Introduction
Acute myocardial infarction (AMI) is a serious
disease with high morbidity and mortality rates, affecting >7
million individuals around the world each year (1). Novel treatment strategies including
coronary intervention technologies and the use of anticoagulant
agents, antiplatelet agents, nitroglycerin receptor blockers and
angiotensin receptor blockers, have been demonstrated to decrease
the acute phase mortality of AMI (2); however, the prevalence of chronic heart
failure in patients with AMI has increased and the long-term
mortality of patients post-AMI remains high (3). Endogenous heart regeneration, including
cardiomyocyte proliferation, resident stem cell niches,
neovascularization, inflammation and extracellular matrix
remodeling, are potential novel pathways that may stimulate repair
following AMI (4).
Endothelial progenitor cells (EPCs) located in bone
marrow primarily express cluster of differentiation (CD)34, CD133
and kinase domain receptors (KDR) (5). These cells may be exported to the
peripheral blood and undergo differentiation into endothelial cells
to support vascular endothelial repair and angiogenesis, which may
be associated with cytokine gradients (6). Due to their potential to repair and
regenerate vascular tissue, EPCs have been postulated as a
potential treatment to improve cardiovascular disease (7).
Tumor necrosis factor (TNF)-related weak inducer of
apoptosis (TWEAK) is a member of the TNF ligand superfamily and
acts by binding to fibroblast growth factor-inducible 14 (Fn14),
the sole receptor of TWEAK, to initiate several intracellular
signaling pathways, including nuclear factor-κB (NF-κB) (5,8). TWEAK
is expressed at low levels in healthy normal tissues; however, it
is overexpressed following tissue injury, which may contribute to
cancer, chronic autoimmune diseases and acute ischemic stroke
(8,9). TWEAK stimulates cancer cell
proliferation, migration and resistance to chemotherapeutic agents
(10). Furthermore, the expression
of pro-angiogenic and pro-inflammatory cytokines is enhanced upon
TWEAK/Fn14 activation (11).
TWEAK/Fn14 signaling also serves a protective role against
intestinal inflammation and prevents colitis-associated cancer via
its proapoptotic effects (12).
Compared with healthy individuals, soluble TWEAK is significantly
elevated in patients with acute MI (AMI) (13). Thus, TWEAK/Fn14 have been suggested
as potential mediators of cardiovascular disease and potential
treatment targets (14).
The aim of the present study was to determine the
effects of TWEAK on EPCs in AMI. The results demonstrated that
TWEAK promotes EPC viability, migration and differentiation,
providing protection against further cardiac injury in mice with
AMI. It was also revealed that TWEAK is associated with the
activation of the NF-κB signaling pathway.
Materials and methods
Reagents
Endothelial cell basal medium-2 (EBM-2) and
endothelial cell growth medium-2 (EGM-2) were purchased from Lonza
Group, Ltd. (Basel, Switzerland). Human TWEAK (cat. no. RAB1765)
and Mouse TWEAK (cat. no. RAB0495) ELISA kits were purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Matrigel Matrix,
and rat anti-mouse antibodies against fluorescein isothiocyanate
(FITC)-conjugated CD34 (cat. no. 553733), phycoerythrin
(PE)-conjugated KDR (cat. no. 555308), PE-conjugated CD45 (cat. no.
561087), and PE-conjugated CD146 (cat. no. 562196) were purchased
from BD Biosciences (Franklin Lakes, NJ, USA). Anti-mouse
FITC-conjugated antibodies against CD133 (cat. no. 85-11-1331-80)
were purchased from eBioscience (San Diego, CA). Ulex
europaeus agglutinin-1 (UEA-1) lectin was obtained from Vector
Laboratories, Inc. (cat. no. B-1065-2; Burlingame, CA, USA) and the
Transwell plate was purchased from Corning, Inc. (Corning, NY,
USA). Bovine serum albumin was purchased from Beyotime Institute of
Biotechnology (Nantong, China). Fetal bovine serum (FBS), TRIzol
and DiI-acLDL were purchased from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). The Masson's trichrome staining kit (cat. no.
D026) and vascular endothelial growth factor A (VEGFA) Assay kit
(cat. no. H044) were obtained from Nanjing Jiancheng Bioengineering
Institute (Nanjing, China) to detect secreted VEGFA in the culture
medium according manufacturer's protocol. The GTVisinTM
anti-mouse/anti-rabbit immunohistochemical analysis kit was
purchased from Gene Company, Ltd. (Hong Kong, China).
Anti-phosphorylated (p)65 (cat. no. 8242), anti-phosphate-p65 (cat.
no. 3033), anti-CD31 (cat. no. 3528), anti-α-smooth muscle actin
(SMA; cat. no. 19245), anti-GAPDH (cat. no. 5174), Alexa
Fluor® 488 conjugated anti-mouse immunoglobulin (Ig)G
(cat. no. 4408), Alexa Fluor® 488 conjugated anti-rabbit
IgG (cat. no. 4412) and horseradish peroxidase conjugated goat
anti-rabbit Immunoglobulin G (1:2,000; cat. no. 7074) antibodies,
as well as RIPA Buffer (10X; cat. no. 9806) were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA). Fn14 small
interfering (si)RNA (cat. no. sc-145209) and anti-VEGFA (1:500;
cat. no. sc-4570) were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Dimethylsulfoxide (DMSO), isopropanol,
ethanol and chloroform were purchased from Sinopharm Chemical
Reagent Co., Ltd. (Shanghai, China).
Patient information
Peripheral blood samples were collected from 25 male
patients with AMI and 25 healthy male volunteers (age, 60–80 years)
were recruited between January and June 2016. The patients were
admitted to Zhongda Hospital (Nanjing, China), diagnosed for the
first time, and did not have a history of AMI, exhibited ST-segment
elevation, serum CK-MB >5 ng/ml and serum Troponin T >0.2
ng/ml. The healthy volunteers were recruited to Zhongda Hospital,
and exhibited normal ST-segments, serum CK-MB <0.6 ng/ml and
serum Troponin T <0.1 ng/ml. The use of human samples was
approved by the Ethics Committee of Zhongda Hospital, Medical
School of Southeast University (Nanjing, China) and written
informed consent was obtained from all patients prior to
enrollment.
EPC isolation and identification
EPCs are derived from the bone marrow under
pathological conditions and are associated with neovascularization
and tissue repair (7). EPCs were
derived from C57Bl/6 mice as previously described (15,16). A
total of 160 10–12-week-old male C57Bl/6 mice (weight, 20–22 g)
were obtained from the Laboratory Animal Center of Southeast
University. The mice were housed in sterilized cages at 21±1°C with
a 12 h light/dark cycle and 55±5% relative humidity, and received
sterilized food and water ad libitum. Animal experiments
were approved by The Animal Care Committee of the Southeast
University. A total of 3 C57Bl/6 mice were sacrificed by cervical
dislocation and soaked in 75% ethanol at room temperature for 10
min. The tibiofibula was removed and the bone marrow was washed
using 1 ml PBS containing 50 U/ml heparin and 0.05 mg/ml DNase
(both Sigma-Aldrich; Merck KGaA). The resulting fluid was
collected, added to lymphocyte separation medium (Yeason, Shanghai,
China) and centrifuged at a 3,000 × g for 30 min at room
temperature. The white intermediate segment was then collected and
washed using PBS. Collected cells were centrifuged at 500 × g for 5
min at room temperature and resuspended with EBM-2 medium
containing 100 U/ml penicillin, 100 U/ml streptomycin and 2%
FBS.
Isolated EPCs were then cultured with EGM-2 and
incubated in a humidified atmosphere containing 5% CO2
at 37°C. The medium was replaced every 3 days. On day 14, cells
were digested with 0.04% collagenase type I (Sigma-Aldrich; Merck
KGaA) and resuspended in PBS. Following an incubation with FcR
blocking reagent (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany)
on ice for 10 min, cells were then stained for 30 min at 4°C with
antibodies against CD34, KDR, CD45, CD133 and CD146 (all 1:200),
then cells were washed three times with 0.01 M PBS for 2 min each
in the dark. To verify positive cells using BD FACSCalibur™ flow
cytometer (BD Biosciences), the results were analyzed using FlowJo
software (version 3.2; Treestar, Inc., Ashland, OR, USA).
Cell viability
EPCs were seeded into a 96-well culture plate at a
density of 5×104 cells/well and treated with 0, 50, 100
and 150 ng/ml TWEAK (cat. no. ab184591; Abcam, Cambridge, MA, USA)
for 24 h at 37°C in incubator. Prior to TWEAK treatment, EPCs were
either transfected with 2 pmol Fn14 siRNA or 2 pmol scramble siRNA
using Lipofectamine® RNAiMAX reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol
for 3 days, and treated with 5 µM Bay 11–7082 (cat. no. S2913;
Selleck Chemicals, Shanghai, China) for 1 h at 37°C or left
untreated. MTT (1 mg/ml, dissolved in PBS solution; 100 µl/well;
Sigma-Aldrich; Merck KGaA) was then added for 4 h at 37°C, followed
by the addition of 100 µl DMSO. The absorbance was determined using
a multiplate reader at a wavelength of 570 nm.
AMI mouse model
The AMI murine model was prepared as previously
described (17). Mice were
anesthetized using intraperitoneal injection of 300 mg/kg chloral
hydrate (18), which was approved by
the Ethics Committee of Zhongda Hospital, Medical School of
Southeast University and the loss of righting reflex was monitored
to ensure that the all mice were fully anesthetized in the
experiments. Mice were artificially ventilated using a
volume-regulated respirator. Hearts were exposed via left
thoracotomy and the left coronary artery was ligated between the
pulmonary artery conus and the left atrium using an 8-0 prolene
suture. Following 30 min, 74 surviving mice were randomly divided
into four groups [AMI, EPCs treatment (1×106 cells in 30
µl PBS), TWEAK pretreated EPCs and sham; n=8/group] or six groups
(AMI, EPC treatment, TWEAK pretreated EPC group, TWEAK pretreated
Fn14 siRNA EPC, TWEAK pretreated Bay 11–7082 EPC and sham;
n=10/group). Mice in the sham group underwent thoracotomy but not
ligation. A total of 30 healthy mice and 30 AMI mice were
sacrificed to detect the content of TWEAK in serum. EPCs
(1×106/30 µl) labeled with DiD dye (cat. no. V22887;
1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.) for 1 h at
room temperature and washed with 0.01 M PBS two times. The cells
were intramyocardially injected into mice to assess the adherence
of EPCs to injured heart tissue. At 30 min following coronary
ligation, 1×106 cells/30 µl in PBS or PBS alone were
administered into the myocardium.
Echocardiography
Transthoracic two-dimensional M mode echocardiograms
and pulsed wave Doppler spectral tracings were produced using a
Toshiba Aplio 80 Imaging System (Toshiba Medical Systems
Corporation, Tochigi, Japan) equipped with a 12 MHz linear
transducer. Echocardiographic studies were performed on mice with a
maintained body temperature of 37°C. M-mode tracings were used to
measure left ventricle (LV) wall thickness, end-systolic dimensions
(ESD) and end-diastolic dimensions (EDD).
Tube formation assay
A 24-well culture plate was placed on ice and 0.289
ml/well chilled Matrigel Matrix (10 mg/ml; BD Biosciences) was
added. The plate was then incubated at 37°C for 30 min and the
remaining matrix was removed. A total of 300 µl cell suspension
(1×105 cells) was added to each well and incubated at
37°C for 24 h.
Transwell assay
EGM-2 medium containing 10% FBS (0.6 ml) was added
to the lower compartment of a Transwell plate with an 8 µm pore
insert. A total of 5×104 cells in EGM-2 serum free
medium were added to the upper compartment and incubated at 37°C
for 12 h. Cells in the lower chamber were fixed with 4%
paraformaldehyde at room temperature for 10 min and stained with 1%
crystal violet in 2% ethanol at room temperature for 20 min. The
number of cells in the lower chamber were counted under a light
microscope (magnification, ×20).
Histology
A histological examination was performed on three
samples of mouse heart from the same position under a light
microscope (magnification, ×20). Samples were fixed with 10%
formalin at room temperature overnight, embedded in paraffin and
sliced into 4 µm thick sections. Slides were processed using a
GTVisinTM anti-mouse/anti-rabbit immunohistochemical analysis kit
according to the manufacturer's protocol.
Masson's trichrome staining
Masson's trichrome staining for 20 min was performed
at room temperature using the aforementioned kit to distinguish
collagen fibers from muscular tissues. Blue staining indicated a
positive result. Staining was performed according to the
manufacturer's protocol.
Western blotting
EPCs were collected following various treatments.
Whole protein was extracted from whole cell lysate using RIPA
buffer and the concentration was measured using a BCA assay. The
protein (20 µg) was separated by 12.5% SDS-PAGE. Proteins were
transferred to polyvinylidene membranes (EMD Millipore, Billerica,
MA, USA) at 300 mA for 90 min. The membranes were then blocked at
room temperature for 1 h with TBST containing 0.1% Tween-20 and 5%
dry milk and incubated overnight with primary antibodies against
p65, phospho-p65, CD31, α-SMA (all 1:1,000) and GAPDH (1:2,000) at
4°C. Membranes were washed in triplicate with TBST and incubated
for 2 h with horseradish peroxidase-conjugated goat anti-rabbit
antibody (1:2,000) at room temperature. The optical densities of
antibody-specific bands were analyzed using a Luminescent Image
Analyzer (Protein Simple, San Jose, CA, USA) and ImageJ software
(version 1.37 for Windows; National Institutes of Health, Bethesda,
MD, USA).
Immunofluorescence
Cells (5×104/ml) cultured with 2 µg/ml
UEA-1 Lectin and 5 µg/ml Dil-acLDL at room temperature for 30 min,
washed three times with PBS and fixed with 4% paraformaldehyde for
30 min. The cells were observed using a confocal microscope
(magnification, ×40) to verify that they were EPCs. DiD-labelled
EPCs (1×106) were injected into AMI mice. After 14 days,
the mice were sacrificed by cervical dislocation. Then, heart
slices were fixed with 4% paraformaldehyde at room temperature
overnight, permeabilized with 0.3% Triton X-100 for 30 min at room
temperature and sliced into 4-µm-thick sections. Following blocking
with 3% bovine serum albumin for 1 h at room temperature, the
sections were incubated with antibodies against CD31 and α-SMA
(1:1,000) at room temperature for 2 h. Slides were washed three
times with PBS and incubated with Alexa Fluor 488-conjugated
secondary antibodies (1:1,000) for 1 h at room temperature. Nuclei
were stained with DAPI (10 µg/ml) for 5 min at room temperature.
Images were acquired using confocal microscopy (magnification,
×40).
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR) analysis
RNA was removed from TWEAK-treated EPCs using 1 ml
of TRIzol and the cDNA was generated using PrimeScript™
RT Master Mix (Takara, Kyoto, Japan) for 15 min at 37°C. The
reaction was terminated by heating the samples at 85°C for 5 sec.
The specific primers of VEGFA (sense 5′-AAAGGCTTCAGTGTGGTCTGAGAG-3′
and antisense 5′-GGTTGGAACCGGCATCTTTATC-3′) and GAPDH (sense
5′-CGACTTCAACAGCAACTCCCACTCTTCC-3′ and antisense
5′-TGGGTGGTCCAGGGTTTCTTACTCCTT-3′; both Shenggong, Shanghai, China)
were mixed with 100 ng cDNA and qPCR was performed using the DyNAmo
SYBR Green 2-step RT-qPCR kit (cat. no. F430L; Finnzymes; Thermo
Fisher Scientific, Inc.). The experimental protocol was as follows:
95°C for 10 min (denaturation), 35 cycles of 95°C for 15 sec, 60°C
for 10 sec, 72°C for 45 sec (amplification and quantification
program), a melting curve program consisting of 15 sec at 95°C
(denaturation), 30 sec at 55°C (annealing) and a melting and
continuous measuring step at 0.5°C/sec up to 85°C, and finally a
cooling step at 40°C. Data collection was performed using the ABI
PRISM 7000 Sequence Detection System (Applied Biosystems; Thermo
Fisher Scientific, Inc.). GAPDH was used as an internal control.
The 2−ΔΔCq method was applied to analyze the relative
changes in VEGFA gene expression (19).
Statistical analysis
Experimental data are expressed as the mean ±
standard deviation of at least three independent experiments.
Statistical analyses were performed using unpaired Student's t-test
or one-way analysis of variance followed by Tukey's test, and SPSS
18.0 statistical software (SPSS, Inc., Chicago, IL, USA). P<0.05
was used to indicate a statistically significant difference.
Results
TWEAK expression in patients and mice
with AMI
It has been demonstrated that TWEAK is upregulated
in tissue injury (20). A
retrospective analysis was performed on blood samples obtained from
25 patients with AMI and 25 healthy volunteers, as well as mice
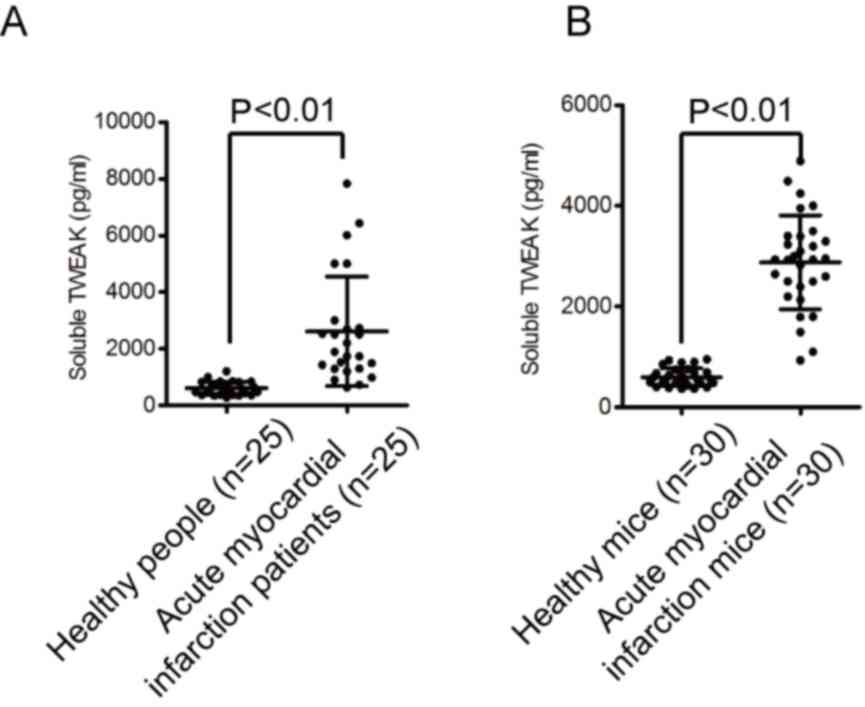
with experimentally induced AMI. As presented in Fig. 1A and B, TWEAK was significantly
upregulated in patients and mice with AMI compared with their
respective healthy controls.
Characterization of cultured EPCs
Murine EPCs derived from the bone marrow were
collected using lymphocyte separation medium. EPCs were verified
using UEA-1 Lectin (early EPCs marker) and Dil-acLDL (late EPCs
marker) double staining, as presented in Fig. 2A. EPCs were further verified using
flow cytometry. It was demonstrated that EPCs express CD34, KDR and
CD146, but not CD45 and CD133 (Fig.
2B). In order to identify the function of EPCs in AMI,
DiD-stained EPCs were injected into mice with AMI. The results
revealed that EPCs can migrate to damaged tissues, and
differentiate into veins (indicated by CD31 expression) and
arteries (indicated by α-SMA expression; Fig. 2C).
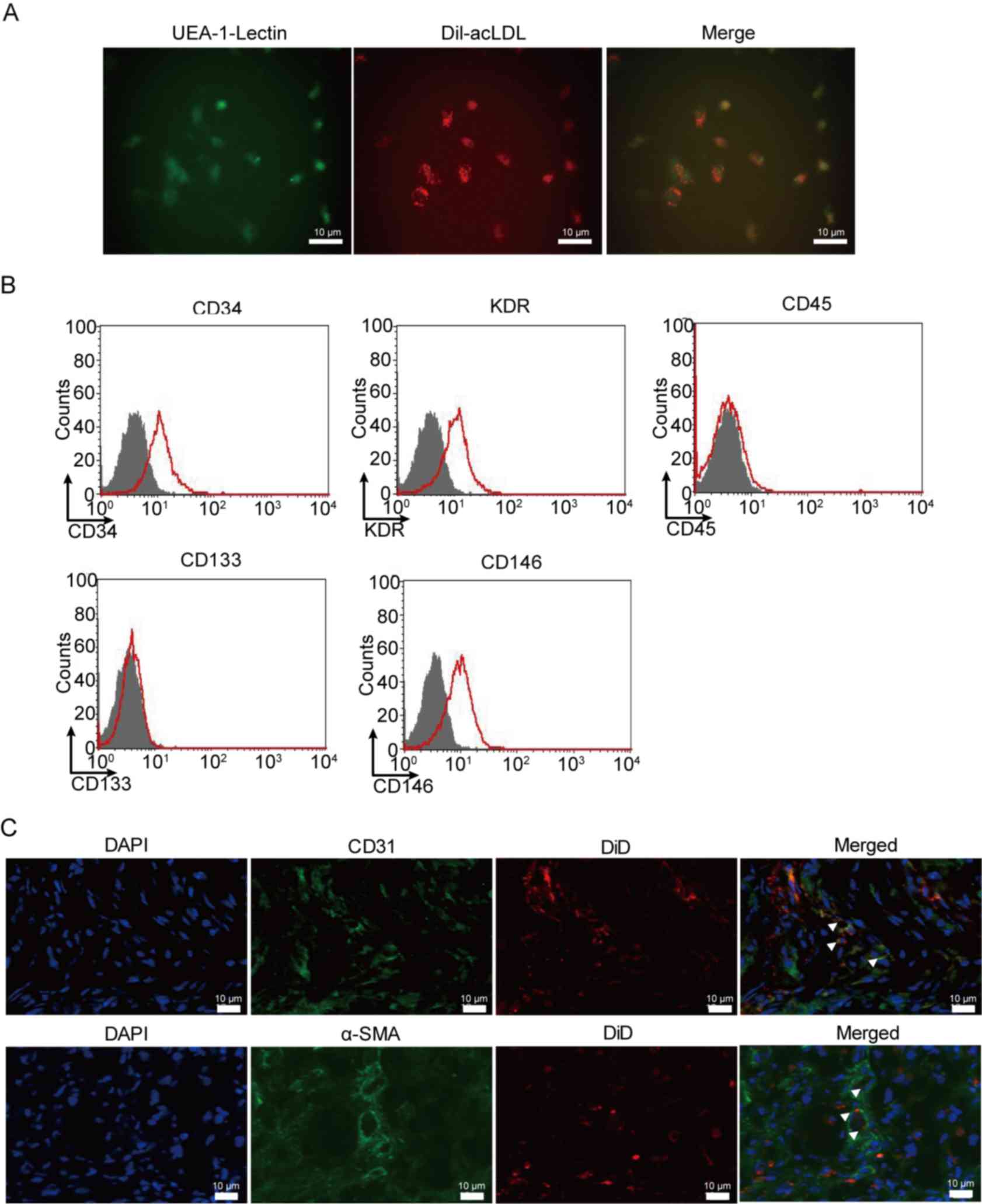 | Figure 2.Phenotypic characterization of
cultured EPCs. EPCs exhibited a change toward mesenchymal
transformation following transplantation in AMI mice. (A) UEA-1
lectin binding (green) and DiI-acLDL molecular probe uptake (red)
were evaluated in early and late EPCs to confirm culture using
photomicrographs. (B) EPCs expressed CD34, KDR and CD146, but were
negative for CD45 and CD133, as assessed using flow cytometry. (C)
Immunofluorescent staining was performed with antibodies against
CD31 and α-SMA to detect the differentiation of EPCs labeled with
DiD in veins and arteries following transplantation in AMI mice.
White triangles indicate the differentiated EPCs. EPCs, EPC,
endothelial progenitor cells; AMI, acute myocardial infarction;
UEA-1, ulex europaeus agglutinin-1; CD, cluster of differentiation;
KDR, kinase domain receptor; SMA, smooth muscle actin. |
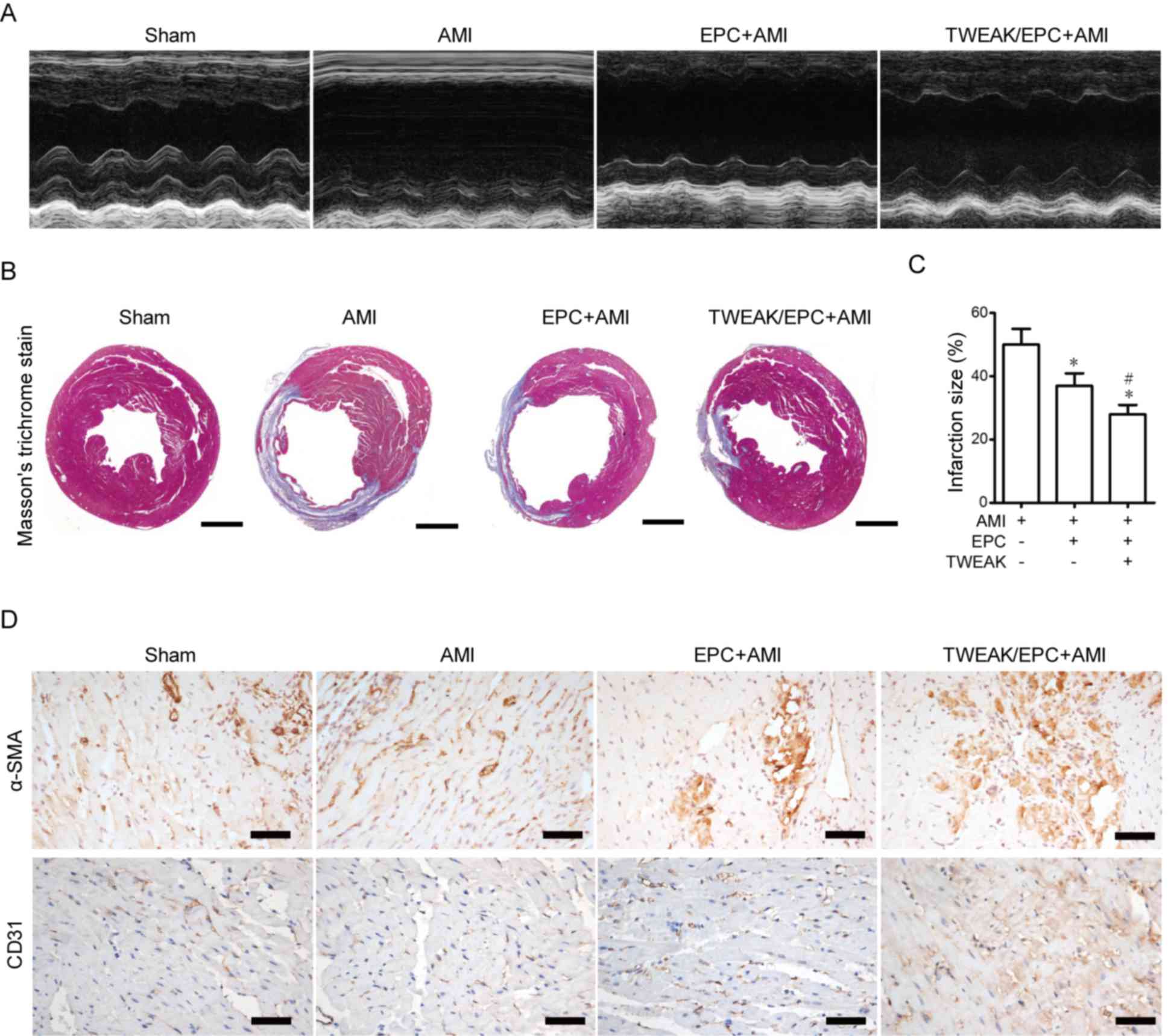
EPCs pre-incubated with TWEAK improve
cardiac function, alleviate AMI and promote vasculogenesis in
murine hearts
EPCs were preincubated with 100 ng/ml TWEAK for 24 h
and injected into the myocardium of mice with AMI to assess whether
TWEAK affects EPCs in AMI. M-mode images indicated that the LV
cavity was dilated in AMI, however this effect was markedly reduced
by treatment with TWEAK-treated EPCs compared with untreated EPCs
(Fig. 3A). The effect of EPCs
pre-treated with TWEAK on various physiological parameters and
cardiac functions in AMI mice are presented in Table I. In contrast to AMI mice, EPC
treatment decreased heart weight/body weight, left ventricular
internal diameter at end-diastole, left ventricular internal
diameter at end-systole (LVIDs), and increased left ventricular
ejection fraction (LVEF) and left ventricular fractional shortening
(LVFS) in AMI mice. Compared with the EPC+AMI group, EPCs
pre-treated with TWEAK were demonstrated to further increase LVEF
and LVFS, while LVIDs was reduced. Masson's trichrome staining
revealed that EPCs pre-treated with TWEAK downregulated collagen
synthesis and infarct size in the AMI and EPC+AMI groups (Fig. 3B and C). Although EPC treatment
appears to exacerbate the effect of AMI, the infarction decreased.
Capillary density in the infarct area was assessed using
immunohistochemical staining. A marked increase in angiogenesis was
observed in untreated and TWEAK-treated EPC groups as CD31 staining
increased; however, the degree of angiogenesis was greatest in
TWEAK pre-treated EPCs compared with the AMI and EPC+AMI groups.
Previous studies have indicated that EPCs may contribute to
arteriogenesis (21,22). The expression of a-SMA was used as a
measure of arteriogenesis. The results demonstrated that
arteriogenesis was greatest in EPCs pre-treated with TWEAK
(Fig. 3D). The results indicate that
TWEAK treatment improves cardiac function, decreases collagen
synthesis and facilitates EPC differentiation in AMI.
 | Table I.Effect of TWEAK-treated EPCs on
physiological parameters and cardiac functions at 14 days following
AMI induction. |
Table I.
Effect of TWEAK-treated EPCs on
physiological parameters and cardiac functions at 14 days following
AMI induction.
|
| Group |
|---|
|
|
|
|---|
| Variable | Sham | AMI | EPC+AMI | TWEAK/EPC+AMI |
|---|
| HW/BW (mg/g) | 5.43±0.21 |
8.12±0.33a |
7.12±0.36c |
6.62±0.46c |
| LVEF (%) | 80.37±0.54 |
27.32±0.23b |
35.48±1.54c |
65.73±0.52d,e |
| LVFS (%) | 50.32±0.82 |
13.15±0.95b |
18.19±0.71c |
25.33±0.22c,e |
| LVIDd (mm) | 3.17±0.60 |
6.17±0.26a |
5.21±0.32c |
4.12±0.51c |
| LVIDs (mm) | 1.55±0.18 |
4.65±0.27a | 3.38±0.25c |
2.60±0.25c,e |
TWEAK promotes migration, tube
formation, viability and VEGFA generation in EPCs
The effect of TWEAK treatment on EPC migration, tube
formation and cell viability was assessed in vitro.
Migrating cells were stained with crystal violet and counted using
a light microscope. As presented in Fig.
4A, the migration of EPCs was dependent on the concentration of
TWEAK. It was also demonstrated that TWEAK promotes tube formation
in a dose-dependent manner (Fig.
4B). Next, cell viability was assessed using an MTT assay. It
was demonstrated that TWEAK treatment increased the viability of
EPCs in a dose-dependent manner (Fig.
4C). To further assess vasculogenesis and tube formation,
levels of VEGFA mRNA and released VEGFA in the medium were detected
following TWEAK treatment. TWEAK was demonstrated to promote VEGFA
expression and increase the release of VEGFA into the medium in a
dose-dependent manner (Fig. 4D).
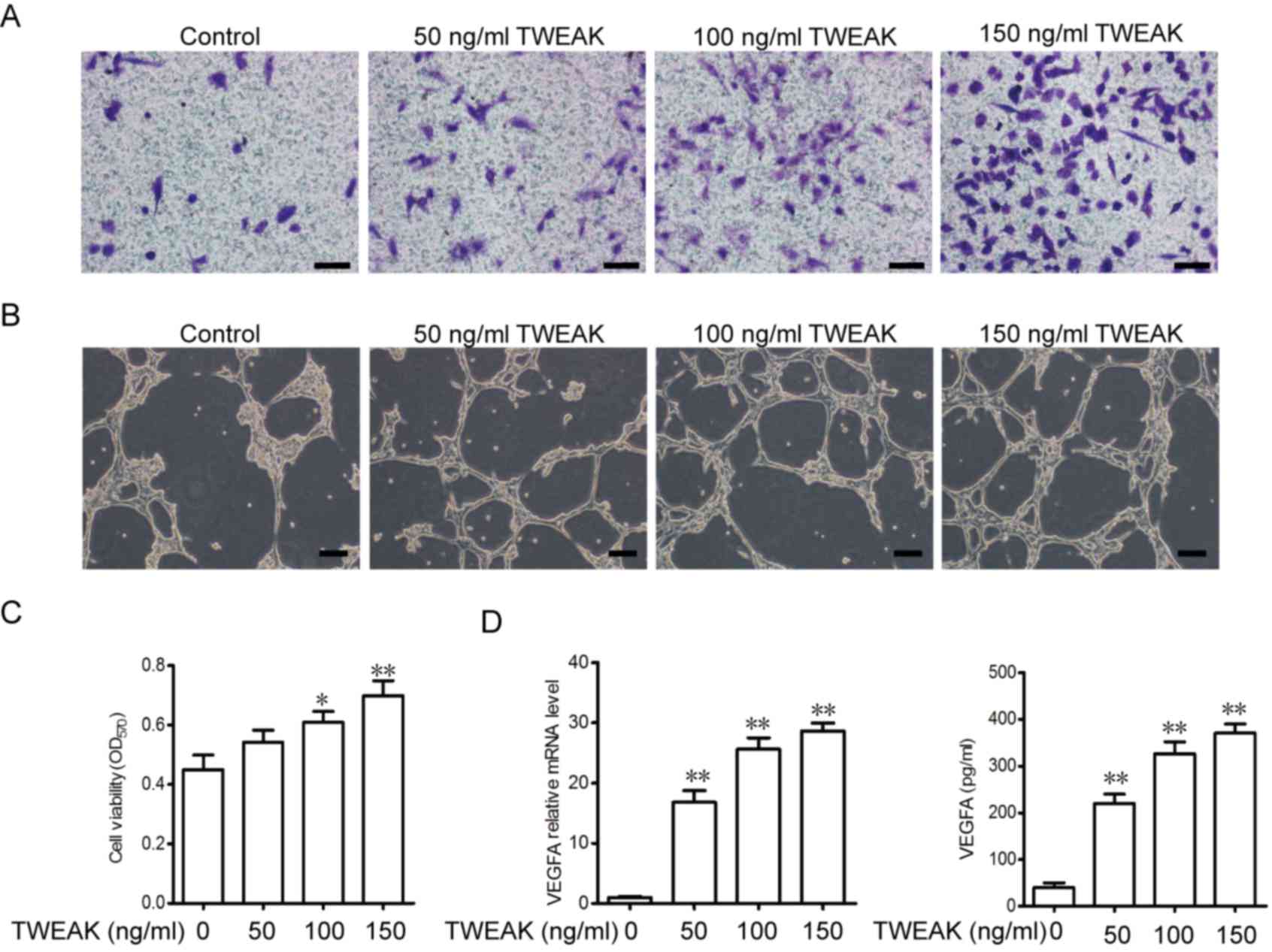 | Figure 4.TWEAK promotes EPC migration, tube
formation and viability, as well as the generation of VEGFA in
EPCs. (A) EPCs were treated with 50, 100 and 150 ng/ml TWEAK and
seeded (5×104/well) into Transwell plates for 12 h to
assess migration (scale bar, 50 µm). (B) A tube formation assay was
performed (scale bar, 100 µm). (C) The viability of EPCs treated
with 50, 100 and 150 ng/ml TWEAK was measured via an MTT assay. (D)
VEGFA mRNA levels and the concentration of VEGFA secreted into
medium following TWEAK treatment were measured using
reverse-transcription quantitative polymerase chain reaction and
ELISA, respectively. *P<0.05 and **P<0.01 vs. the control (0
ng/ml TWEAK) group. TWEAK, tumor necrosis factor-related weak
inducer of apoptosis; EPCs, EPC, endothelial progenitor cells;
VEGFA, vascular endothelial growth factor A. |
TWEAK promotes EPC migration, tube
formation and viability via the Fn14-NF-κB pathway
Fn14 siRNA and Bay 11–7082, an inhibitor of the
NF-κB pathway, were used to determine whether Fn14-NF-κB signaling
is associated with the mechanism of TWEAK-mediated EPC-induced
cardiac protection. As presented in Fig.
5A, Fn14-siRNA and Bay 11–7082-treated EPCs that received 100
ng/ml TWEAK for 24 h exhibited decreased migration and tube
formation when compared with TWEAK-treated EPCs group (Fig. 5B). However, compared with scramble
EPCs, Fn14 siRNA or Bay 11–7082-treated EPCs in the absence of
TWEAK demonstrated no marked difference in migration or tube
formation. Furthermore, the results demonstrated that
TWEAK-mediated EPC viability is inhibited by Fn14 depletion or
NF-κB inhibition (Fig. 5C). TWEAK
treatment alone also upregulated activated p65 and downstream
VEGFA; however, further Fn14-siRNA or Bay 11–7082 treatment
significantly decreased the expression of phosphorylated p65 and
VEGFA (Fig. 5D-F). The results
indicated that the TWEAK-Fn14-NF-κB pathway serves a role in EPC
migration, vasculogenesis and viability.
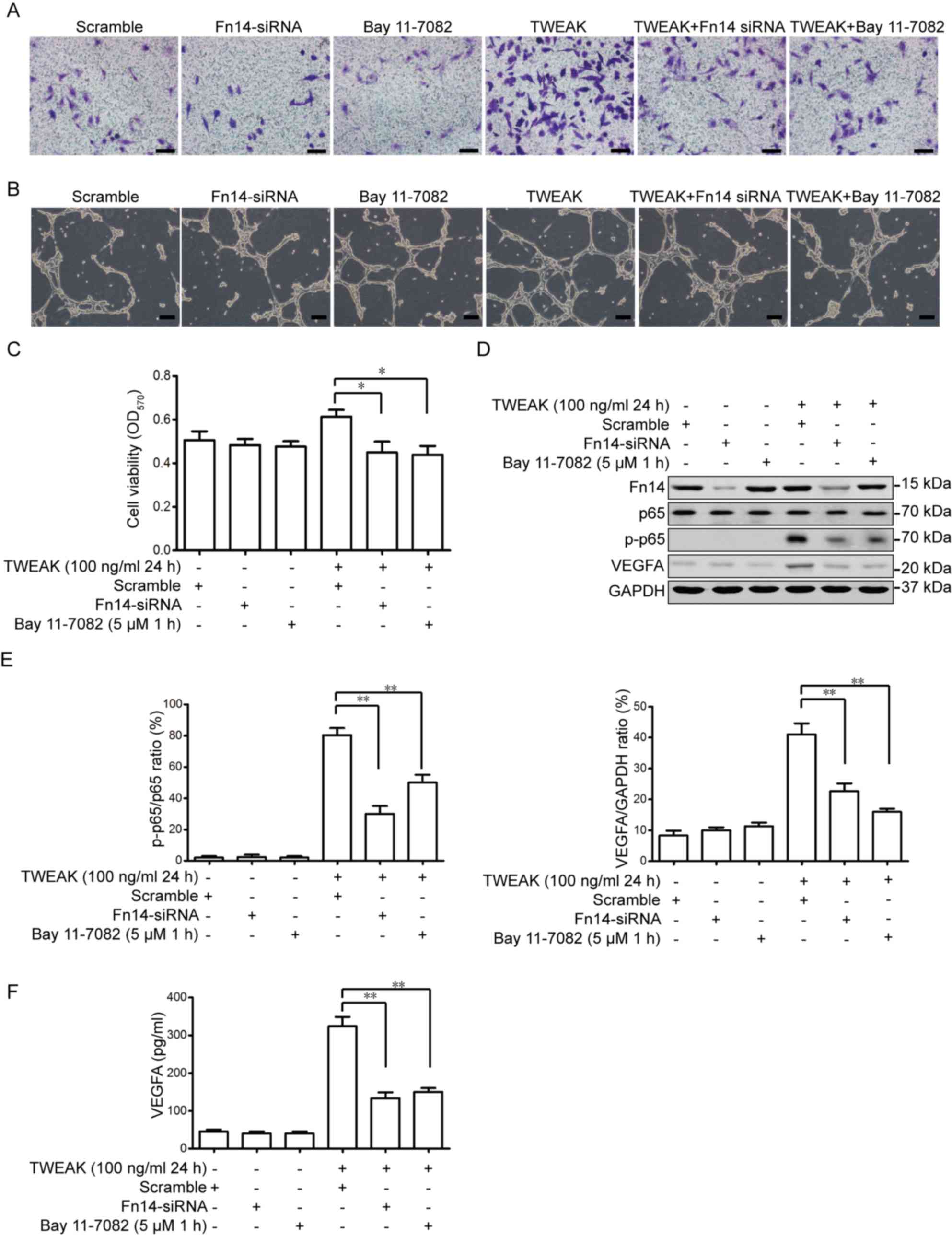 | Figure 5.TWEAK promotes EPC migration, tube
formation and viability via the Fn14-NF-κB pathway. EPCs were
pre-treated with Fn14-siRNA or Bay 11–7028 for 48 or 1 h,
respectively. (A) EPCs then received 100 ng/ml TWEAK treatment for
24 h and were seeded (5×104 cells/well) into Transwell
plates for 12 h (scale bar, 50 µm). (B) A tube formation assay was
performed (scale bar, 100 µm). (C) The viability of EPCs were
detected using an MTT assay. (D) The expression of Fn14, p65, p-p65
and VEGFA were measured using western blotting. (E) Quantitative
analysis of p-p65 and VEGFA was calculated from three independent
experiments. (F) Secreted VEGFA in the medium were assessed using
ELISA. *P<0.05 and **P<0.01 vs. the TWEAK+scramble group.
TWEAK, tumor necrosis factor-related weak inducer of apoptosis;
EPCs, EPC, endothelial progenitor cells; siRNA, small interfering
RNA; Fn14, fibroblast growth factor-inducible 14; p,
phosphorylated; VEGFA, vascular endothelial growth factor A. |
EPCs pretreated with TWEAK alleviate
AMI via the Fn14-NF-κB pathway
To further ascertain whether the protective effect
of TWEAK on EPC transplantation is mediated by the Fn14 and NF-κB
pathway, EPCs were treated with Fn14-siRNA and Bay 11–7028,
followed by TWEAK. M-mode images revealed that the anterior and
posterior walls of the heart were thinner in mice with AMI compared
with normal mice (Fig. 6A).
Furthermore, the LVEF and LVFS were significantly reduced following
the induction of MI (Fig. 6A). EPCs
pretreated with TWEAK for 24 h prior to transplantation were
demonstrated to protect cardiac function in mice with AMI via the
Fn14-NF-κB pathway. Fn14-siRNA and Bay 11–7028 treatment increased
the ESD and EDD, while the LVEF and LVFS were decreased (Fig. 6A). Inhibiting Fn14 or NF-κB signaling
alleviated the protective effect of TWEAK on a number of
physiological parameters and cardiac functions (Table II). Furthermore, Masson's trichrome
staining was enhanced in Fn14-siRNA and Bay 11–7028-treated EPCs of
AMI mice compared with TWEAK-pretreated EPCs of AMI mice (Fig. 6B). AMI was assessed via the
histological examination of infarct size and it was demonstrated
that Fn14 or NF-κB inhibition significantly alleviated the
protective effect of TWEAK-pretreated EPCs (Fig. 6C). Fn14 depletion or NF-κB pathway
inhibition in TWEAK-pretreated EPCs slightly decreased the
expression of α-SMA-positive (Fig.
6D) and CD31-positive (Fig. 6E)
microvessels in vivo compared with TWEAK-pretreated EPCs. As
such, the TWEAK mediated NF-κB pathway in EPCs has a protective
effect in mice with AMI.
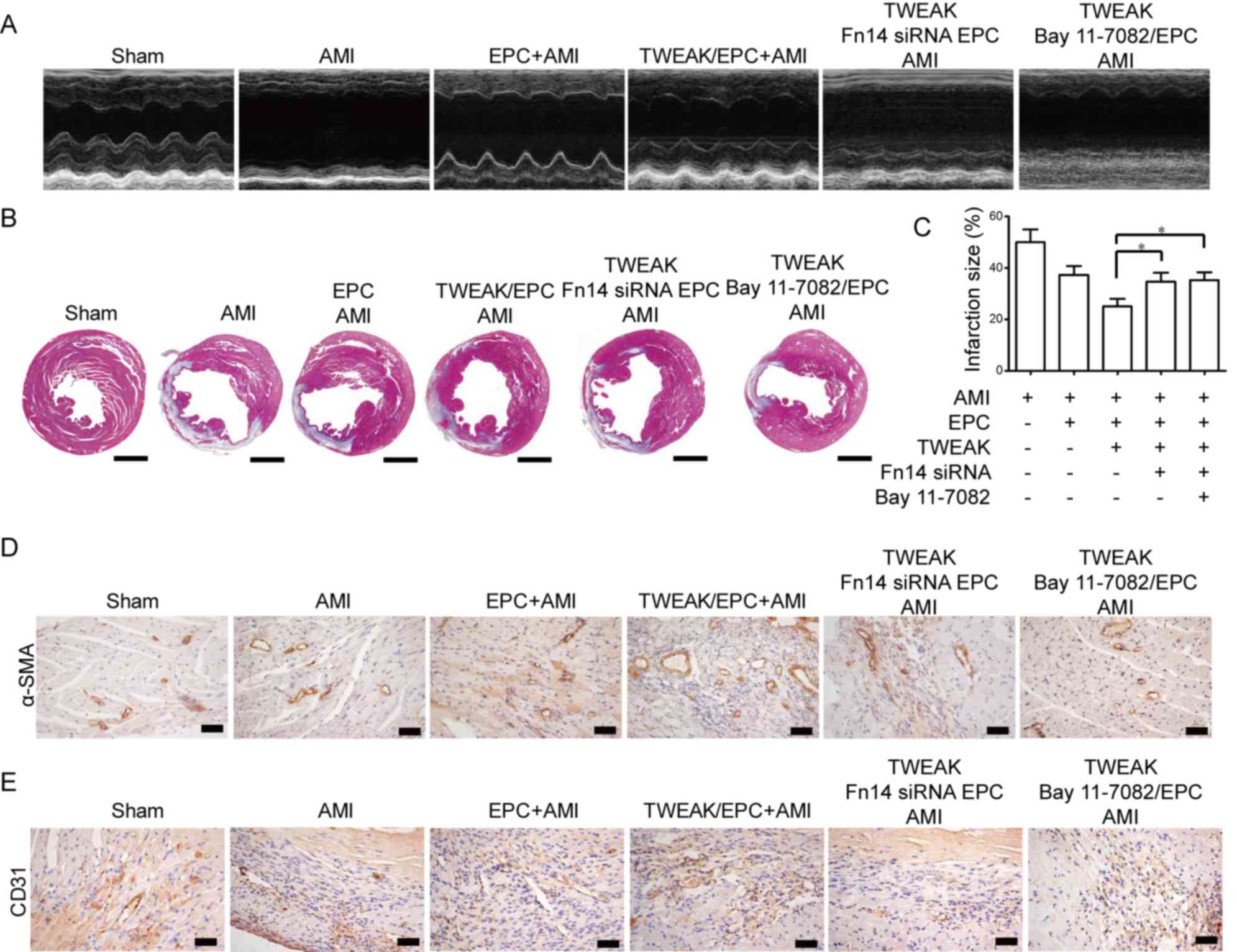 | Figure 6.EPCs pretreated with TWEAK alleviate
acute myocardial infarction via the Fn14 and NF-κB pathway.
Fn14-siRNA or Bay 11–7028 pretreated EPCs that subsequently
received TWEAK were transplanted into mice with AMI. (A)
Echocardiographic images of heart structure were then taken. (B)
Masson's trichrome staining (blue) was performed to discriminate
collagen fibers from murine heart tissue on histological slides
(scale bar, 0.2 cm). (C) Histological examination of murine AMI
size (each, n=6). *P<0.05. (D) α-SMA and (E) CD31 were measured
using immunohistochemistry in each group (scale bar, 50 µm). EPCs,
EPC, endothelial progenitor cells; TWEAK, tumor necrosis
factor-related weak inducer of apoptosis; Fn14, fibroblast growth
factor-inducible 14; siRNA, small interfering RNA; NF-κB, nuclear
factor-κB; AMI, acute myocardial infarction; SMA, smooth muscle;
CD, cluster of differentiation. |
| Table II.Role of Fn14 or NF-κB signaling in
TWEAK and its effect on physiological parameters and cardiac
function, 14 days following AMI induction. |
Table II.
Role of Fn14 or NF-κB signaling in
TWEAK and its effect on physiological parameters and cardiac
function, 14 days following AMI induction.
| Variable | Sham | AMI | EPC+AMI | TWEAK/EPC+AMI | TWEAK/Fn14 siRNA
EPC+AMI | TWEAK/Bay
11-7082/EPC+AMI |
|---|
| HW/BW (mg/g) | 5.37±0.73 | 7.64±0.73 | 6.81±0.38 | 6.21±0.84 | 6.93±0.78 | 7.14±0.64 |
| LVEF (%) | 79.32±0.42 | 31.02±0.38 | 40.21±0.91 | 65.33±0.84 | 43.21±0.43a |
44.29±0.91a |
| LVFS (%) | 48.13±0.42 | 12.25±0.55 | 21.27±0.37 | 31.48±0.42 | 20.28±0.78a |
19.32±0.58a |
| LVIDd (mm) | 3.28±0.43 | 6.86±0.32 | 5.13±0.57 | 4.01±0.32 | 5.23±0.89 | 5.31±0.38a |
| LVIDs (mm) | 1.41±0.17 | 5.27±0.38 | 4.01±0.46 | 2.51±0.47 |
4.32±0.78a | 4.21±0.58a |
Discussion
EPC transplantation may be a promising strategy for
the treatment of patients with AMI. It has been reported that EPCs
bind to UEA-1 and uptake acetylate low-density lipoprotein in
vivo after 4–7 days (23). A
number of hematopoietic cells have exhibited similar capabilities
(15). In the present study, the
expression of CD34, KDR, CD45, CD133 and CD146 was used to assess
EPCs. The results demonstrated that cultured EPCs migrate to
injured tissues and differentiate into blood vessels in
vivo. However, there are many additional factors that promote
EPC activation and differentiation, including cytokines, hypoxia,
exercise and Olmesartan (24,25).
VEGFA promotes vessel formation, while VEGFA and
stromal cell-derived factor (SDF-)1 released from injured vascular
tissues bind to the VEGF receptor (R) and C-X-C motif chemokine
receptor 4 (CXCR4) to recruit more EPCs (26). SDF-1 binds to CXCR4, activating
phosphoinositide 3-kinase (PI3K) and promoting the generation and
release of nitrous oxide (NO) via the Akt-mediated phosphorylation
of endothelial nitrous oxide synthase (eNOS) (27). VEGFA also promotes eNOS
phosphorylation, followed by NO production to promote EPC growth
and migration (28,29). When EPCs migrate to injured tissues,
adhesion molecules including P-selectin, E-selectin and
intracellular adhesion molecule 1 expressed on endothelial cells
bind to P-selectin glycoprotein ligand 1 and β2 integrins expressed
on EPCs to promote movement across the endothelium into the stroma
(30,31). In addition to VEGFR and CXCR4, other
factors and signaling pathways may be associated with the promotion
of EPC growth and migration.
TWEAK is a proinflammatory factor that is released
under pathological conditions and is upregulated in patients with
AMI (5). It was upregulated in AMI
patients. In order to assess the effect of TWEAK, EPCs were treated
with 100 ng/ml soluble TWEAK and transplanted into AMI mice. The
results demonstrated that EPC transplantation repairs injured
myocardial tissue, significantly improves cardiac function and
promotes the differentiation of EPCs to form vessels.
TWEAK binds to Fn14 and initiates downstream
signaling via the TNF receptor-associated factor (32). TWEAK/Fn14 promotes the nuclear
translocation of NF-κB and increases the downstream expression of
genes, including regulated upon activation normal T cell expressed
and secreted (RANTES) and monocyte chemoattractant protein-1
(33,34). Furthermore, TWEAK/FN14
signaling-mediated NF-κB pathway activation contributes to the
metastasis of prostate cancer (34).
However, it has been reported that TWEAK aggravates ventricular
damage following AMI by directly inhibiting oxidative
phosphorylation of peroxisome proliferator-activated receptor-γ
coactivator 1α and cardiomyocytes (35). TWEAK treatment has also been
demonstrated to accelerate collagen synthesis and heart fibroblast
proliferation via NF-κB activation (36). In renal tubule epithelial cells,
TWEAK activates extracellular signal related kinase (ERK), PI3K and
NF-κB signaling to accelerate their proliferation (37). Furthermore, ERK and PI3K inhibitors
block the proliferation of renal tubular epithelial and myocardial
cells (37). Inhibiting NF-κB
signaling also prevents TWEAK-induced proliferation of renal
tubular epithelial cells (37).
NF-κB signaling is also critical for EPCs. It has been demonstrated
that TGF-β-induced protein increases levels of notch ligands,
including Jagged-1 and delta-like protein 1, to facilitate EPC
differentiation and angiogenesis through activated NF-κB signaling
(38). NF-κB inhibition has been
demonstrated to decrease human endothelial cell growth and
survival, as well as inhibiting the migration of gastric cancer
cells (39,40). In addition, VEGFA generation is
regulated by NF-κB signaling to promote angiogenesis (41).
Given that Fn14 and NF-κB signaling serve roles in
EPCs, Fn14-siRNA and Bay 11–7028 were used to assess whether TWEAK
facilitates the viability and differentiation of EPC via these
pathways to alleviate AMI in mice. The results revealed that
inhibiting Fn14 and NF-κB signaling downregulates the migration and
tube formation of TWEAK pretreated EPCs in vitro.
Additionally, it was demonstrated that downregulating the Fn14 or
NF-κB pathways in TWEAK-pretreated EPCs promotes heart failure and
increase infarct size in vivo, therefore decreasing the
protective effect of EPCs. Downregulation of these pathways also
decreased the expression of α-SMA and CD31. However, whether other
signaling pathways serve a role in the regulation of TWEAK-mediated
vasculogenesis remains to be elucidated.
In conclusion, the results of the present study
demonstrated that the TWEAK/Fn14 mediated activation of NF-κB
signaling promotes EPC viability, migration and vasculogenesis
in vitro, as well as enhancing the protective effect of EPCs
on injured murine AMI heart tissue in vivo. Thus, the
present study indicated the beneficial effects and possible
mechanisms of TWEAK in EPCs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Nature Science Foundation of the People's Republic of China (grant
no. 81400225 for ZS; grant no. 81470401 for YY; grant no. 81400219
for ZC; grant no. 81500204 for XP) and the Jiangsu Provincial
Medical Youth Talent, Jiangsu, China (grant no. QNRC2016815).
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZLS conceived and designed the experiments. CWJ
wrote the manuscript. CWJ, BL, ZPC, XDP and GLY performed all of
the experiments. YRH analyzed the data. YYY and GSM contributed in
collecting clinical tissue samples.
Ethics approval and consent to
participate
The use of human samples was approved by the Ethics
Committee of Zhongda Hospital, Medical School of Southeast
University (Nanjing, China) and written informed consent was
obtained from all patients prior to blood collection. The use of
animals was approved by The Animal Care Committee of Southeast
University (Nanjing, China).
Patient consent for publication
Patients provided written informed consent for the
publication of any associated data from their blood samples.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Reed GW, Rossi JE and Cannon CP: Acute
myocardial infarction. Lancet. 389:197–210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Korhonen MJ, Robinson JG, Annis IE,
Hickson RP, Bell JS, Hartikainen J and Fang G: Adherence tradeoff
to multiple preventive therapies and all-cause mortality after
acute myocardial infarction. J Am Coll Cardiol. 70:1543–1554. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nahrendorf M, Pittet MJ and Swirski FK:
Monocytes: Protagonists of infarct inflammation and repair after
myocardial infarction. Circulation. 121:2437–2445. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cahill TJ, Choudhury RP and Riley PR:
Heart regeneration and repair after myocardial infarction:
Translational opportunities for novel therapeutics. Nat Rev Drug
Discov. 16:699–717. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bossen C, Ingold K, Tardivel A, Bodmer JL,
Gaide O, Hertig S, Ambrose C, Tschopp J and Schneider P:
Interactions of tumor necrosis factor (TNF) and TNF receptor family
members in the mouse and human. J Biol Chem. 281:13964–13971. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Urbich C and Dimmeler S: Endothelial
progenitor cells: Characterization and role in vascular biology.
Circ Res. 95:343–353. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tompkins BA, Natsumeda M, Balkan W and
Hare JM: What is the future of cell-based therapy for acute
myocardial infarction. Circ Res. 120:252–255. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Winkles JA: The TWEAK-Fn14
cytokine-receptor axis: Discovery, biology and therapeutic
targeting. Nat Rev Drug Discov. 7:411–425. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Burkly LC: TWEAK/Fn14 axis: The current
paradigm of tissue injury-inducible function in the midst of
complexities. Semin Immunol. 26:229–236. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roos A, Dhruv HD, Mathews IT, Inge LJ,
Tuncali S, Hartman LK, Chow D, Millard N, Yin HH, Kloss J, et al:
Identification of aurintricarboxylic acid as a selective inhibitor
of the TWEAK-Fn14 signaling pathway in glioblastoma cells.
Oncotarget. 8:12234–12246. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu G, Zeng W and Xia Y: TWEAK/Fn14
signaling in tumors. Tumour Biol. 39:10104283177146242017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Di Martino L, Dave M, Menghini P, Xin W,
Arseneau KO, Pizarro TT and Cominelli F: Protective role for
TWEAK/Fn14 in regulating acute intestinal inflammation and
colitis-associated tumorigenesis. Cancer Res. 76:6533–6542. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chorianopoulos E, Jarr K, Steen H,
Giannitsis E, Frey N and Katus HA: Soluble TWEAK is markedly
upregulated in patients with ST-elevation myocardial infarction and
related to an adverse short-term outcome. Atherosclerosis.
211:322–326. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mustonen E, Säkkinen H, Tokola H,
Isopoussu E, Aro J, Leskinen H, Ruskoaho H and Rysä J: Tumour
necrosis factor-like weak inducer of apoptosis (TWEAK) and its
receptor Fn14 during cardiac remodelling in rats. Acta Physiol
(Oxf). 199:11–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Garikipati VNS and Kishore R: Endothelial
progenitor cells: procedure for cell isolation and applications.
Methods Mol Biol. 1553:85–89. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ingram DA, Mead LE, Tanaka H, Meade V,
Fenoglio A, Mortell K, Pollok K, Ferkowicz MJ, Gilley D and Yoder
MC: Identification of a novel hierarchy of endothelial progenitor
cells using human peripheral and umbilical cord blood. Blood.
104:2752–2760. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nagaya N, Nishikimi T, Yoshihara F, Horio
T, Morimoto A and Kangawa K: Cardiac adrenomedullin gene expression
and peptide accumulation after acute myocardial infarction in rats.
Am J Physiol Regul Integr Comp Physiol. 278:R1019–R1026. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liao ZJ, Liang RS, Shi SS, Wang CH and
Yang WZ: Effect of baicalin on hippocampal damage in kainic
acid-induced epileptic mice. Exp Ther Med. 12:1405–1411. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grimholt RM, Urdal P, Klingenberg O and
Piehler AP: Rapid and reliable detection of α-globin copy number
variations by quantitative real-time PCR. BMC Hematol. 14:42014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Devarapu SK, Grill JF, Xie J, Weidenbusch
M, Honarpisheh M, Vielhauer V, Anders HJ and Mulay SR: Tumor
necrosis factor superfamily ligand mRNA expression profiles differ
between humans and mice during homeostasis and between various
murine kidney injuries. J Biomed Sci. 24:772017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pelliccia F, Cianfrocca C, Rosano G,
Mercuro G, Speciale G and Pasceri V: Role of endothelial progenitor
cells in restenosis and progression of coronary atherosclerosis
after percutaneous coronary intervention: a prospective study. JACC
Cardiovasc Interv. 3:78–86. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alobaid N, Alnaeb ME, Sales KM, Seifalian
AM, Mikhailidis DP and Hamilton G: Endothelial progenitor cells and
their potential clinical applications in peripheral arterial
disease. Endothelium. 12:243–250. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Du F, Zhou J, Gong R, Huang X, Pansuria M,
Virtue A, Li X, Wang H and Yang XF: Endothelial progenitor cells in
atherosclerosis. Front Biosci (Landmark Ed). 17:2327–2349. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schier R, El-Zein R, Cortes A, Liu M,
Collins M, Rafat N, Teschendorf P, Wu HK, Heymach J, Mehran R and
Riedel B: Endothelial progenitor cell mobilization by preoperative
exercise: A bone marrow response associated with postoperative
outcome. Br J Anaesth. 113:652–660. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gong X, Shao L, Fu YM and Zou Y: Effects
of olmesartan on endothelial progenitor cell mobilization and
function in carotid atherosclerosis. Med Sci Monit. 21:1189–1193.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu L, Duda DG, di Tomaso E, Ancukiewicz M,
Chung DC, Lauwers GY, Samuel R, Shellito P, Czito BG, Lin PC, et
al: Direct evidence that bevacizumab, an anti-VEGF antibody,
up-regulates SDF1α, CXCR4, CXCL6 and neuropilin 1 in tumors from
patients with rectal cancer. Cancer Res. 69:7905–7910. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cho BS, Zeng Z, Mu H, Wang Z, Konoplev S,
McQueen T, Protopopova M, Cortes J, Marszalek JR, Peng SB, et al:
Antileukemia activity of the novel peptidic CXCR4 antagonist
LY2510924 as monotherapy and in combination with chemotherapy.
Blood. 126:222–232. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stellos K and Gawaz M: Platelets and
stromal cell-derived factor-1 in progenitor cell recruitment. Semin
Thromb Hemost. 33:159–164. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tilling L, Chowienczyk P and Clapp B:
Progenitors in motion: Mechanisms of mobilization of endothelial
progenitor cells. Br J Clin Pharmacol. 68:484–492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oh IY, Yoon CH, Hur J, Kim JH, Kim TY, Lee
CS, Park KW, Chae IH, Oh BH, Park YB and Kim HS: Involvement of
E-selectin in recruitment of endothelial progenitor cells and
angiogenesis in ischemic muscle. Blood. 110:3891–3899. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chavakis E, Aicher A, Heeschen C, Sasaki
K, Kaiser R, El Makhfi N, Urbich C, Peters T, Scharffetter-Kochanek
K, Zeiher AM, et al: Role of beta2-integrins for homing and
neovascularization capacity of endothelial progenitor cells. J Exp
Med. 201:63–72. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Burkly LC: Regulation of tissue responses:
The TWEAK/Fn14 pathway and other TNF/TNFR superfamily members that
activate non-canonical NFκB signaling. Front Immunol. 6:922015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chorianopoulos E, Heger T, Lutz M, Frank
D, Bea F, Katus HA and Frey N: FGF-inducible 14-kDa protein (Fn14)
is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and
mediates nuclear factor-kappaB activation by TWEAK. Basic Res
Cardiol. 105:301–313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yin J, Liu YN, Tillman H, Barrett B,
Hewitt S, Ylaya K, Fang L, Lake R, Corey E, Morrissey C, et al:
AR-regulated TWEAK-FN14 pathway promotes prostate cancer bone
metastasis. Cancer Res. 74:4306–4317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jarr KU, Eschricht S, Burkly LC, Preusch
M, Katus HA, Frey N and Chorianopoulos E: TNF-like weak inducer of
apoptosis aggravates left ventricular dysfunction after myocardial
infarction in mice. Mediators Inflamm. 2014:1319502014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen HN, Wang DJ, Ren MY, Wang QL and Sui
SJ: TWEAK/Fn14 promotes the proliferation and collagen synthesis of
rat cardiac fibroblasts via the NF-κB pathway. Mol Biol Rep.
39:8231–8241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sanz AB, Sanchez-Niño MD, Izquierdo MC,
Jakubowski A, Justo P, Blanco-Colio LM, Ruiz-Ortega M, Egido J and
Ortiz A: Tweak induces proliferation in renal tubular epithelium: A
role in uninephrectomy induced renal hyperplasia. J Cell Mol Med.
13:3329–3342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maeng YS, Choi YJ and Kim EK: TGFBIp
regulates differentiation of EPC
(CD133+c-kit+lin−cells) to EC
through activation of the Notch signaling pathway. Stem Cells.
33:2052–2062. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Singh RP, Dhanalakshmi S, Agarwal C and
Agarwal R: Silibinin strongly inhibits growth and survival of human
endothelial cells via cell cycle arrest and downregulation of
survivin, Akt and NF-κB: Implications for angioprevention and
antiangiogenic therapy. Oncogene. 24:11882005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Duan H, Chen L, Qu L, Yang H, Song SW, Han
Y, Ye M, Chen W, He X and Shou C: Mycoplasma hyorhinis infection
promotes NF-κB-dependent migration of gastric cancer cells. Cancer
Res. 74:5782–5794. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tirziu D, Jaba IM, Yu P, Larrivée B, Coon
BG, Cristofaro B, Zhuang ZW, Lanahan AA, Schwartz MA, Eichmann A
and Simons M: Endothelial nuclear factor-κB-dependent regulation of
arteriogenesis and branching. Circulation. 126:2589–2600. 2012.
View Article : Google Scholar : PubMed/NCBI
|