD
536 and then exposed to H/R for 4 h at 37°C.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's protocol. Single-strand cDNA was
synthesized from total RNAs using the Reverse Transcription System
(Promega Corp., Madison, WI, USA) at 70°C for 10 min. cDNA was
amplified by qPCR with SYBR Green using a StepOne Plus real-time
PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Thermocycling conditions were as follows: 10 min polymerase
activation at 94°C followed by 40 cycles at 95°C for 15 sec and
60°C for 60 sec. The following primers were used: Shh, forward
5′-TTCTGTGAAAGCAGAGAACTCC-3′ and reverse
5′-GGGACGTAAGTCCTTCACCA-3′; Shh, forward 5′-AGTGGACATCACCACGTCTG-3′
and reverse 5′-CACCGAGTTCTCTGCTTTCA-3′; GAPDH: Forward
5′-TGTCCGTCGTGGATCTGAC-3′ and reverse 5′-CCTGCTTCACCACCTTCTTG-3′.
Products were separated on 2% agarose gels and results were
normalized against GAPDH and quantified using SynGene software
(1.6.1; Syngene Europe, Cambridge, UK) using the 2−ΔΔCq
method. Experiments were performed in triplicate.
Western blotting
H9C2 myocardial cells were lysed in lysis buffer (50
mM Tris-base, 0.5 M NaCl, 1 mM EDTA, 1% NP40, 1% Glycerol, 1 mM
β-mercaptoethanol, proteinase k inhibitor) from each experiment (30
mg per lane) and separated by 10% SDS-PAGE. Proteins were then
transferred to a nitrocellulose membrane. Membranes were then
incubated for 1 h at room temperature with 3% non-fat dried milk in
PBS followed by incubation with the following primary antibodies:
Anti-Shh (sc-373779), anti-p-mTOR (sc-101738), anti-SOD (sc-17767),
anti-catalase (sc-50508; all 1:5,000; all Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), anti-p53 (ab17990), anti-eNOS (ab5589),
anti-Gli-1 (ab49314; all 1:5,000; all Abcam, Cambridge, UK),
anti-total Akt (9272S; 1:5,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA), and anti-GAPDH (ab37168; 1:10,000; Abcam) at 4°C
overnight. Membranes were washed and subsequently incubated with
the horseradish peroxidase-conjugated secondary antibodies [goat
anti-rabbit IgG (sc-2004), 1:5,000; goat anti-mouse IgG (sc-2005),
1:5,000; Santa Cruz Biotechnology Inc.] for 1 h at room
temperature. The membranes were developed using an enhanced
chemiluminescence system (Thermo Fisher Scientific, Inc.). The band
intensities were normalized to GAPDH and quantified using SynGene
software (1.6.1; Syngene Europe). Experiments were performed in
triplicate.
Cell viability assay
In the present study, H9C2 myocardial cells that had
not been exposed to hypoxia served as the negative control group
(group 0). Hypoxia-treated cells were pre-incubated in an
atmosphere containing 5% CO2 in 1% FBS serum-starvation
medium for 12 h at 37°C. Following treatment, H9C2 myocardial cells
were washed with PBS and incubated in fresh DMEM medium containing
1 g/l MTT (BioVision, Inc., Milpitas, CA, USA) for 4 h at 37°C and
MTT crystals were dissolved in dimethyl sulfoxide. MTT was removed
and the absorption was measured at 490 nm using an ELISA
reader.
Apoptosis assay
Cells were harvested and centrifuged at 5,000 × g
for 5 min at 4°C, following which the supernatant was aspirated.
Normal or apoptotic cells were distinguished using the staining
buffer (Sigma-Aldrich; Merck KGaA), which was then mixed with 2 µl
Annexin-V and PI (Alexa Fluor® 488 Annexin V/Dead Cell
Apoptosis kit; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Cells were analyzed using a flow cytometer
and ~20,000 counts were acquired from each sample (BD FACSuite™
software; version, 1.0; Becton-Dickinson; BD Biosciences, Franklin
Lakes, NJ, USA).
Caspase-3 activity assay
Caspase-3 activity was analyzed using a caspase-3
assay kit (Caspase 3 Assay kit; Sigma-Aldrich; Merck KGaA). H9C2
myocardial cells were seeded in 96-well plates at a density of
5×104 cells/well. Cells were trypsinized, washed with
PBS and centrifuged (1,000 × g; 5 min; 4°C). The caspase-3 assay
buffer was added, cells were centrifuged (1,000 × g; 5 min; 4°C)
and the supernatant was transferred to another tube. The cell
lysates were mixed with the caspase-3 assay buffer in each well and
then incubated for 30 min in the dark at 37°C. The relative
fluorescence of each well was detected using a fluorescence plate
reader at 450 nm (A450) within 30 min.
Monitoring ROS generation
Dichlorofluorescein dye (non-fluorescent CM-H2DCFDA)
is able to diffuse through the cell membrane and the fluorescence
intensity is indicative of intracellular ROS contents. ROS levels
in H9C2 myocardial cells were measured using flow cytometry
following incubation under H/R conditions, with or without Shh
activator, Shh inhibitor or Akt inhibitor. Cells were trypsinized,
washed and re-suspended in Hanks' Balanced Salt Solution for 48 h.
Cells were subsequently incubated with 5 µM CM-H2DCFDA for 30 min
at 37°C in a humidified atmosphere containing 5% CO2.
DCFDA florescence was measured using a BD FACSCanto Flow cytometer
at 520 nm. At least 10,000 events were acquired.
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical significance was assessed using unpaired two-tailed
Student's t-test for comparisons between two groups or using
one-way analysis of variance followed by Dunnett's multiple
comparison for more than three groups using. Analyses were
performed using SPSS 19.0 statistical software (IBM Corp., Armonk,
NY, USA) and P<0.05 was considered to indicate a statistically
significant difference.
Results
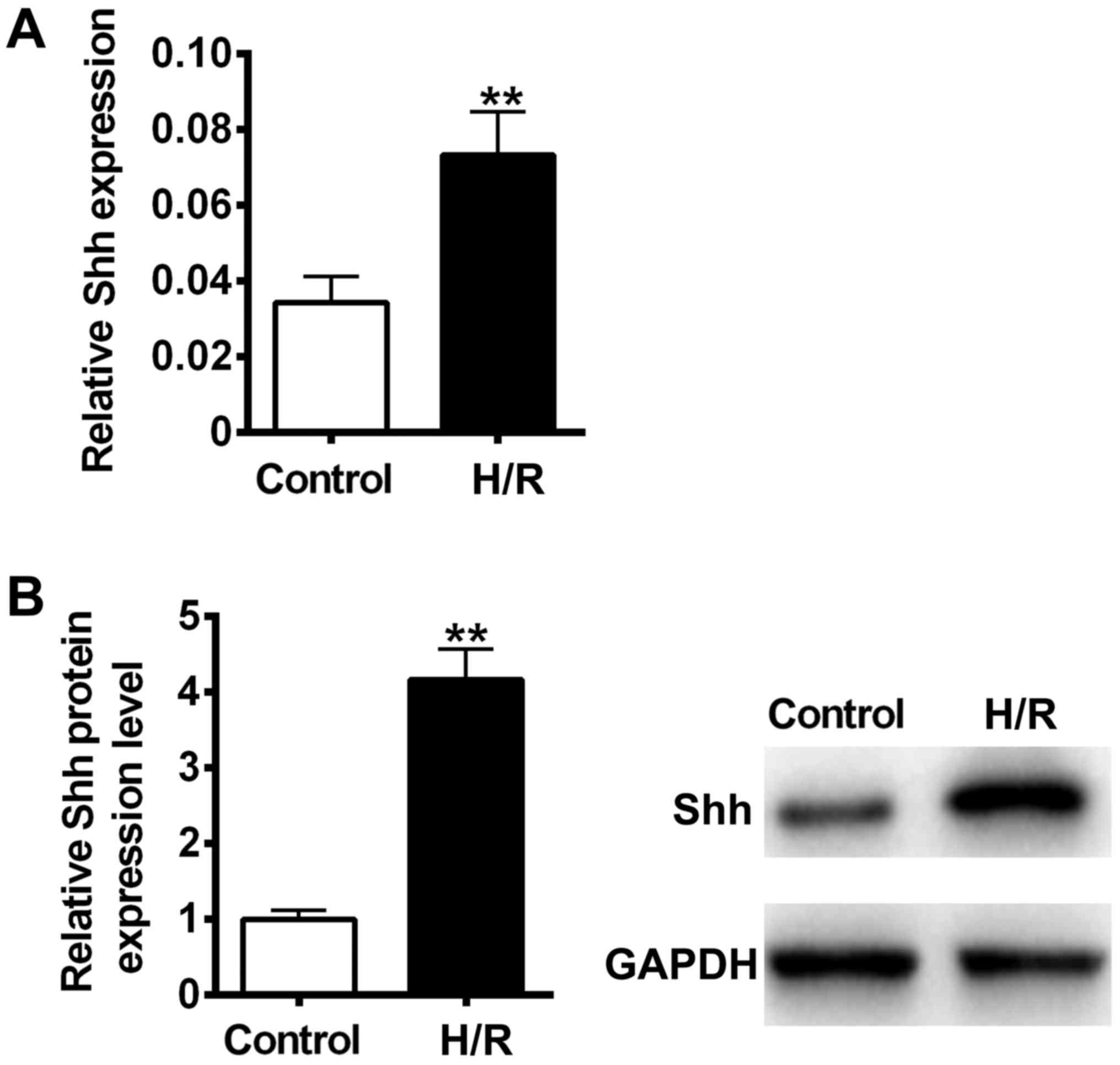
H/R induced Shh expression in the H9C2
myocardial cell model
It has previously been reported that cellular
hypoxia and reoxygenation are significant elements of
ischemia-reperfusion injury (28).
To determine whether H/R could activate the Shh signaling pathway
in H9C2 myocardial cells, Shh mRNA and protein expression was
measured using RT-qPCR and immunoblotting. The results revealed
that Shh mRNA and protein levels were significantly increased
following H/R treatment compared with the control (P<0.01;
Fig. 1). These results suggest that
the Shh signaling pathway may participate in H/R-induced cellular
injury in H9C2 myocardial cells.
Shh signaling is activated to protect
against H/R-induced apoptosis
It has previously been reported that the
morphological nuclear changes associated with apoptosis are
triggered by the activation of caspase proteins (29). In the caspase family, caspase-3
serves as the executor of apoptosis (30). To determine whether the Shh signaling
pathway serves a role in H/R-induced caspase-3 cleavage, H9C2
myocardial cells were exposed to H/R conditions and treated with
the Shh activator purmorphamine or Shh inhibitor cyclopamine. The
results revealed that combined treatment with H/R and purmorphamine
significantly ameliorated H/R-induced caspase-3 cleavage compared
with the control (P<0.01; Fig.
2A). However, caspase-3 cleavage significantly increased
following treatment with H/R and cyclopamine compared with the H/R
group (P<0.01; Fig. 2A).
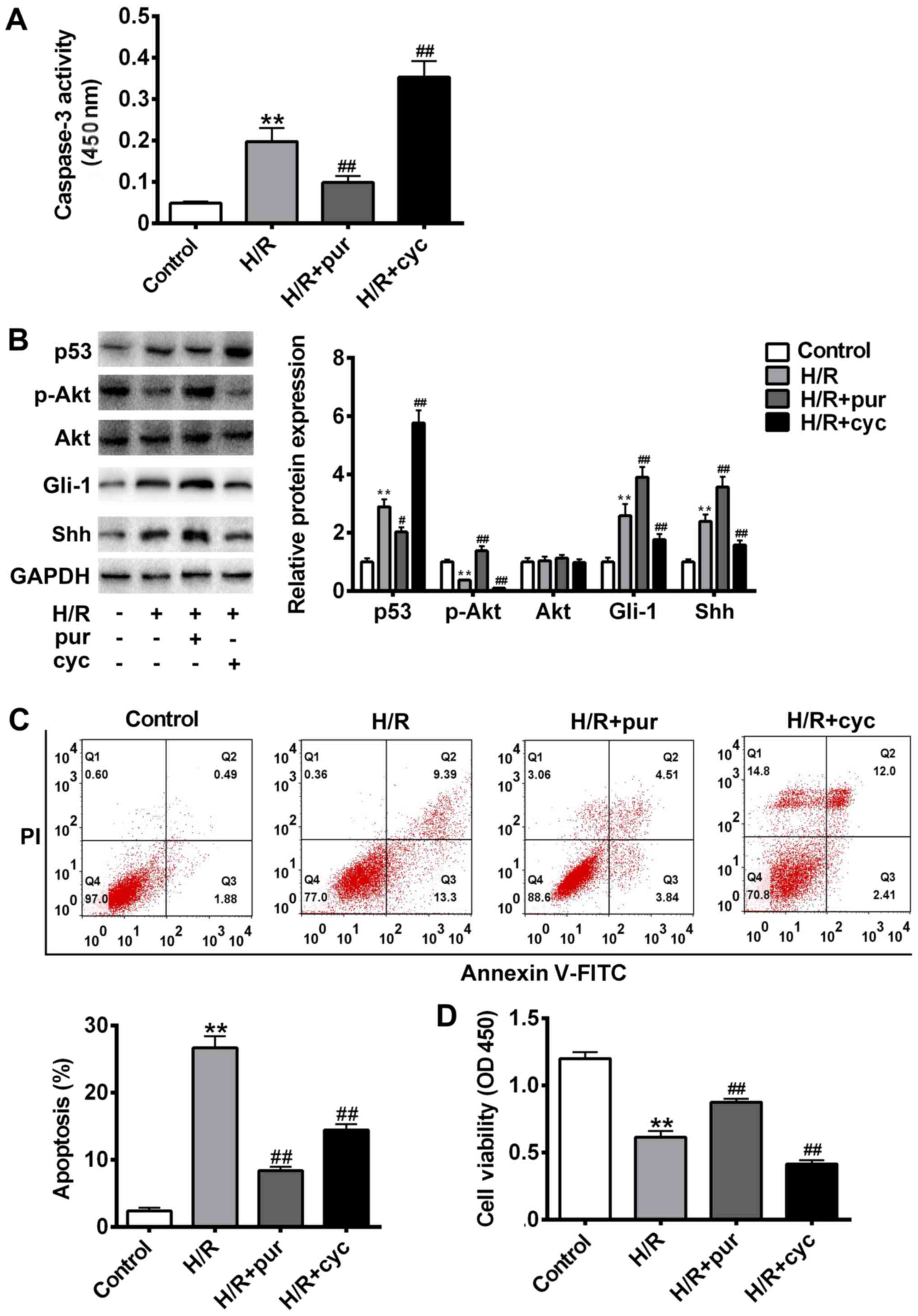 | Figure 2.Effects of the Shh signaling pathway
on H/R-induced apoptosis. Cells were treated with H/R only, H/R +
20 mM pur or HR + 10 µΜ cyc. (A) Cleaved caspase-3 expression was
assessed using a kit and the absorbance was measured at 450 nm. (B)
p53, Gli-1, Akt, p-Akt, Shh and GAPDH protein expression was
measured using western blotting. (C) Apoptosis was assessed using
Annexin V-FITC and PI staining with a flow cytometer. (D) Cell
viability was assessed using an MTT assay and measured at 450 nm.
Data are presented as the mean ± standard deviation. **P<0.01
vs. control group; #P<0.05 and ##P<0.01
vs. H/R group. Shh, sonic hedgehog; H/R, hypoxia reoxygenation;
pur, purmorphamine; cyc, cyclopamine; Gli-1, glioma-associated
oncogene 1; Akt, protein kinase B; p, phosphorylated; FITC,
fluorescein isothiocyanate; PI, propidium iodide; OD, optical
density. |
To assess the role of Shh signaling in H/R-induced
apoptosis, cell apoptosis was measured using flow cytometry with
Annexin V-FITC and PI staining (Fig.
2B), while the expression of p53, Gli-1 and p-Akt was measured
using western blotting (Fig. 2C).
Compared with the untreated control group, cell apoptosis was
significantly increased following H/R treatment (P<0.01). The
expression of p-Akt was significantly decreased following H/R
treatment compared with the control (P<0.01; Fig. 2C); however co-treatment with
purmorphamine reversed this effect (P<0.01 Fig. 2C). Immunohistochemistry revealed that
the expression of Gli1 was upregulated following combined treatment
with H/R and purmorphamine compared with the control (Fig. 2), suggesting that purmorphamine
inhibits H/R-induced apoptosis and activates the Akt pathway.
However, Gli1 upregulation could be blocked by cyclopamine.
Compared with the H/R alone group, cell viability was significantly
increased following treatment with purmorphamine (P<0.01;
Fig. 2D). Collectively, these
results suggest that the Shh activator purmorphamine decreases
H/R-induced apoptosis.
Shh signaling is critical for
H/R-induced apoptosis via the PI3K/AKT/mTOR pathway
The Akt/mTOR pathway serves a critical protective
role against apoptosis, and so it was investigated whether the
mechanism of purmorphamine involves this pathway. It H9C2
myocardial cells were pre-treated with H/R and then treated with
purmorphamine alone or purmorphamine and Akt inhibitor AZD 5363
(Fig. 3).
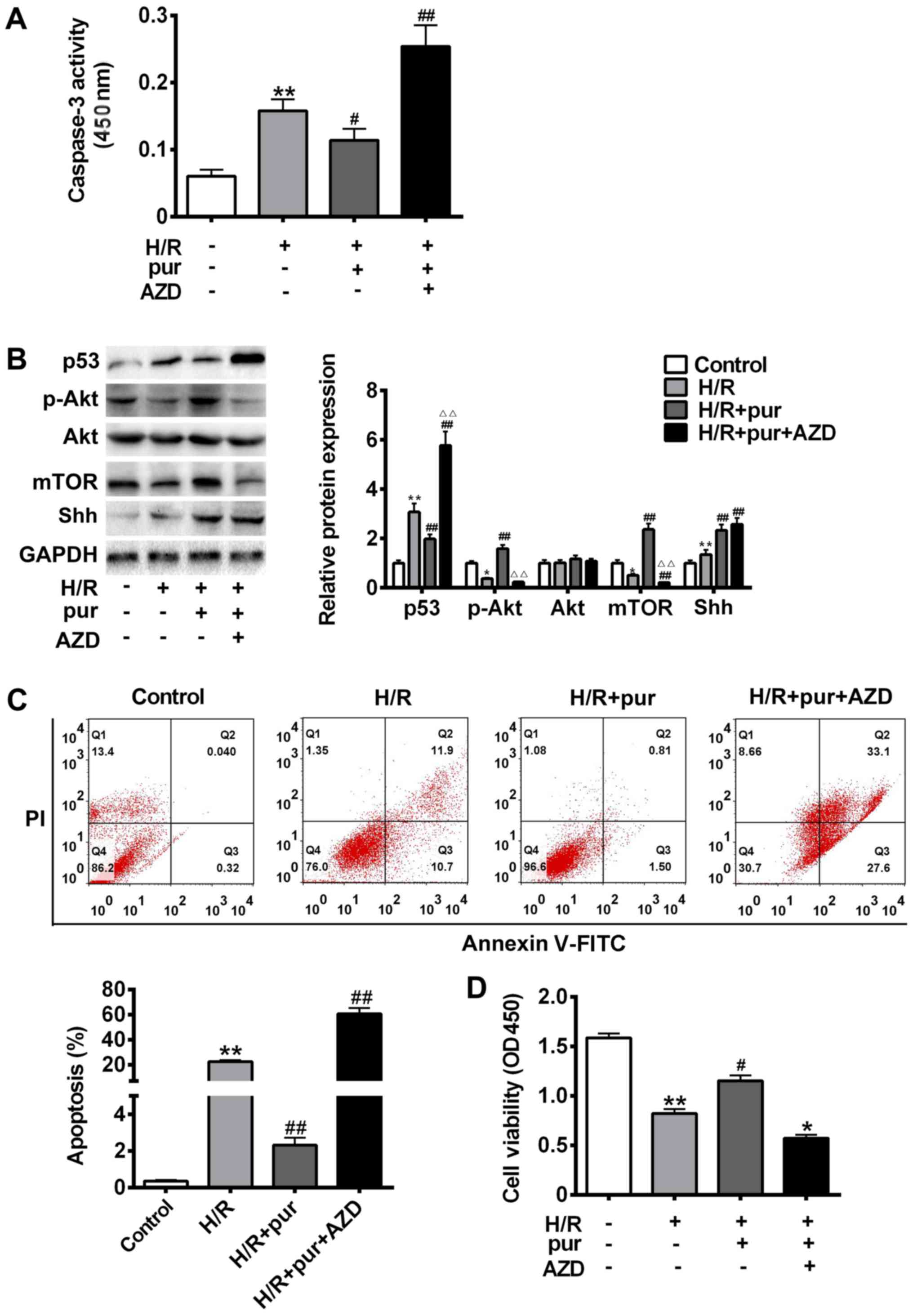 | Figure 3.Effects of Shh and Akt signaling on
H/R-induced apoptosis. Cells were treated with H/R only, H/R + 20
mM pur for 24 h or H/R + 5 µΜ Akt inhibitor AZD 5363 for 24 h. (A)
Cleaved caspase-3 expression was assessed using a kit and the
absorbance was measured at 450 nm. (B) p53, mTOR, Akt, p-Akt, Shh
and GAPDH protein expression was measured using western blotting.
(C) Apoptosis was assessed using Annexin V-FITC and PI staining
with a flow cytometer. (D) Cell viability was assessed using an MTT
assay and measured at 450 nm. Data are presented as the mean ±
standard deviation. **P<0.01 vs. control group;
#P<0.05 and ##P<0.01 vs. H/R group.
Shh, sonic hedgehog; H/R, hypoxia reoxygenation; pur,
purmorphamine; Akt, protein kinase B; AZD, AZD 5363; Gli-1,
glioma-associated oncogene 1; p, phosphorylated; FITC, fluorescein
isothiocyanate; PI, propidium iodide; OD, optical density. |
Treatment with purmorphamine treatment alone
significantly protected H9C2 myocardial cells from H/R-induced
apoptosis (P<0.05; Fig. 3C).
However, AZD 5363 inhibited the expression of p-Akt and mTOR,
leading to a significant increase in caspase-3 cleavage (P<0.05;
Fig. 3A) compared with the
purmorphamine alone group (Fig.
3B-C). p53 protein expression was also significantly
upregulated in H9C2 myocardial cells treated with purmorphamine and
AZD 5363 compared with the H/R group (P<0.01; Fig. 3B). Cell viability was significantly
decreased compared with the H/R group following co-treatment with
purmorphamine and AZD 5363 (P<0.05; Fig. 3D). These results suggest that Shh may
serve a protective role against H/R injury by activating the
PI3K/Akt pathway.
Shh activation restored oxidative
damage induced by H/R
The effects of Shh signaling activation in
H/R-induced oxidative stress were investigated. Intracellular ROS
production was assessed after H/R treatment using flow cytometry
(Fig. 4A). H9C2 myocardial cells
co-treated with H/R and purmorphamine showed a significant decrease
in fluorescence intensity compared with control cells treated with
H/R alone (P<0.05). In addition, a significant overall increase
in ROS production was observed in the cyclopamine group
(P<0.01).
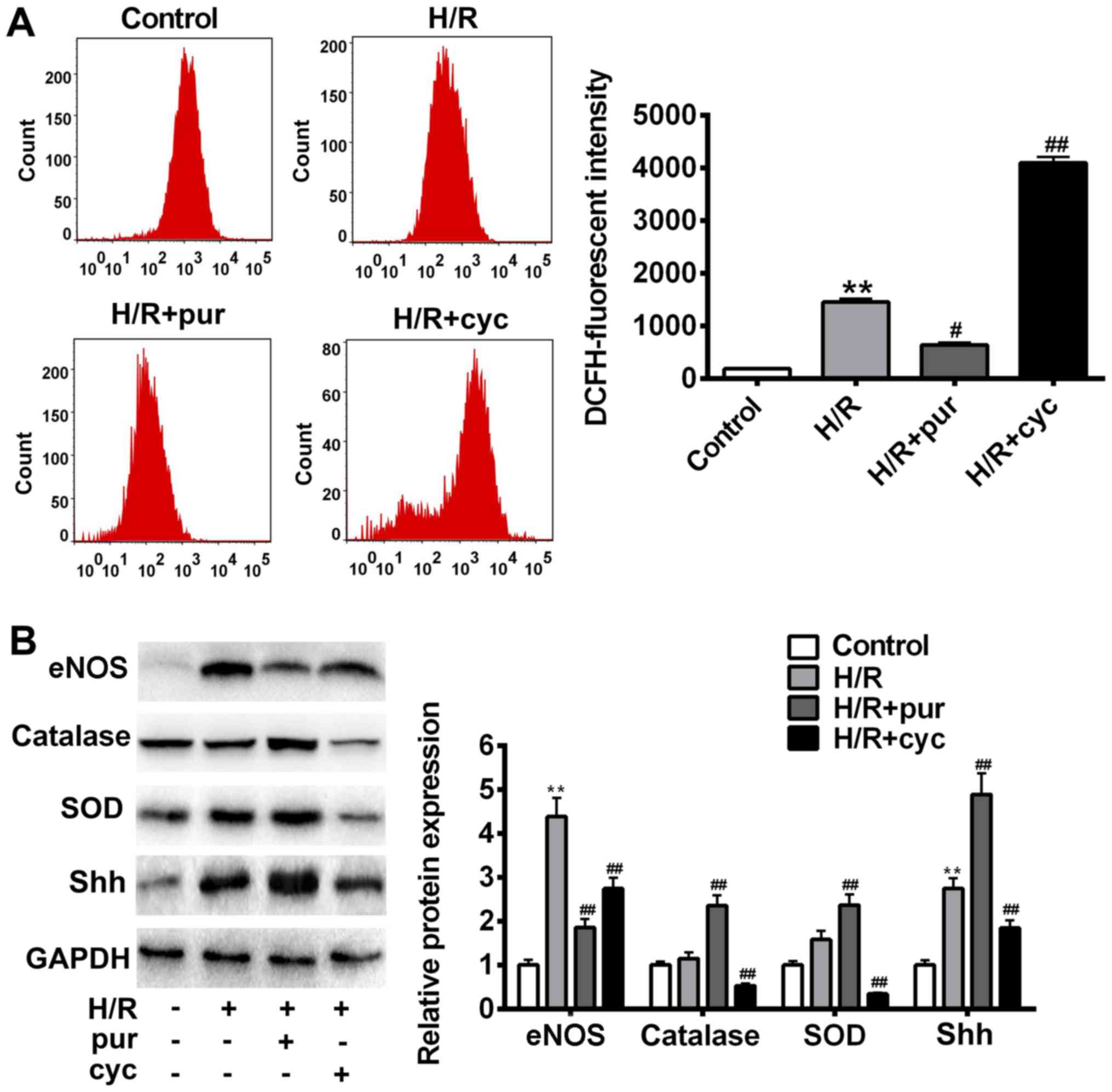 | Figure 4.Effects of Shh activation on ROS
production following H/R treatment. (A) Intracellular ROS
accumulation was determined by measuring the DCF-derived
fluorescence following incubation with DCFH-D. (B) eNOS, catalase,
SOD, Shh and GAPDH expression was assessed using western blotting.
Data are presented as the mean ± standard deviation. *P<0.05 and
**P<0.01 vs. control group; #P<0.05 and
##P<0.01 vs. H/R group. Shh, sonic hedgehog; ROS,
reactive oxygen species; H/R, hypoxia reoxygenation; p,
phosphorylated; eNOS, endothelial nitric oxide synthase; SOD,
superoxide dismutase; NO, nitric oxide. |
In order to identify candidate antioxidant enzymes
for H/R treatment, intracellular SOD, catalase and eNOS levels were
measured using western blotting (Fig.
4B). It was demonstrated that H/R caused cellular oxidative
stress and increased eNOS expression (Fig. 4B). Purmorphamine significantly
reversed H/R-induced cell damage and inhibited eNOS expression
(P<0.05; Fig. 4B). Similarly,
co-treatment with purmorphamine resulted in a significant increase
in intracellular SOD and catalase compared with the H/R group
(P<0.01; Fig. 4B). In comparison
with the H/R group, eNOS expression significantly increased
following cyclopamine treatment (P<0.05; Fig. 4B). These data suggest that the
activation of Shh signaling results in ROS scavenging, thereby
protecting cells from H/R-induced oxidative stress.
Discussion
It has previously been reported that the Shh
signaling pathway acts as a key mediator of cardioprotection in
cardiomyocytes (31,32). Hypoxic injury after reoxygenation is
a significant cause of cellular injury in the myocardial tissue
(33,34). In the present study, it was first
determined whether the Shh signaling pathway could be activated by
H/R. The results demonstrated that the expression of Shh and
downstream factors was increased following H/R treatment, which is
consistent with a previous study in which activation of the Shh
signaling pathway has been reported to be associated with hypoxic
conditions (35). Shh activators and
inhibitors were used to enhance or suppressed the expression of
Shh. The results indicated that combined treatment with H/R and Shh
activator reversed H/R-induced apoptosis, while the opposite was
observed with the Shh inhibitor. These results suggest that the Shh
signaling pathway may serve an important role in a H9C2 myocardial
cell model of H/R-induced cellular injury.
Cellular H/R generally typically results in cell
death due to necrosis or apoptosis (36). Shh signaling contributes to cell
survival and is able to partially ameliorate stress-induced
apoptosis in cells (37). It has
been reported that the activation of Shh signaling promotes
coronary neovascularization and protects myocardial tissues from
ischemia (38,39). However, the role of Shh signaling in
H/R induced cellular injury remains unclear.
In the present study, the effects of SHH signaling
activation on H/R-induced cell apoptosis were assessed. Apoptosis
was measured using a number of assays, including annexin V-binding,
caspase-3 activity and p53 expression. Subsequently, the effects of
Shh signaling activation via the PI3K/Akt pathway were also
investigated. In the present study, the PI3K/Akt pathway was
revealed to contribute to cell viability and inhibit cell
apoptosis. However, treatment with the Akt inhibitor disrupted the
protective effect of Shh signaling in H/R-induced cell injury. In
the present study, it was speculated that the PI3K/Akt pathway may
be a downstream target of the Shh pathway. Given that Shh signaling
and the PI3K/Akt pathway are associated with cell survival, it was
postulated that stimulating PI3K/Akt with insulin-like growth
factor-I potentiated Gli might be essential for Shh signaling
(40,41). PI3K/Akt activation allowed cells to
combat oxidative stress, while specific inhibitors of the PI3K/Akt
pathway blocked the Shh-mediated protective effects in H/R
conditions.
Oxidative stress is a major cause of cellular injury
and has been reported in many diseases, including cancer,
neurodegeneration and cardiovascular and cerebrovascular diseases
(42,43). Apoptosis may be activated by
increased intracellular ROS production (44,45),
which typically occurs after H/R injury. Oxidative stress
contributes to mitochondrial permeability and release of Cyto c
(46), while reoxygenation-induced
cardiomyocyte apoptosis is associated with the activation of
caspases-3 and Cyto c (28,47). Inhibiting ROS production in H/R
injury requires the protection of various reperfused tissues using
anti-oxidant enzymes, including SOD (48,49).
Anti-oxidant systems function as ROS scavengers that limit the
damage caused by reoxygenation-induced cellular injury (28,50).
Cellular ROS are produced via mitochondrial electron transport
complexes under hypoxia pre-exposure conditions (51). Interestingly, the number of
mitochondrial enzymes was decreased following H/R injury,
indicating the downregulation of anti-oxidant defenses in hypoxia,
which in turn may result in increased ROS production by
reoxygenated mitochondria. In the present study, it was revealed
that the Shh activator could significantly ameliorate H/R-induced
cell damage and inhibit the expression of eNOS. These data suggest
that the activation of Shh signaling protects cardiomyocytes from
oxidative stress. Previous studies have demonstrated that
extracellular SOD and catalase are able to completely prevent
reoxygenation injury (52). In the
present study, SOD and catalase were upregulated in response to
pre-treatment with the Shh activator. Furthermore, it has been
reported that Shh expression stimulates cellular SOD and catalase
expression, resulting in cardiac protection against oxidative
stress (53,54).
In summary, activation of the Shh signaling pathway
significantly increases the expression of cellular anti-oxidant
factors and protects H9C2 myocardial cells against H/R-induced
oxidative stress. The Shh signaling pathway regulates the PI3K/Akt
pathway to attenuate H/R-induced apoptosis and enhance the activity
of cellular antioxidant enzymes to combat oxidative stress. The
results of the present study provide a novel insight into the
protective effects of the Shh signaling pathway and may serve as a
basis for the development of effective treatments for
cardiovascular disease.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RZ designed the study. JM collected the data. HL and
ZQ analyzed the data and ZQ drafted the manuscript. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Namura S, Zhu J, Fink K, Endres M,
Srinivasan A, Tomaselli KJ, Yuan J and Moskowitz MA: Activation and
cleavage of caspase-3 in apoptosis induced by experimental cerebral
ischemia. J Neurosc. 18:3659–3668. 1998. View Article : Google Scholar
|
|
2
|
Kopelman PG: Obesity as a medical problem.
Nature. 404:635–643. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Van Gaal LF, Mertens IL and Christophe E:
Mechanisms linking obesity with cardiovascular disease. Nature.
444:875–880. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abel ED, Litwin SE and Sweeney G: Cardiac
remodeling in obesity. Physiol Rev. 88:389–419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reiter RJ and Tan DX: Melatonin: A novel
protective agent against oxidative injury of the
ischemic/reperfused heart. Cardiovasc Res. 58:10–19. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dhalla NS, Temsah RM and Netticadan T:
Role of oxidative stress in cardiovascular diseases. J Hypertens.
18:655–673. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Braunwald E and Kloner RA: Myocardial
reperfusion: A double-edged sword? J Clin Invest. 76:1713–1719.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu W, Lee WL, Wu YY, Chen D, Liu TJ, Jang
A, Sharma PM and Wang PH: Expression of constitutively active
phosphatidylinositol 3-kinase inhibits activation of caspase 3 and
apoptosis of cardiac muscle cells. J Biol Chem. 275:40113–40119.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chang J, Xie M, Shah VR, Schneider MD,
Entman ML, Wei L and Schwartz RJ: Activation of Rho-associated
coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays
an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci
USA. 103:14495–14500. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Narula J, Pandey P, Arbustini E, Haider N,
Narula N, Kolodgie FD, Dal Bello B, Semigran MJ, Bielsa-Masdeu A,
Dec GW, et al: Apoptosis in heart failure: Release of cytochrome c
from mitochondria and activation of caspase-3 in human
cardiomyopathy. Proc Natl Acad Sci USA. 96:8144–8149. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants, and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Raha S and Robinson BH: Mitochondria,
oxygen free radicals, disease and ageing. Trends Biochem Sci.
25:502–508. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Radhakrishnan Y, Maile LA, Ling Y, Graves
LM and Clemmons DR: Insulin-like growth factor-I stimulates
Shc-dependent phosphatidylinositol 3-kinase activation via
Grb2-associated p85 in vascular smooth muscle cells. J Biol Chem.
283:16320–16331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rosner D, Stoneman V, Littlewood T,
McCarthy N, Figg N, Wang Y, Tellides G and Bennett M:
Interferon-gamma induces Fas trafficking and sensitization to
apoptosis in vascular smooth muscle cells via a PI3K- and
Akt-dependent mechanism. Am J Pathol. 168:2054–2063. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D,
Li P, Qiu X, Wen S, Xiao RP and Tang J: Dysregulation of HSG
triggers vascular proliferative disorders. Nat Cell Biol.
6:872–883. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Campbell M and Trimble ER: Modification of
PI3K- and MAPK-dependent chemotaxis in aortic vascular smooth
muscle cells by protein kinase CbetaII. Circ Res. 96:197–206. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ueda S, Masutani H, Nakamura H, Tanaka T,
Ueno M and Yodoi J: Redox control of cell death. Antioxid Redox
Signal. 4:405–414. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Torres M and Forman HJ: Redox signaling
and the MAP kinase pathways. Biofactors. 17:287–296. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma XM and Blenis J: Molecular mechanisms
of mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maiese K, Chong ZZ, Hou J and Shang YC:
Oxidative stress: Biomarkers and novel therapeutic pathways. Exp
Gerontol. 45:217–234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hooper JE and Scott MP: Communicating with
hedgehogs. Nat Rev Mol Cell Bio. 6:306–317. 2005. View Article : Google Scholar
|
|
23
|
Riobo NA and Manning DR: Pathways of
signal transduction employed by vertebrate Hedgehogs. Biochem J.
403:369–379. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sharma N, Nanta R, Sharma J, Gunewardena
S, Singh KP, Shankar S and Srivastava RK: PI3K/AKT/mTOR and sonic
hedgehog pathways cooperate together to inhibit human pancreatic
cancer stem cell characteristics and tumor growth. Oncotarget.
6:32039–32069. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ghanizadeh A: Malondialdehyde, Bcl-2,
superoxide dismutase and glutathione peroxidase may mediate the
association of sonic hedgehog protein and oxidative stress in
autism. Neurochem Res. 37:899–901. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sims JR, Lee SW, Topalkara K, Qiu J, Xu J,
Zhou Z and Moskowitz MA: Sonic hedgehog regulates
ischemia/hypoxia-induced neural progenitor proliferation. Stroke.
40:3618–3626. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Surace EM, Balaggan KS, Tessitore A,
Mussolino C, Cotugno G, Bonetti C, Vitale A, Ali RR and Auricchio
A: Inhibition of ocular neovascularization by hedgehog blockade.
Mol Ther. 13:573–579. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li C and Jackson RM: Reactive species
mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol
Cell Physiol. 282:C227–C241. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kobayashi T, Masumoto J, Tada T, Nomiyama
T, Hongo K and Nakayama J: Prognostic significance of the
immunohistochemical staining of cleaved caspase-3, an activated
form of caspase-3, in gliomas. Clin Cancer Res. 13:3868–3874. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ueda K, Takano H, Niitsuma Y, Hasegawa H,
Uchiyama R, Oka T, Miyazaki M, Nakaya H and Komuro I: Sonic
hedgehog is a critical mediator of erythropoietin-induced cardiac
protection in mice. J Clin Invest. 120:2016–2029. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Paulis L, Fauconnier J, Cazorla O, Thireau
J, Soleti R, Vidal B, Ouillé A, Bartholome M, Bideaux P, Roubille
F, et al: Activation of Sonic hedgehog signaling in ventricular
cardiomyocytes exerts cardioprotection against ischemia reperfusion
injuries. Sci Rep. 5:79832015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schäfer C, Ladilov Y, Inserte J, Schäfer
M, Haffner S, Garcia-Dorado D and Piper HM: Role of the reverse
mode of the Na+/Ca2+ exchanger in reoxygenation-induced
cardiomyocyte injury. Cardiovascu Res. 51:241–250. 2001. View Article : Google Scholar
|
|
34
|
Buja LM: Myocardial ischemia and
reperfusion injury. Cardiovascu Pathol. 14:170–175. 2005.
View Article : Google Scholar
|
|
35
|
Keith B and Simon MC: Hypoxia-inducible
factors, stem cells, and cancer. Cell. 129:465–472. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Saikumar P, Dong Z, Weinberg JM and
Venkatachalam M: Mechanisms of cell death in hypoxia/reoxygenation
injury. Oncogene. 17:3341–3349. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mazumdar T, DeVecchio J, Shi T, Jones J,
Agyeman A and Houghton JA: Hedgehog signaling drives cellular
survival in human colon carcinoma cells. Cancer Res. 71:1092–1102.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kusano KF, Pola R, Murayama T, Curry C,
Kawamoto A, Iwakura A, Shintani S, Ii M, Asai J, Tkebuchava T, et
al: Sonic hedgehog myocardial gene therapy: Tissue repair through
transient reconstitution of embryonic signaling. Nat Med.
11:1197–1204. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lavine KJ, White AC, Park C, Smith CS,
Choi K, Long F, Hui CC and Ornitz DM: Fibroblast growth factor
signals regulate a wave of Hedgehog activation that is essential
for coronary vascular development. Genes Dev. 20:1651–1666. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Riobó NA, Lu K, Ai X, Haines GM and
Emerson CP Jr: Phosphoinositide 3-kinase and Akt are essential for
Sonic Hedgehog signaling. Proc Natl Acad Sci USA. 103:4505–4510.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Koh SH, Kim SH, Kwon H, Park Y, Kim KS,
Song CW, Kim J, Kim MH, Yu HJ, Henkel JS and Jung HK:
Epigallocatechin gallate protects nerve growth factor
differentiated PC12 cells from oxidative-radical-stress-induced
apoptosis through its effect on phosphoinositide 3-kinase/Akt and
glycogen synthase kinase-3. Brain Res Mol Brain Res. 118:72–81.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mariani E, Polidori M, Cherubini A and
Mecocci P: Oxidative stress in brain aging, neurodegenerative and
vascular diseases: An overview. J Chromatogr B Analyt Technol
Biomed Life Sci. 827:65–75. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Uttara B, Singh AV, Zamboni P and Mahajan
RT: Oxidative stress and neurodegenerative diseases: A review of
upstream and downstream antioxidant therapeutic options. Curr
Neuropharmacol. 7:65–74. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Matés JM and Sánchez-Jiménez FM: Role of
reactive oxygen species in apoptosis: Implications for cancer
therapy. Int J Biochem Cell Biol. 32:157–170. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Giorgio M, Trinei M, Migliaccio E and
Pelicci PG: Hydrogen peroxide: A metabolic by-product or a common
mediator of ageing signals? Nat Rev Mol Cell Biol. 8:722–728. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ott M, Robertson JD, Gogvadze V,
Zhivotovsky B and Orrenius S: Cytochrome c release from
mitochondria proceeds by a two-step process. Proc Natl Acad Sci
USA. 99:1259–1263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kumar D and Jugdutt B: Apoptosis and
oxidants in the heart. J Lab Clin Med. 142:288–297. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang W, Wang M, Xie HY, Zhou L, Mseng XQ,
Shi J and Zheng S: Role of reactive oxygen species in mediating
hepatic ischemia-reperfusion injury and its therapeutic
applications in liver transplantation. Transplant Proc.
39:1332–1337. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dhalla NS, Elmoselhi AB, Hata T and Makino
N: Status of myocardial antioxidants in ischemia-reperfusion
injury. Cardiovasc Res. 47:446–456. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yamada J, Yoshimura S, Yamakawa H, Sawada
M, Nakagawa M, Hara S, Kaku Y, Iwama T, Naganawa T, Banno Y, et al:
Cell permeable ROS scavengers, Tiron and Tempol, rescue PC12 cell
death caused by pyrogallol or hypoxia/reoxygenation. Neurosci Res.
45:1–8. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Vanden Hoek TL, Becker LB, Shao Z, Li C
and Schumacker PT: Reactive oxygen species released from
mitochondria during brief hypoxia induce preconditioning in
cardiomyocytes. J Biol Chem. 273:18092–18098. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sorescu D and Griendling KK: Reactive
oxygen species, mitochondria, and NAD(P)H oxidases in the
development and progression of heart failure. Congest Heart Fail.
8:132–140. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Al-Ayadhi LY: Relationship between Sonic
hedgehog protein, brain-derived neurotrophic factor and oxidative
stress in autism spectrum disorders. Neurochem Res. 37:394–400.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ghanizadeh A, Akhondzadeh S, Hormozi M,
Makarem A, Abotorabi-Zarchi M and Firoozabadi A:
Glutathione-related factors and oxidative stress in autism, a
review. Curr Med Chem. 19:4000–4005. 2012. View Article : Google Scholar : PubMed/NCBI
|