Introduction
Hypertension is one of the most important risk
factors for cardiovascular disease. Epidemiologic studies have
shown that patients with essential hypertension (EH) accounted for
90–95% of patients with hypertension (1). Although the etiology and pathogenesis
of EH has not been determined, factors such as genetics and
environment may have some impact on the occurrence and development
of EH (2). Among the factors, the
renin-angiotensin system (RAS) plays a crucial role in the
pathogenesis of EH (3). Renin is the
first rate-limiting enzyme in the RAS (4). Angiotensin II (Ang II) is a peptide
hormone that mediates vasoconstriction and increases blood pressure
(BP) by binding to the Ang II type 1 receptor (AT1R) and is
involved in the development of hypertension (5).
The extracellular calcium-sensing receptor (CaSR) is
a class C G protein-coupled receptor (GPCR), which is part of a
superfamily of seven transmembrane domain receptors, the primary
function of which is to regulate calcium homeostasis. The CaSR is
mainly expressed in the thyroid, parathyroid, kidneys, and bone
(6,7). There is also functional expression in
the vascular wall, including vascular smooth muscle and endothelial
cells (8–10). Increased extracellular
Ca2+ (Ca2+]o) induces binding to
the CaSR and activates the G-protein-phospholipase C (PLC)-inositol
1,4,5-trisphophate (IP3) receptor pathway, triggers an
elevation in intracellular Ca2+
([Ca2+]i), and is involved in the occurrence
and development of hypertension (6).
Studies on the relationship between calcium and BP have shown that
proper intake of Ca2+ can effectively lower BP (11). This conclusion has been confirmed in
animal experiments (12). Some
researchers have also shown that elevated Ca2+ leads to
vasodilation in vitro (13).
Unfortunately, despite these findings, the molecular mechanisms
underlying this association are not fully understood.
NPSR568 (R568), a calcimimetic, is an allosteric
agonist of the CaSR. The mechanism of action of R568 is to increase
the sensitivity of the CaSR to [Ca2+]o, and
shift the calcium concentration response curve to the left. It is
currently believed that the binding site of R568 to the CaSR is at
the 7th transmembrane region of the receptor. The CaSR may be
involved in BP regulation, and has become a current focus of
research. Ogata et al (14)
argued that R568 reduces BP in uremic rats and SHRs, but has no
effect on normotensive rats. Rybczynska et al (15,16)
reported that administration of NPS2143, an allosteric inhibitor of
the CaSR, increased BP in normotensive rats; however, in rats with
surgically removed parathyroid glands or an AT1R (e.g., losartan)
in the presence of a calcium channel blocker or antagonist, no
effect of elevated BP was observed. Atchison et al (11) and Ortiz-Capisano et al
(17) suggest that the CaSR is
expressed in juxtaglomerular cells, and that activation of the CaSR
activates the ryanodine receptor (RyR) via the PLC/IP3
pathway to increase [Ca2+]i and inhibit cAMP
formation, thereby inhibiting renin release; meanwhile, Maillard
et al (18) also proved that
the calcimimetic R568 can regulate renin release via CaSR. Our
previous research also proved that reduced expression of the CaSR
is associated with increased proliferation and remodeling of
vascular smooth muscle cells (VSMCs) and promotes the development
of EH via activation of the cAMP-RAS pathway (9).
The goal of this study was to determine the effect
of CaSR activation on BP in spontaneously hypertensive rats (SHRs)
and to partially elucidate the mechanism of action of the CaSR in
regulating BP from the perspective of the RAS. Thus, we
hypothesized that a R568-activated CaSR may lower BP and improve
thoracic aortic proliferation and remodeling through the RAS
pathway.
Materials and methods
Animals
For this study, 42 male SHRs and age-matched male
WKY (8-week old, 180–220 g, purchased from Vital River Laboratory
Animal Science and Technology Co., Ltd, Beijing. License Number:
SCXK2012-0001) were randomly divided into 4 groups: Group 1, WKY+NS
(n=21); group 2, WKY+R568 (n=21); group 3, SHR+NS (n=21); and group
4, SHR+R568 group (n=21). R568 was dissolved in normal saline (NS)
at a dose of 1.2 mg/kg/day in group 2/4, and was administered by
intraperitoneal (i.p.) injection. At R568 treatment at 0, 4, and 8
weeks, rats were equivalent to 8, 12, and 16 weeks of age. Seven
rats in each group were randomly selected and sacrificed to detect
the corresponding indexes. In group 1/3, the rats received NS i.p.,
and treated as mentioned above. All animals were housed at a
constant room temperature, humidity, and light cycle (alternating
12 h light/dark cycle), and had access to food and drinking water
ad libitum. All experimental procedures in this study were
approved by the Institutional Animal Research Committee of Shihezi
Medical University, and all animals received humane care in
compliance with the Guide for the Care and Use of Laboratory
Animals published by the National Institutes of Health (NIH
Publication 86–23, revised 1986).
Evaluation of EH
In the experiment, systolic blood pressure (SBP),
diastolic blood pressure (DBP), mean arterial pressure [MAP;
(MAP=DBP+1/3 {SBP-DBP})] were measured once a week. At the same
time of the week, the BP of rats was measured by tail-cuff method
[BP-98A-L; Softron, Tokyo, Japan] (19), the room temperature was kept at a
controlled temperature of 30°C, and repeated measurements were
taken 3 times and averaged. At 0, 4, and 8 weeks with this
treatment and after SBP, DBP, and MAP measurements, the rats were
anesthetized, blood was extracted from the abdominal aorta and
isolated plasma, then stored at −80°C for an enzyme-linked
immunosorbent assay (ELISA). Rats were sacrificed and the thoracic
aorta was isolated, 0.5 cm of the thoracic aorta was cut, placed in
10% neutral buffered formalin, and paraffin-embedded for
histopathologic evaluation (hematoxylin and eosin and Masson
staining) and immunohistochemistry analysis. The remaining part of
the thoracic aorta tissue were stored at −80°C for subsequent
protein extracted for western blotting and aorta homogenate for
ELISA assay.
Histologic evaluation
Thoracic aorta tissue was fixed in 10% formalin for
more than 48 h, then dehydrated, embedded, sectioned, and stained
with hematoxylin and eosin or Masson's trichrome. We then randomly
selected three fields in each slice, calculated the parameters of
the thoracic aorta vascular cross-sections by measuring the wall
thickness, cross-sectional vessel area and wall area, total vessel
wall thickness, and the percentages of medial wall thickness-to-the
external diameter (WT%), cross-sectional vessel wall area-to-the
total area (WA%), and collagen area-to-total vessel wall area
(CA%). We captured the thoracic aorta images with an Olympus BX40
microscope and analyzed using Image-Pro Plus 6.0 software (Media
Cybernetics Inc., Buckinghamshire, UK).
Immunohistochemistry analysis
Tissue sections were deparaffinized, washed, and
antigen was retrieved with 0.01 M sodium citrate buffer (pH=6.0) in
a pressure cooker. The sections were incubated with primary mouse
polyclonal antibodies against CaSR (1:50; Abcam, MA, USA), calponin
(1:600; Boster, Wuhan, China), smooth muscle actin α (α-SMA, 1:400;
Boster), proliferating cell nuclear antigen (PCNA, 1:200; Boster),
and primary rabbit monoclonal antibody against osteopontin (OPN,
1:100; Abcam) overnight at 4°C. Slides were washed three times in
PBS (pH 7.4–7.6) and incubated with the corresponding secondary
horseradish peroxidase-conjugated antibody (Invitrogen, Beijing,
China) for 30 min at 37°C. A diaminobenzidine/peroxidase substrate
was used to generate a brown signal. Negative controls were omitted
with primary antibodies. To quantify protein levels, positive
staining in tissue sections was further analyzed by Integral
Optical Density (IOD)/vascular wall area using Image-Pro Plus 6.0
software. The same microscope and camera set was used for all
images.
Western blot analysis
Total protein was extracted from the thoracic aorta
and quantified for protein concentration. The extracted protein was
subjected to sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred proteins to 0.45-mm
SequiBlot PVDF membranes. After incubation with 5% non-fat milk,
then incubated overnight at 4°C with primary antibodies against
CaSR (1:1,000; Abcam), PCNA (1:250; Boster), α-SMA (1:400; Boster),
calponin (1:300; Boster), OPN (1:1,000; Abcam), renin (1:250;
Bioss, Beijing, China), AT1R (1:1,000; Abcam,), and β-actin
(1:1,000; Boster). After being washed with TBST, the membranes were
incubated with fluorescence conjugated goat anti-mouse or
anti-rabbit IgGs (1:20,000; Boster) for approximately 2 h at room
temperature. Protein was visualized using an enhanced
chemiluminescence system (ECL, Pierce; Thermo Fisher Scientific,
Inc., Waltham, MA, USA USA). The intensity of protein bands was
quantified using Bio-Rad Quantity One software (Bio-Rad, Hercules,
CA, USA) and the levels of protein detected were normalized to
β-actin levels.
Enzyme-linked immunosorbent assay
An ELISA kit (Wuhan Elabscience Biotechnology,
Wuhan, China) was used to determine the levels of CaSR, cAMP,
renin, and Ang II from plasma and thoracic aortic tissue
homogenate, and the specific measurement method was according to
the manufacturer's instructions. A microplate reader was used to
read the absorbance at 450 nm (Bio-Rad Model 3550-UV).
Drugs
R-568 has a chiral carbon atom and acts
stereospecifically on CaSR. R568 was purchased from Tocris
Bioscience Co., Ltd. (Minneapolis, MN, USA). The hydrophobic
compounds were dissolved in normal saline at a dose of 1.2
mg/kg/day as a stock solution.
Statistical analysis
Quantitative data were expressed as the mean ± SEM.
For multiple experimental groups, we used one way analysis of
variance (ANOVA) or Kruskal-Wallis test, then a Bonferroni post hoc
test was used after ANOVA. For all analysis results, P<0.05 was
considered statistically significant. Statistical analysis was
performed using the SPSS 17.0 software (SPSS, Inc., Chicago, IL,
USA).
Results
Effects of R568 on SBP, DBP, and mean
arterial pressure (MAP) in SHRs
To investigate the effect of R568 on BP in SHRs, we
measured the changes in SBP, DBP, and MAP using the tail-cuff
method. Results showed that the BP of SHRs at different ages was
higher than age-matched WKY rats (P<0.05), and the BP of the
SHR16 group was significantly higher than the SHR8w group
(P<0.05; Fig. 1). During 8 weeks
of treatment, R568 reduced the SBP of SHRs at 16 weeks (P<0.05;
Fig 1A), while R568 had little
effect on the DBP and MAP (Fig. 1B and
C).
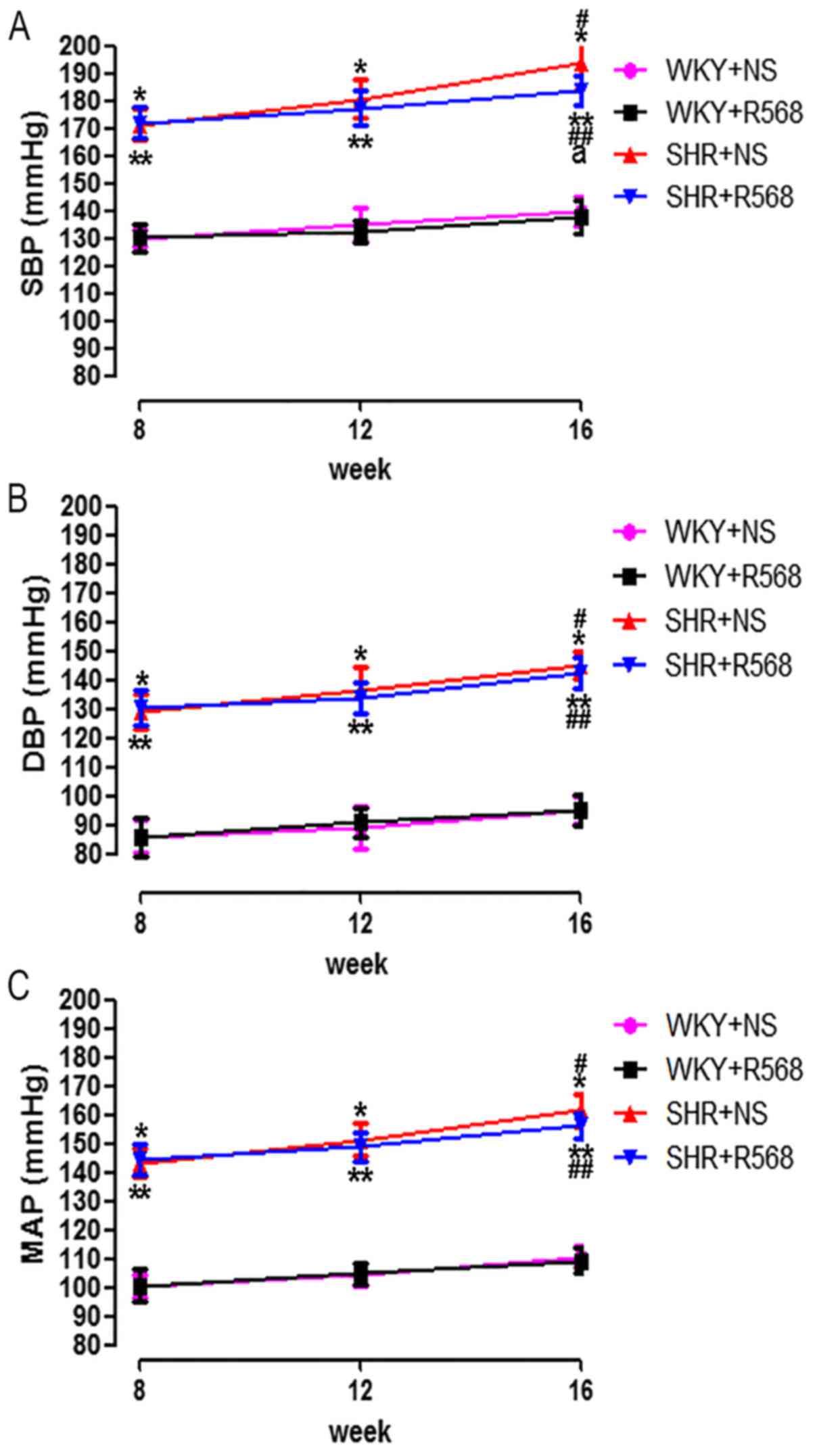 | Figure 1.Comparison of SBP, DBP, and MAP
between different groups of rats. (A) Systolic blood pressure; (B)
diastolic blood pressure; (C) mean arterial pressure. Data
are means ± standard deviation (n=7). SHR+NS groups vs. the
age-matched WKY+NS groups, *P<0.05; SHR16w+NS groups vs.
SHR8w+NS groups, #P<0.05; SHR+R568 groups vs. the
age-matched WKY+R568 groups, **P<0.05; SHR16w+R568 groups vs.
SHR8w+R568 groups, ##P<0.05; SHR+R568 groups vs.
SHR+NS groups, aP<0.05. WKY, Wistar Kyoto rats; SHR,
spontaneously hypertensive rats; NS, normal saline; R568, NPSR568.
SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP,
mean arterial pressure. |
R568 attenuates aortic media
remodeling in SHRs
The morphology of the thoracic aorta was determined
with HE and Masson staining (Fig.
2). Compared with WKY rats, thoracic aorta endothelial cells
were injured and the aortic media was thickened (increased WT% and
WA%, P<0.05; Fig. 2A, C and D),
and abnormal collagen accumulated (increase in CA%, P<0.05;
Fig. 2B and E) as the BP increased.
The above indicators in the SHR-16 group changes were more apparent
than SHR-8 group (P<0.05; Fig.
2). After R568 treatment, 16w SHRs can reduce thoracic aortic
proliferation and remodeling (P<0.05; Fig. 2).
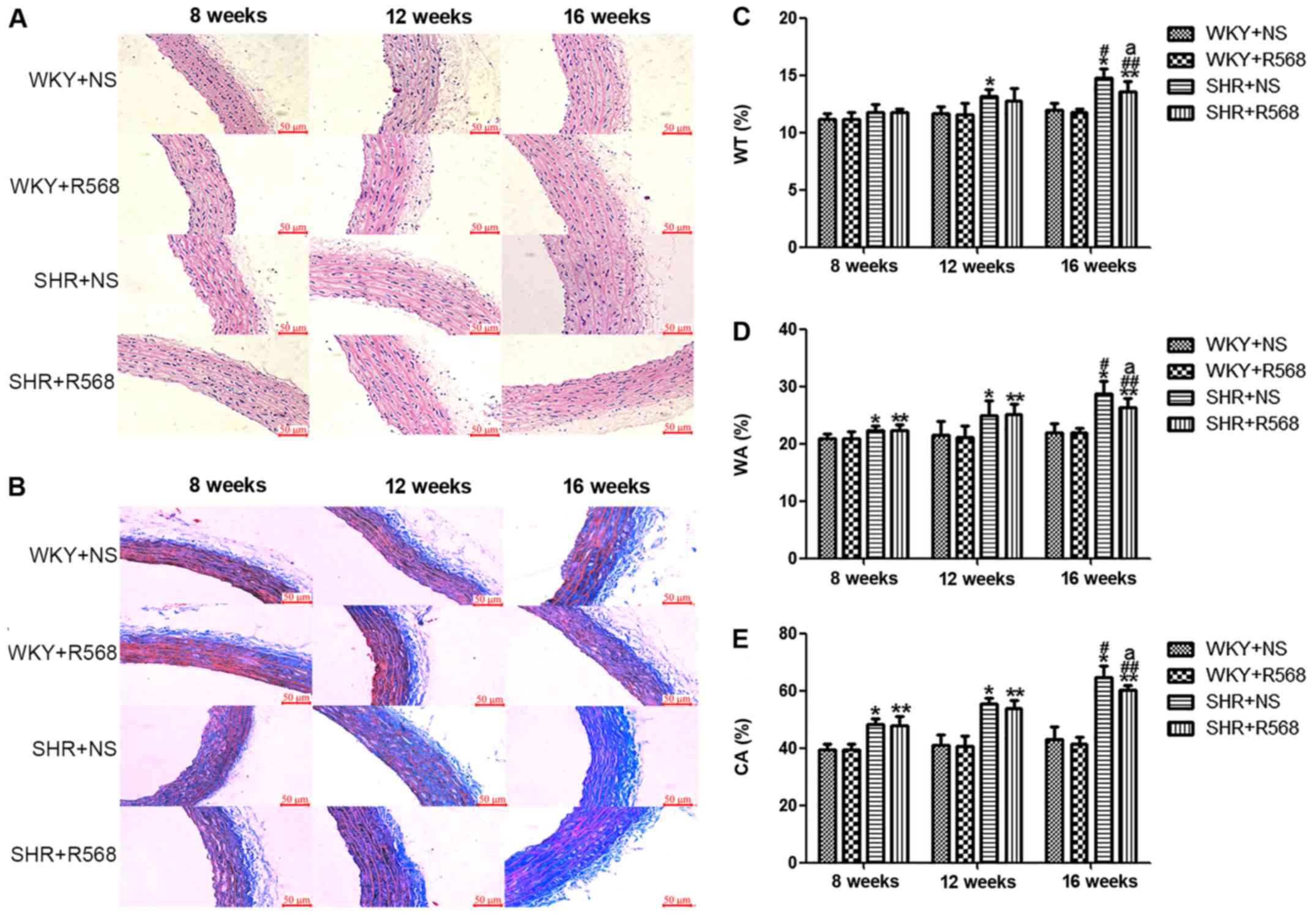 | Figure 2.Changes of proliferation and
remodeling indices in thoracic aortic smooth muscle of rats. (A) HE
staining. (B) Masson staining. (C) WT%, the wall thickness
percentage of the external diameter. (D) WA%, total vessel wall
area as a percentage of the total area (WA%). (E) CA%, total area
occupied by collagen as a percentage of the total vessel wall area.
Data are means ± standard deviation (n=7). SHR+NS groups vs. the
age-matched WKY+NS groups, *P<0.05; SHR16w+NS groups vs.
SHR8w+NS groups, #P<0.05; SHR+R568 groups vs. the
age-matched WKY+R568 groups, **P<0.05; SHR16w+R568 groups vs.
SHR8w+R568 groups, ##P<0.05; SHR+R568 vs. SHR+NS
groups, aP<0.05. WKY, Wistar Kyoto rats; SHR,
spontaneously hypertensive rats; NS, normal saline; R568, NPSR568;
WT%, the wall thickness percentage of the external diameter; WA%,
total vessel wall area as a percentage of the total area (WA%);
CA%, total area occupied by collagen as a percentage of the total
vessel wall area. Scale bar, 50 µM. |
R568 inhibits proliferation and
remodeling in thoracic aortas of SHRs
Immunohistochemistry and western blotting were used
to analyze marker protein changes in hypertensive aortas. Compared
with the age-matched WKY rats, two contractile/differentiated
phenotype marker proteins (calponin and α-SMA) were decreased in
the media of thoracic aortas in SHRs (P<0.05), and the
expression of OPN, a synthetic/dedifferentiated phenotype marker
protein, and PCNA was significantly increased (P<0.05; Figs. 3 and 4). The same trend was observed when
comparing the SHR16w group with the SHR8w group (P<0.05);
however, activation of the CaSR by R568 increased the expression of
calponin and α-SMA, and reduced the expression of OPN and PCNA
protein in the thoracic aortas of SHR16w group (P<0.05; Figs. 3 and 4). R568 had no significant effect on the
expression of proliferative and remodeling proteins in WKY
groups.
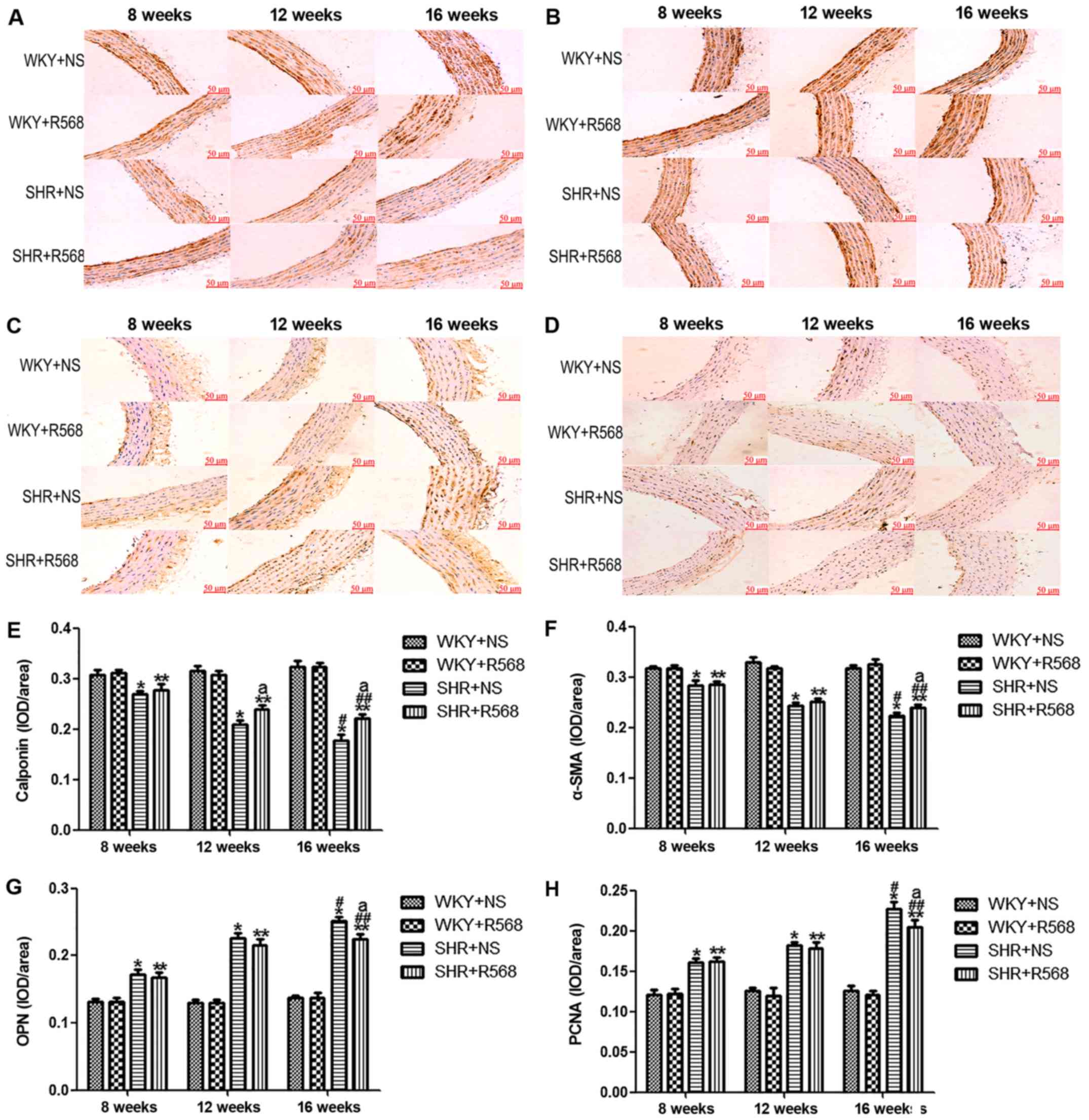 | Figure 3.Immunohistochemical analysis of
proliferating remodeling protein expression in the thoracic aorta
of rats. Representative images of (A) Calponin; (B) α-SMA, smooth
muscle α-actin; (C) OPN, osteopontin; (D) PCNA, proliferating cell
nuclear antigen. Quantification of (E) Calponin; (F) α-SMA, smooth
muscle α-actin; (G) OPN, osteopontin; (H) PCNA, proliferating cell
nuclear antigen. Data are means ± standard deviation (n=7). SHR+NS
groups vs. the age-matched WKY+NS groups, *P<0.05; SHR16w+NS
groups vs. SHR8w+NS groups, #P<0.05; SHR+R568 groups
vs. the age-matched WKY+R568 groups, **P<0.05; SHR16w+R568
groups vs. SHR8w+R568 groups, ##P<0.05; SHR+R568
groups vs. SHR+NS groups, aP<0.05. WKY, Wistar Kyoto
rats; SHR, spontaneously hypertensive rats; NS, normal saline;
R568, NPSR568; α-SMA, smooth muscle α-actin; OPN, osteopontin;
PCNA, proliferating cell nuclear antigen. Scale bar, 50 µM. |
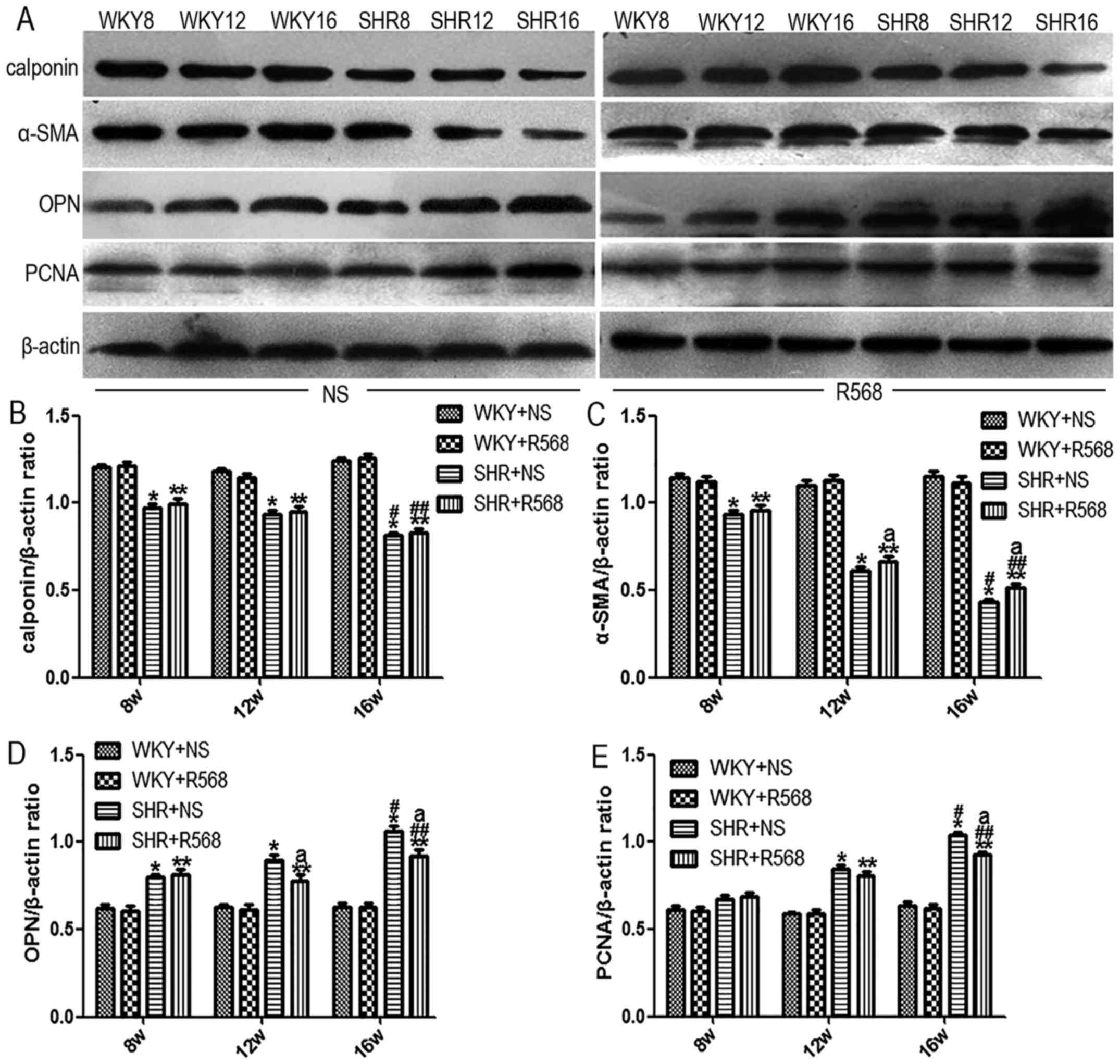 | Figure 4.Western blot analysis of
proliferating remodeling protein expression in the thoracic aorta
of rats. (A) Western blot analysis of the protein expression. (B)
Calponin. (C) α-SMA, smooth muscle α-actin. (D) OPN, osteopontin.
(E) PCNA, proliferating cell nuclear antigen. Data are means ±
standard deviation (n = 7). SHR+NS groups vs. the age-matched
WKY+NS groups, *P<0.05; SHR16w+NS groups vs. SHR8w+NS groups,
#P<0.05; SHR+R568 groups vs. the age-matched WKY+R568
groups, **P<0.05; SHR16w+R568 groups vs. SHR8w+R568 groups,
##P<0.05; SHR+R568 groups vs. SHR+NS groups,
aP<0.05. WKY, Wistar Kyoto rats; SHR, spontaneously
hypertensive rats; NS, normal saline; R568, NPSR568; α-SMA, smooth
muscle α-actin; OPN, osteopontin; PCNA, proliferating cell nuclear
antigen. |
R568 reverses the low expression of
the CaSR in rat thoracic aortas
Western blotting and immunohistochemistry showed
that expression of the CaSR in SHRs was lower than age-matched WKY
rats (P<0.05). Compared with SHR16w group and SHR8w group,
expression of the CaSR protein in the thoracic aortas of rats
increased as the BP increased in SHRs (P<0.05; Fig. 5A-D); however, R568 reversed
expression of the CaSR in SHRs at 16 weeks (P<0.05; Fig. 5A-D). R568 had no significant effect
on CaSR protein expression in thoracic aortas of WKY rats.
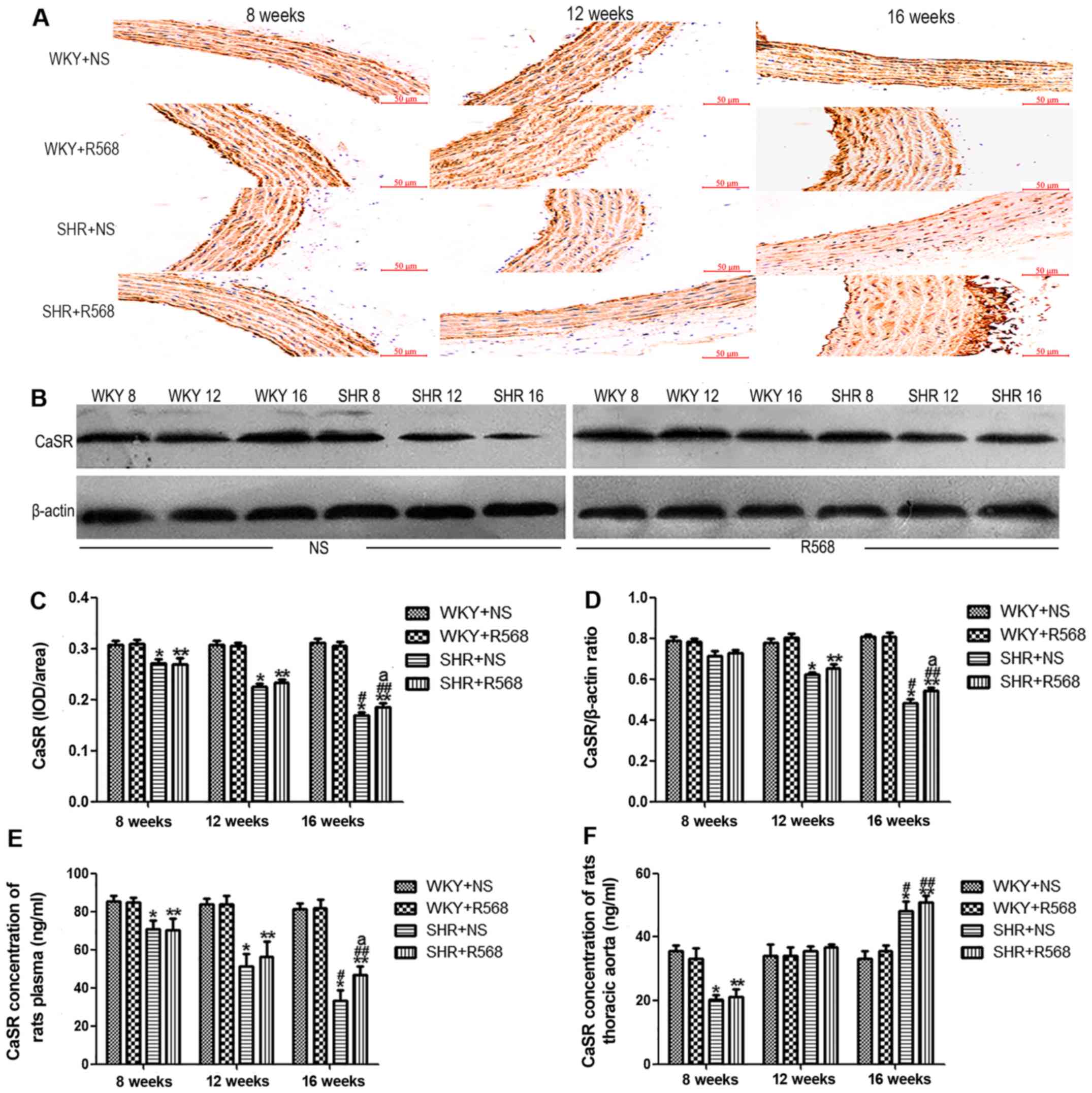 | Figure 5.Detection of CaSR protein expression
and content in plasma and thoracic aorta of rats. (A)
Immunohistochemical analysis. (B) Western blotting analysis. (C)
Densitometric analysis of A. (D) Densitometric analysis of B. (E)
CaSR level in rat plasma (as detected by ELISA). (F) CaSR content
in thoracic aorta (as detected by ELISA). Data are means ± standard
deviation (n=7). SHR+NS groups vs. the age-matched WKY+NS groups,
*P<0.05; SHR16w+NS groups vs. SHR8w+NS groups,
#P<0.05; SHR+R568 groups vs. the age-matched WKY+R568
groups, **P<0.05; SHR16w+R568 groups vs. SHR8w+R568 groups,
##P<0.05; SHR+R568 groups vs. SHR+NS groups,
aP<0.05. WKY, Wistar Kyoto rats; SHR, spontaneously
hypertensive rats; NS, normal saline; R568, NPSR568; CaSR;
calcium-sensing receptor. Scale bar, 50 µM. |
R568 reverses the low expression of
the CaSR in plasma of SHRs and has no significant effect on aortic
homogenates
The concentration of the CaSR in plasma and thoracic
aortas of rats was determined with an enzyme-linked immunosorbent
assay (ELISA). The CaSR concentration in SHRs plasma was
significantly lower than age-matched WKY rats (P<0.05; Fig. 5E). The same trend was observed in
comparing SHR16w group with SHR8w group (P<0.05; Fig. 5E). Interestingly, the content of the
CaSR in SHR-16 group thoracic aortas was significantly higher than
in WKY-16 group (P<0.05; Fig.
5F). After treatment with R568, the CaSR concentration was
significantly elevated in the plasma of SHR16w group (P<0.05;
Fig. 5E). However, R568 had no
significant effect on the CaSR content in the thoracic aortas
(Fig. 5F).
R568 inhibits renin and AT1R protein
increases while decreasing renin and Ang II levels in the SHR
thoracic aorta
Western blotting and ELISA results showed that
compared with age-matched WKY rats, renin, AT1R protein expression,
and renin and Ang II concentration in thoracic aortas were
increased in SHRs; the above indicators increased more
significantly in SHR16w compared with those in SHR8w group
(P<0.05; Fig. 6). R568 treatment
reduced renin and AT1R protein expression, and renin and Ang II
levels in the thoracic aortas of SHR16w group (P<0.05; Fig. 6). R568 had no significant effect on
RAS-related protein expression in the thoracic aorta of WKY rats
(Fig. 6).
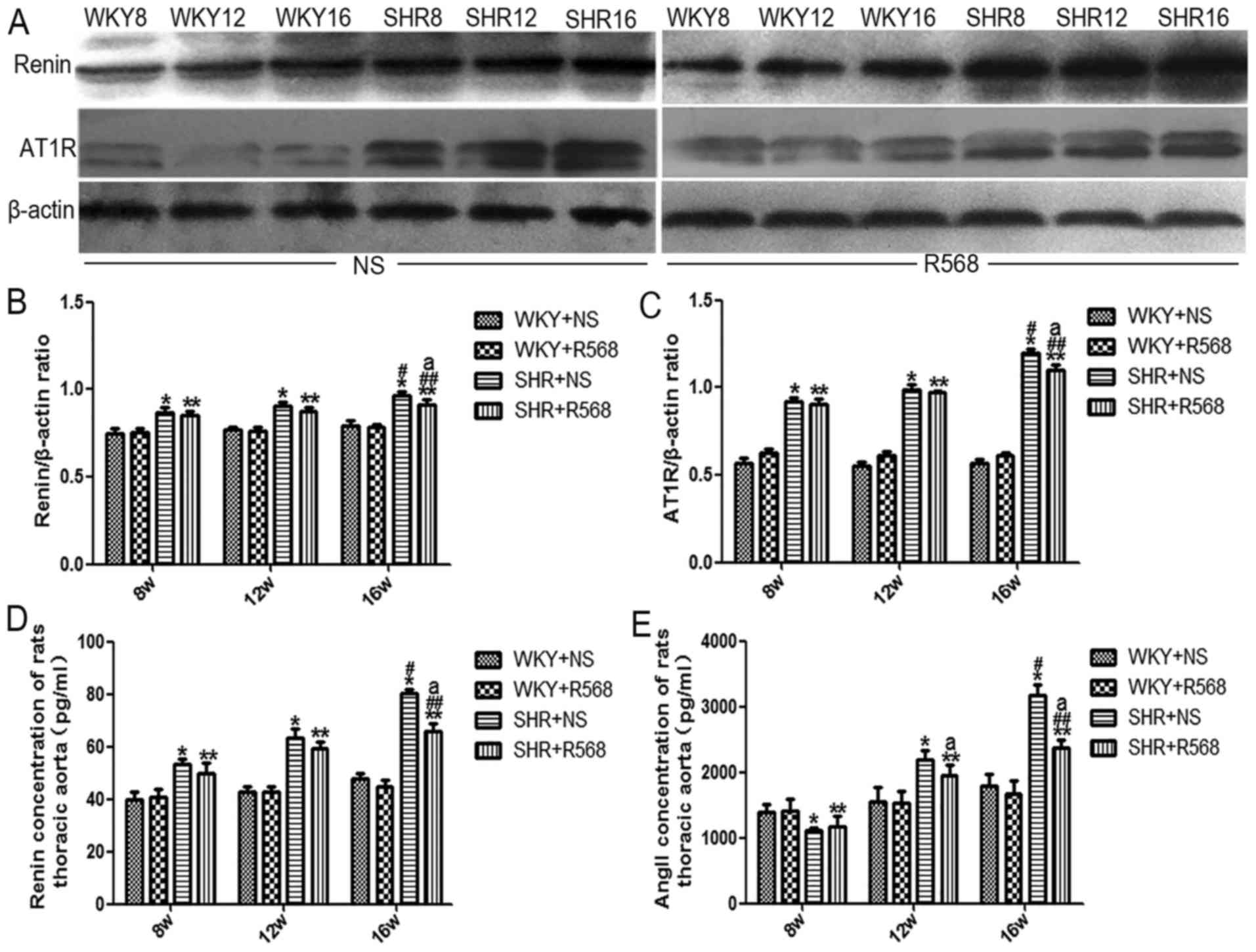 | Figure 6.Determination of renin, AT1R protein
expression and renin, Ang II levels in thoracic aorta of rats. (A)
Western blotting analysis of Renin and AT1R expression. (B)
Densitometric analysis of Renin expression. (C) Densitometric
analysis of AT1R expression. (D) ELISA detection of renin
concentration. (E) ELISA detection of Ang II concentration. Data
are means ± standard deviation (n=7). SHR+NS groups vs. the
age-matched WKY+NS groups, *P<0.05; SHR16w+NS groups vs.
SHR8w+NS groups, #P<0.05; SHR+R568 groups vs. the
age-matched WKY+R568 groups, **P<0.05; SHR16w+R568 groups vs.
SHR8w+R568 groups, ##P<0.05; SHR+R568 groups vs.
SHR+NS groups, aP<0.05. WKY, Wistar Kyoto rats; SHR,
spontaneously hypertensive rats; NS, normal saline; R568, NPSR568;
Ang II, Angiotensin II; AT1R, Ang II type 1 receptor. |
R568 has no significant effect on
cAMP, renin, and Ang II levels in the plasma of rats
Our ELISA results showed that plasma levels of cAMP
and Ang II increased and the renin levels decreased with increasing
BP in SHRs compared with age-matched WKY rats (P<0.05; Fig. 7); the same trend can be observed
compared with SHR16w and SHR8w groups (P<0.05; Fig. 7). There were no significant changes
in plasma cAMP, renin, and Ang II levels in SHRs and WKY rats after
treatment with R568 for 8 weeks (Fig.
7).
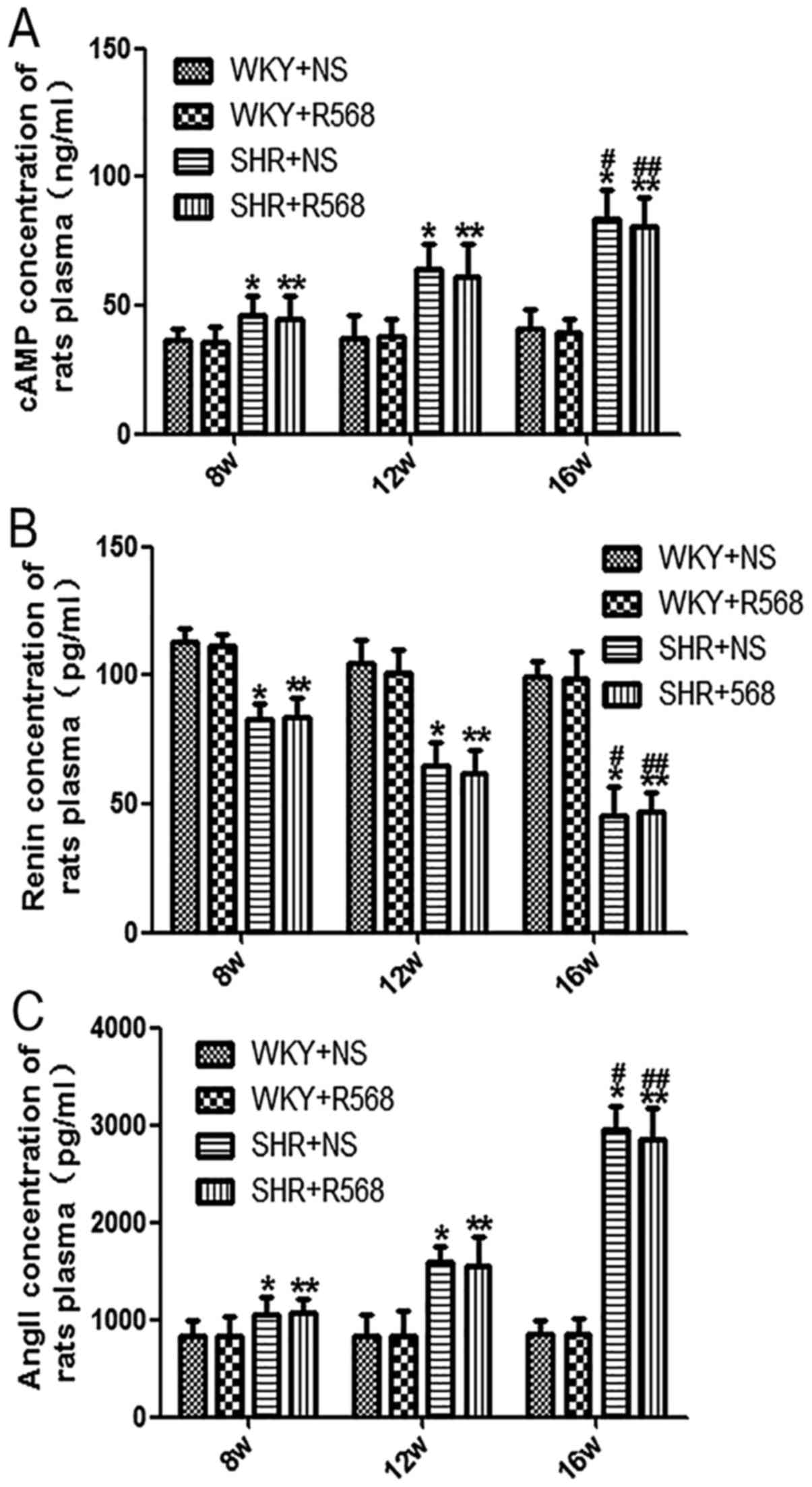 | Figure 7.Determination of renin, Ang II and
cAMP levels in plasma of rats. ELISA was used to detect the levels
of (A) renin, (B) Ang II and (C) cAMP. Data are means ± standard
deviation (n=7). SHR+NS groups vs. the age-matched WKY+NS groups,
*P<0.05; SHR16w+NS groups vs. SHR8w+NS groups,
#P<0.05; SHR+R568 groups vs. the age-matched WKY+R568
groups, **P<0.05; SHR16w+R568 groups vs. SHR8w+R568 groups,
##P<0.05. WKY, Wistar Kyoto rats; SHR, spontaneously
hypertensive rats; NS, normal saline; R568, NPSR568; Ang II,
Angiotensin II; cAMP, cyclic adenosine monophosphate. |
Discussion
CaSR activation has a protective effect on EH and
hypertension-induced aortic smooth muscle proliferation and
remodeling. By analyzing the effects of a R568-activated CaSR on
rat BP, aortic remodeling, and RAS, we demonstrated the following:
i) decreased CaSR expression is involved in hypertension
development; ii) low CaSR expression can activate circulating and
vascular local RAS through the cAMP pathway and promote the
development of hypertension; and iii) the CaSR may regulate BP by
regulating local RAS activity. These data provide new insight into
the regulatory mechanism of the CaSR on EH from the perspective of
the RAS. To date, this is the first study to explore CaSR-regulated
EH mechanisms through the RAS pathway.
The CaSR, a member of the GPCR superfamily, exerts
biological effects by regulating the PLC-IP3 signaling
pathway to induce an increase in [Ca2+]i
(7). Recent studies have suggested
that the CaSR agonist, R568, can reduce BP, but the underlying
mechanism is not clear. Maillard et al (18) suggested that the calcimimetic R568
can regulate renin release via CaSR, and renin plays an important
role in the occurrence of EH. Ogata et al (14) compared the effects of NPSR568 and
parathyroidectomy on the progression of renal failure and showed
that R568 decreased BP in uremic rats and SHRs, but had no effect
on normotensive rats. Rybczyńska et al (20) also observed the effect of intravenous
administration of R568 on the MAP of SHRs and WKY rats in the
presence or absence of thyroparathyroidectomy and found that in the
presence of parathyroid, calcimimetic R568 lowered the BP of SHRs,
but has no effect on BP in WKY rats. In the current study, we first
used SHRs and WKY rats of different ages to dynamically observe the
anti-hypertensive effect of the CaSR agonist, R568. The results
showed that R568 activation of CaSR reduced BP in SHRs but had no
significant effect on BP in normotensive rats. Therefore, we
believe that low CaSR expression is involved in the development of
hypertension. Different views have also been proposed by other
researchers. Nakagawa et al (21) observed the changes in MAP after acute
injection of femoral vein R568 and the enantiomer, S568, in
Sprague-Dawley (SD) rats. The results showed that R568 exerts an
acute, CaSR-independent antihypertensive effect through
vasodilation and negative degeneration at concentrations exceeding
those required to modulate PTH secretion. Because S568 has little
effect on the CaSR, Nakagawa et al (21) suggested that the effect of R568 on
lowering BP may not be CaSR-mediated. From this point of view, the
researchers believe that R568 is a phenylalkylamine compound, its
effect on CaSR is basically stereotactic, and both isomers R568 and
S568 are equivalent in blocking voltage or ligand-gated
Ca2+ influx (21). The
lack of stereoselectivity and the hypotensive effect of R568
suggest that exclusion of voltage-gated Ca2+ channels,
except for CaSR-mediated activity, is still required; however, this
remains to be demonstrated (21,22).
As an adaptive response, the progression of
hypertension is often accompanied by changes in the structure and
function of blood vessels [i.e., vascular remodeling (VR)], which
is an independent risk factor for the increased incidence of
cardiovascular events in hypertension (23). The central part of VR is the
proliferation of VSMCs, migration to the sub-endocardium, and
secretion of the extracellular matrix (24). VSMCs play an important role in
maintaining BP, but progression of hypertension leads to phenotypic
changes in VSMCs. The conversion of VSMCs from a contractile
phenotype to a synthetic phenotype leads to thickening of the
vessel wall. In this experiment, we determined the expression of
SMAα and calponin (two contractile/differentiated phenotypic
markers) (25), OPN (a
synthetic/dedifferentiated phenotypic marker) (26), and PCNA (an important marker of cell
proliferation) (27) in the thoracic
aortas of rats. Protein expression in thoracic aortas in WKY rats
and SHRs was detected with immunohistochemistry and western
blotting, respectively. We observed that the expression of α-SMA
and calponin decreased, while OPN and PCNA increased significantly
with increasing age in SHRs; however, these changes were inhibited
by the CaSR agonist, R568. HE and Masson staining showed that the
WA%, WT%, and CA% of SHR thoracic aortas were significantly
increased compared with WKY rats, which was reduced by R568. The
above results indicate that R568 improved the proliferation and
remodeling of thoracic aortas in SHRs.
Ziegelstein et al (28) suggested that the CaSR is expressed in
aortic endothelial cells; whereas Smajilovic et al (29) demonstrated that the CaSR also exists
in aortic VSMCs and that the CaSR can affect the proliferation of
aortic VSMCs. Our previous study also put forward a similar view;
expression of the CaSR protein in rat thoracic aorta VSMCs and the
CaSR content were decreased with the increase in BP (9). In our experiments, similar results were
obtained. We found that reduction in the CaSR could be reversed
after treatment with R568 for 8 weeks in the plasma and thoracic
aortas of SHRs rather than in thoracic aorta homogenates; however,
the opposite result was observed in thoracic aorta homogenates when
compared with SHR16w and WKY16w. Moreover, the same reverse trend
was detected in CaSR protein expression in the thoracic aortas when
SHR16w were compared with SHR8w. Unfortunately, no strong evidence
has been found to explain this phenomenon. We presume this finding
may be a compensatory manifestation for the increase of BP in SHRs,
which provides a direction for further research in the future.
Taken together, our results partially demonstrate that the CaSR
agonist (R568) may play a protective role in EH, and EH evokes
proliferation and remodeling of thoracic aortic cells in SHRs.
There is a complex network of contacts between the
RAS, VR, and hypertension. Studies suggest that CaSR regulation of
BP may involve six related pathways, but the molecular mechanism
underlying this association has not been fully elucidated (30). To investigate the mechanism
underlying the CaSR in hypertensive vascular remodeling, we
selected the RAS pathway for further study, which is closely
related to the development of clinical hypertension. Renin, which
is closely linked to hypertension, is the first rate-limiting
enzyme in the RAS, and cAMP plays a key role in this system
(4). Alderman et al (31) divided hypertension into three types
(high, low, and intermediate renin). At present, low renin
hypertension (LRH), a subtype of EH, has been unanimously approved
by researchers (32). The majority
of EH patients in China are LRH (33). Angiotensinogen produces angiotensin I
(Ang I) under the action of renin and angiotensin converting enzyme
(ACE) acts on Ang I to produce angiotensin II (Ang II) (34). Ang II is the main active substance of
the RAS, and binds to Ang II type 1 receptor (AT1R), thus causing
vasoconstriction and cell proliferation (35). AT1R mediates the biological effects
of Ang II and plays an important role in the regulation of BP
(36). RAS is an important body
fluid regulating system in the body. Circulating RAS mainly exists
in plasma, and local RAS exists in the heart, aorta, brain,
kidneys, and many other tissues (37). Recent studies have shown that
intracellular RAS plays a more important role in the function and
regulation of local tissues under certain cell types or pathologic
conditions (38,39). Local RAS operates independently of
circulating RAS and many of the adverse effects after AT1R
activation are caused by locally-generated Ang II (40). Blaine et al (41) believes that the vascular local RAS,
through the independent regulation of vascular function, promotes
the occurrence of hypertension. Unger et al (42) also suggests that Angiotensin
converting enzyme inhibitors (ACEIs), such as captopril, reduce BP
in SHRs by inhibiting local RAS activity. In our experiments, it
was observed that renin, Ang II, and AT1R in thoracic aortas
increased with the increase in BP, whereas R568 inhibited the
increase in these factors. In circulating blood, renin was lower in
SHRs, whereas Ang II and cAMP exhibited the opposite trend, which
is consistent with our previous study (9). Unfortunately, R568 treatment failed to
significantly alter renin and Ang II levels in plasma. Li et
al (43) believe that in the
early stage of hypertension, circulatory RAS is activated and
involved in the initial rise of BP, but after 8 weeks, circulating
blood RAS is decreased, while BP continues to rise; at which time
RAS in the heart, brain, kidneys, and other local tissues actively
participate in EH and target organ damage maintenance, which is
consistent with our point of view. Meanwhile, Ferrario et al
(44) also noted that the effects of
RAS blockers on Ang II concentrations in circulating plasma and
local tissues were different, suggesting that local RAS
intervention may be viable in the prevention and treatment of
related diseases. As mentioned above, our results suggested that
the CaSR agonist (R568) is viable to suppress local RAS activity to
lower BP and improve VSMC proliferation and remodeling.
The limitations of our experiments were a lack of
inhibition of the CaSR activity with NPS2314 or Calhex231, and
further observations regarding the relationship between BP,
thoracic aortic proliferation, and remodeling and RAS activity are
needed. Despite these limitations, our study partially explains the
basic mechanism by which the CaSR mediates BP through the RAS
pathway.
In summary, our results indicate that low expression
of the CaSR is involved in the development of hypertension by cAMP
pathway activation of circulatory and vascular local RAS.
Regulation of the CaSR on BP in SHRs, but not WKY rats, may be
through regulation of local RAS activity. The clinically used ACEIs
and Ang II receptor blockers lower BP by inhibiting RAS activity,
but have significant side effects. We found that R568 can reduce BP
by activating the CaSR to regulate RAS activity, thus preventing
the side effects of these drugs. Continued studies are being
conducted in our laboratory to further determine the mechanism by
which the CaSR regulates BP at the molecular level. Our findings
provide new insight into the pathogenesis of this complex disease
and suggest that the CaSR may be a valuable pharmacologic target in
patients with hypertension.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 31560287)
and the Xinjiang Graduate Student Research Innovation Project
(grant no. XJGRI2017049).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
RS and FH contributed to the conception and design
of the study. RS drafted the manuscript. WZ and NT performed the
data analyses. HZ, LW and YL performed Masson's staining and
immunohistochemical analysis. YZ and TZ performed and analyzed
western blotting and ELISA. FH reviewed the manuscript and gave
final approval to the submitted and final versions. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Shihezi Medical University (Shihezi, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lloyd-Jones D, Adams R, Carnethon M, De
Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund
K, et al: Heart disease and stroke statistics-2009 update: A report
from the American Heart Association Statistics Committee and Stroke
Statistics Subcommittee. Circulation. 119:480–486. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Osborn JW, Fink GD, Sved AF, Toney GM and
Raizada MK: Circulating angiotensin II and dietary salt: Converging
signals for neurogenic hypertension. Curr Hypertens Rep. 9:228–235.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Das M, Pal S and Ghosh A: Angiotensin
converting enzyme gene polymorphism (insertion/deletion) and
hypertension in adult asian indians: A population-based Study from
Calcutta, India. Hum Biol. 80:303–312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Churchill PC: Second messengers in renin
secretion. Am J Physiol. 249:F175–F184. 1985.PubMed/NCBI
|
|
5
|
Kang G, Lee YR, Joo HK, Park MS, Kim CS,
Choi S and Jeon BH: Trichostatin a modulates angiotensin II-induced
vasoconstriction and blood pressure via inhibition of p66shc
activation. Korean J Physiol Pharmacol. 19:467–472. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brown EM and Macleod RJ: Extracellular
calcium sensing and extracellular calcium signaling. Nat Rev Mol
Cell Biol. 4:530–538. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang R, Xu C, Zhao W, Zhang JK, Yang B and
Wu L: Calcium and polyamine regulated calcium-sensing receptors in
cardiac tissues. Eur J Biochem. 270:2680–2688. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Molostvov G, James S, Fletcher S, Bennett
J, Lehnert H, Bland R and Zehnder D: Extracellular calcium-sensing
receptor is functionally expressed in human artery. Am J Physiol
Renal Physiol. 293:F946–F955. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qu YY, Jing H, Wang LM, Na T, Hua Z, Liu
YM, Zhen L, Qian F and Fang H: Reduced expression of the
extracellular calcium-sensing receptor (CaSR) is associated with
activation of the renin-angiotensin system (RAS) to promote
vascular remodeling in the pathogenesis of essential hypertension.
PLoS One. 11:e01574562016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weston AH, Absi M, Ward DT, Ohanian J,
Dodd RH, Dauban P, Petrel C, Ruat M and Edwards G: Evidence in
favor of a calcium-sensing receptor in arterial endothelial cells:
studies with calindol and Calhex 231. Cir Res. 97:391–398. 2005.
View Article : Google Scholar
|
|
11
|
Atchison DK, Ortiz-Capisano MC and
Beierwaltes WH: Acute activation of the calcium-sensing receptor
inhibits plasma renin activity in vivo. Am J Physiol Regul Integr
Comp Physiol. 299:R1020–R1026. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ayachi S: Increased dietary calcium lowers
blood pressure in the spontaneously hypertensive rat. Metabolism.
28:1234–1238. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cow D: Some reactions of surviving
arteries. J Physiol. 42:125–143. 1911. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ogata H, Ritz E, Odoni G, Amann K and Orth
SR: Beneficial effects of calcimimetics on progression of renal
failure and cardiovascular risk factors. J Am Soc Nephrol.
14:959–967. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rybczynska A, Lehmann A, Jurskajasko A,
Boblewski K, Orlewska C, Foks H and Drewnowska K: Hypertensive
effect of calcilytic NPS 2143 administration in rats. J Endocrinol.
191:189–195. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rybczynska A, Jurskajasko A, Boblewski K,
Lehmann A and Orlewska C: Blockade of calcium channels and AT1
receptor prevents the hypertensive effect of calcilytic NPS 2143 in
rats. J Physiol Pharmacol. 61:163–170. 2010.PubMed/NCBI
|
|
17
|
Ortiz-Capisano MC, Reddy M, Mendez M,
Garvin JL and Beierwaltes WH: Juxtaglomerular cell CaSR stimulation
decreases renin release via activation of the PLC/IP(3) pathway and
the ryanodine receptor. Am J Physiol Renal Physiol. 304:F248–F256.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maillard MP, Tedjani A, Perregaux C and
Burnier M: Calcium-sensing receptors modulate renin release in vivo
and in vitro in the rat. J Hypertens. 27:1980–1987. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Widdop RE and Li XC: A simple versatile
method for measuring tail cuff systolic blood pressure in conscious
rats. Clin Sci (Lond). 93:191–194. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rybczyńska A, Boblewski K, Lehmann A,
Orlewska C, Foks H, Drewnowska K and Hoppe A: Calcimimetic NPS
R-568 induces hypotensive effect in spontaneously hypertensive
rats. Am J Hypertens. 18:364–371. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakagawa K, Parekh N, Koleganova N, Ritz
E, Schaefer F and Schmitt CP: Acute cardiovascular effects of the
calcimimetic R-568 and its enantiomer S-568 in rats. Pediatr
Nephrol. 24:1385–1389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nemeth EF: Calcimimetic and calcilytic
drugs: Just for parathyroid cells? Cell Calcium. 35:283–289. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Renna NF, de Las Heras N and Miatello RM:
Pathophysiology of vascular remodeling in hypertension. Int J
Hypertens. 2013:8083532013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lan TH, Huang XQ and Tan HM: Vascular
fibrosis in atherosclerosis. Cardiovasc Pathol. 22:401–407. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Beamish JA, He P and Marchant RE:
Molecular regulation of contractile smooth muscle cell phenotype:
Implications for vascular tissue engineering. Tissue Eng Part B
Rev. 16:467–491. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weber GF, Lett GS and Haubein NC:
Osteopontin is a marker for cancer aggressiveness and patient
survival. Br J Cancer. 103:861–869. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yi B, Cui J, Ning JN, Wang GS, Qian GS and
Lu KZ: Over-expression of PKGIα inhibits hypoxia-induced
proliferation, Akt activation and phenotype modulation of human
PASMCs: The role of phenotype modulation of PASMCs in pulmonary
vascular remodeling. Gene. 492:354–360. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ziegelstein RC, Xiong Y, He C and Hu Q:
Expression of a functional extracellular calcium-sensing receptor
in human aortic endothelial cells. Biochem Biophys Res Commun.
342:153–163. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Smajilovic S, Hansen JL, Christoffersen
TEH, Lewin E, Sheikh SP, Terwilliger EF, Brown EM, Haunso S and
Tfelt-Hansen J: Extracellular calcium sensing in rat aortic
vascular smooth muscle cells. Biochem Biophys Res Commun.
348:1215–1223. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smajilovic S, Yano S, Jabbari R and
Tfelt-Hansen J: The calcium-sensing receptor and calcimimetics in
blood pressure modulation. Br J Pharmacol. 164:884–893. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alderman MH, Cohen HW, Sealey JE and
Laragh JH: Plasma renin activity levels in hypertensive persons:
Their wide range and lack of suppression in diabetic and in most
elderly patients. Am J Hypertens. 17:1–7. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Everett CM, Turner B and Lobo M: Posterior
reversible encephalopathy syndrome in (low renin) essential
hypertension. J R Soc Med. 100:522–523. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yuan W, Pan W, Kong J, Zheng W, Szeto FL,
Wong KE, Cohen R, Klopot A, Zhang Z and Li YC:
1,25-dihydroxyvitamin D3 suppresses renin gene transcription by
blocking the activity of the cyclic AMP response element in the
renin gene promoter. J Biol Chem. 282:29821–29830. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Joshi HB, Newns N, Stainthorpe A,
MacDonagh RP, Keeley FX Jr and Timoney AG: Ureteral stent symptom
questionnaire: Development and validation of a multidimensional
quality of life measure. J Urol. 169:1060–1064. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
de Gasparo M, Catt KJ, Inagami T, Wright
JW and Unger T: International union of pharmacology. XXIII. The
angiotensin II receptors. Pharmacol Rev. 52:415–472.
2000.PubMed/NCBI
|
|
36
|
Mogi M, Iwai M and Horiuchi M: New
insights into the regulation of angiotensin receptors. Curr Opin
Nephrol Hypertens. 18:138–143. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Danser AH: Local renin-angiotensin
systems. Mol Cell Biochem. 157:211–216. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Singh VP, Le B, Bhat VB, Baker KM and
Kumar R: High-glucose-induced regulation of intracellular ANG II
synthesis and nuclear redistribution in cardiac myocytes. Am J
Physiol Heart Circ Physiol. 293:H939–H948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Singh VP, Le B, Khode R, Baker KM and
Kumar R: Intracellular angiotensin ii production in diabetic rats
is correlated with cardiomyocyte apoptosis, oxidative stress and
cardiac fibrosis. Diabetes. 57:3297–3306. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hunyady L and Catt KJ: Pleiotropic AT1
receptor signaling pathways mediating physiological and pathogenic
actions of angiotensin II. Mol Endocrinol. 20:953–970. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Blaine EH, Schorn TW and Boger J:
Statine-containing renin inhibitor. Dissociation of blood pressure
lowering and renin inhibition in sodium-deficient dogs.
Hypertension. 6:I111–I118. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Unger T, Ganten D, Lang RE and Schölkens
BA: Is tissue converting enzyme inhibition a determinant of the
antihypertensive efficacy of converting enzyme inhibitors? Studies
with the two different compounds, Hoe498 and MK421, in
spontaneously hypertensive rats. J Cardiovasc Pharmacol. 6:872–880.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li J, Gao X, Zhang B, Kang L, Guo Z and
Fan G: Antagonistic effect of traditional Chinese medicine on RAS
changes in experimental hypertensive rats. Chin med New Drugs Clin
Pharmacol. 15:68–70. 2004.(In Chinese).
|
|
44
|
Ferrario CM, Jessup J, Chappell MC,
Averill DB, Brosnihan KB, Tallant EA, Diz DI and Gallagher PE:
Effect of angiotensin-converting enzyme inhibition and angiotensin
II receptor blockers on cardiac angiotensin-converting enzyme 2.
Circulation. 111:2605–2610. 2005. View Article : Google Scholar : PubMed/NCBI
|














