Introduction
Osteoporosis (OP) is a common metabolic skeletal
disease in the elderly population featured by osteopenia and
microstructure, mainly due to the imbalance between bone resorption
and bone formation, resulting in increased morbidity and high cost
of health care. Current existing therapeutic agents specific to OP
are mainly antiresorptive drugs that repress bone resorptive action
of osteoclasts. Largely owing to that the mechanisms of excessive
bone resorption have been thoroughly explored (1). However, the medicinal efficacy on bone
mass and strength of patients remains limited. Thus, it is
imperative to address this public health concern, and identify its
underlying molecular mechanisms.
The cellular and molecular processes involved in
pathophysiology of OP are relatively complicated regulatory
networks, involving numerous genes, factors and several pathways.
In molecular regulatory networks, it is considered that alterations
of gene expression elicit abnormal protein function and pathways
turbulence, thus occurrence and development of disease. It is
well-known that substantial genes were regulated by transcription
factors (TFs), recognized as the master regulators binding to DNA
sequences specifically and thereby modulating gene transcription.
The transcriptional regulation elicited by TFs is of importance to
normal function of the organism, with transcriptional regulation
central to cell cycle growth and survive (2), cell differentiation (3), cell adhesion (4) and cell homeostasis (5). However, TF failure leads to nearly 1/3
of human developmental diseases ascribed to TF misregulations
(6,7). Thus, TF prediction is a pivotal step to
comprehending sophisticated regulatory molecular networks. To date,
effective screening methods of TFs related to OP remained largely
lacking. OP mediated by osteoblasts is not able to balance bone
absorption mediated by osteoclasts, leading to decrease in bone
mass and low bone mineral density. Osteoblasts, as bone-forming
cells, derive from mesenchymal stem cells (MSCs) which is the
source for bone remodeling, besides, the differentiated osteoblasts
are decreased from MSCs with aging concerning series of molecular
mechanisms (8). However, research on
osteoblast action involving underlying mechanisms are relatively
ignored.
Therefore, the present study was designed to
investigate the mechanistic TFs for OP progression using
bioinformatics methods based on gene expression data from the MSC
of OP patients. TF targets enrichment analysis was performed for
the chosen differentially expressed genes (DEGs). Then, analysis of
TF impact factors (IFs) was conducted for DEGs. Moreover, influence
of TF network analysis was executed for DEGs. Finally, the obtained
TFs based on TF targets prediction and influence of TFs targets or
TF network were analyzed comprehensively to achieve the key TFs,
which might contribute to future therapy of OP.
Materials and methods
Microarray data collection and
preprocessing
The gene expression profile data GSE35956 were
extracted from the study of Benisch et al (9), which was based on Gene Expression
Omnibus database. Human mesenchymal stem cells (hMSC) of elderly
patients (79–94 years) suffering from OP were isolated from femoral
heads after low-energy fracture of the femoral neck. A total of 5
OP patients (elderly) and 5 control group (middle-aged) were
included. Then, the raw data were normalized and converted into
expression values by the robust multi-array average (RMA) algorithm
(www.bioconductor.org) and R statistical
software (version 3.0.0; R Project for Statistical Computing,
http://www.r-project.org/).
DEG analysis
Multiple linear regression package, limma package
(10,11) was used to determine the DEGs between
the OP patients and normal controls. Multiple testing corrections
were performed using the Bayesian method. Only genes with logarithm
of fold change (|logFC|) ≥2 and P-value <0.05 were chosen as the
DEGs. To ensure the stability of the object in the screening
process, all the DEGs were chosen if their number was over 300 with
their |logFC| beyond 2 while the top 300 DEGs were chosen if their
the number was below 300 with their |logFC| beyond 2.
TF genes or TF target prediction
It is known that each TF, as a protein, has a
corresponding regulatory gene, which mainly binds to specific DNA
sequences and thus exerts its regulatory role in incomputable
biological process. Hence, TF gene was identified first based on
mapping DEGs to TF databases including ITFP (12), Tissue-specific regulatory circuits
(13), TRRUST (14) databases involved in TFs genes, their
downstream regulated genes (target genes) and binding sites. Then,
once no TF genes existed, we speculated potential effect of TF by
analyzing whether TF targets contained DEGs using Fisher's exact
test, which could reflect that the more TF targets were enriched,
the more crucial the TFs were. Noticeably, the abundant TF targets
appeared due to likelihood that TF targets were widely regulated by
TFs such as TFs involved in regulating cell cycle. Thus, such TFs
were needed for the following study combined with disease
state.
The influence of TF analysis
IFs of TFs were assessed by calculating G-score
according to its metric. By comprehensively considering the G-score
(the forum was shown as follow), average G-score and numbers of
regulated genes, the IF (expressed in P-value) of TF was gained.
The lower of this value is, the bigger of TF influence is.
According to the P-value, the crucial TFs targets were
predicted.
GSx=|LSx|(-log10PSx)
The influence of TF network
analysis
Considering the possibility that TFs could form a
co-expression relationship with its target genes, to assess the
importance of TFs, Search Tool for the Retrieval of Interacting
Genes/Proteins (STRING) and the afore-mentioned 3 databases were
used to calculate the influence of TFs on local network
neighborhood using the weighted sum method (15). Subsequently, G-score of network was
calculated and the crucial TF targets were determined by its
ranking.
Identification of optimal set of
TFs
To select the optimal set of TFs with the greatest
combined influence, TFs with maximum coverage of DEGs were
determined. Top TFs obtained based on above third methods (TF
targets enrichment analysis, TF targets influences analysis, TF
network influences analysis) were integrated to obtain the optimal
crucial TFs.
Results
Data preprocessing and DEG
identification
After data preprocessing, a total of 20,514 genes
were obtained from 5 OP patients (elderly) and 5 control groups
(middle-aged). A total of 300 DEGs were obtained, namely, LYG2,
PPEF1, TTC16, LOC100134368, LOC100505716, CKM, LOC100506272,
ADAMTS7, MAB21L2, NR0B1, which were all upregulated in OP group
(Table I).
 | Table I.The top 10 DEGs. |
Table I.
The top 10 DEGs.
| Gene name | logFC | AveExpr | t | P-value | adj.P.Val |
|---|
| LYG2 | 3.822863546 | 4.898607116 | 11.79654581 | 3.09E-07 | 0.006343212 |
| PPEF1 | 2.967917113 | 5.306881431 | 8.769700405 | 4.84E-06 | 0.049630867 |
| TTC16 | 3.371397328 | 5.527493053 | 8.198929385 | 8.85E-06 | 0.054473657 |
|
LOC100134368 | 2.877190274 | 5.538742477 | 7.729046328 | 1.49E-05 | 0.054473657 |
|
LOC100505716 | 3.098691591 | 5.203848263 | 7.677271035 | 1.58E-05 | 0.054473657 |
| CKM | 3.47319398 | 6.247940415 | 7.670112434 | 1.59E-05 | 0.054473657 |
|
LOC100506272 | 2.368835964 | 4.8244862 | 7.19255559 | 2.78E-05 | 0.081533396 |
| ADAMTS7 | 1.372895995 | 7.074901655 | 6.9525671 | 3.72E-05 | 0.092051022 |
| MAB21L2 | 3.654174352 | 9.046481165 | 6.727015578 | 4.91E-05 | 0.092051022 |
| NR0B1 | 1.911083095 | 5.997674202 | 6.593036972 | 5.82E-05 | 0.092051022 |
TF genes or TF targets prediction
The result of TF genes enrichment analysis suggested
that no DEGs were identified as TF genes. In the following, we
analyzed whether TF targets were contained in DEGs using Fisher's
exact test. As shown in Table II,
165 TFs targets including CKM, CD74, ADAMTS, ADAMTS7, NPAS1 were
enriched, correspondingly, the top 10 TFs were WT1, ZBTB7A, CACBP,
ZNF281, ZBTB47, ZNF219, PATZ1, TFAP2C, HGS, and KLF16.
| Table II.Top 10 TF enrichment analysis of
DEGs. |
Table II.
Top 10 TF enrichment analysis of
DEGs.
| TF_name | TF_P-value | FDR_p | num_genes |
|---|
| WT1 | 6.52E-08 | 7.48E-05 | 97 |
| ZBTB7A | 1.83E-05 | 0.007169601 | 48 |
| CACBP | 2.16E-05 | 0.007169601 | 118 |
| ZNF281 | 2.50E-05 | 0.007169601 | 111 |
| ZBTB47 | 3.30E-05 | 0.00756938 |
6 |
| ZNF219 | 3.96E-05 | 0.00756938 | 100 |
| PATZ1 | 0.000106106 | 0.015445541 | 79 |
| TFAP2C | 0.000128968 | 0.015445541 | 112 |
| HGS | 0.000132363 | 0.015445541 |
5 |
| KLF16 | 0.000153328 | 0.015445541 | 107 |
| EGR | 0.000157328 | 0.015445541 | 127 |
The influence of TF analysis
According to the G-score, average G-score, numbers
of regulated genes and the P-value of TF comprehensively, 87 TF
targets from DEGs were obtained, correspondingly, the top 10
crucial TFs were FKBP8, SP1, LMO4, KLF3, KHDRBS1, ZFPM1, EML3,
ZNF580, ZBTB45, and WDTC1 (Table III).
| Table III.Top 10 TFs identified from DEGs based
on their influence. |
Table III.
Top 10 TFs identified from DEGs based
on their influence.
| TF_gene | G-score | ave_score | num_genes | rank_p |
|---|
| FKBP8 | 1.629660153 | 1.629660153 | 1 | 0 |
| SP1 | 2063.253105 | 0.206263432 | 10003 | 0 |
| LMO4 | 5.595214111 | 1.398803528 | 4 | 0.02001 |
| KLF3 | 5.506357538 | 1.376589384 | 4 | 0.02405 |
| KHDRBS1 | 2.696028355 | 1.348014178 | 2 | 0.02983 |
| ZFPM1 | 1.310340162 | 1.310340162 | 1 | 0.03837 |
| EML3 | 1.28108726 | 1.28108726 | 1 | 0.04572 |
| ZNF580 | 2.548392738 | 1.274196369 | 2 | 0.04752 |
| ZBTB45 | 2.477553068 | 1.238776534 | 2 | 0.05746 |
| WDTC1 | 1.178355662 | 1.178355662 | 1 | 0.07665 |
The influence of TF network
analysis
Based on the G-score of network ranking, 178 TF
targets were identified from DEGs, correspondingly, the top 10
influential TFs included FOXO1, KLF16, RXRA, RARA, HNF4A, CEBPB,
ESR1, SOX8, ZNF219, and SP1 (Table IV).
| Table IV.Top 10 TFs identified from DEGs based
on their G-scores of TF network. |
Table IV.
Top 10 TFs identified from DEGs based
on their G-scores of TF network.
| TF_gene | G_net_score |
|---|
| FOXO1 | 328.9369128 |
| KLF16 | 323.1693838 |
| RXRA | 312.8356811 |
| RARA | 310.4836752 |
| HNF4A | 300.1282162 |
| CEBPB | 297.0888622 |
| ESR1 | 291.421492 |
| SOX8 | 291.0030097 |
| ZNF219 | 290.6270745 |
| SP1 | 285.5734732 |
| NFIC | 283.8264335 |
Optimal set of TF identification
Ultimately, the above third methods were integrated
with the greatest combined influence to attain the optimal set of
TFs with maximum coverage of DEGs. The PPI network analysis based
on the G-score of network efficacy is better than others methods
due to the number of the 178 TF targets was more than the number of
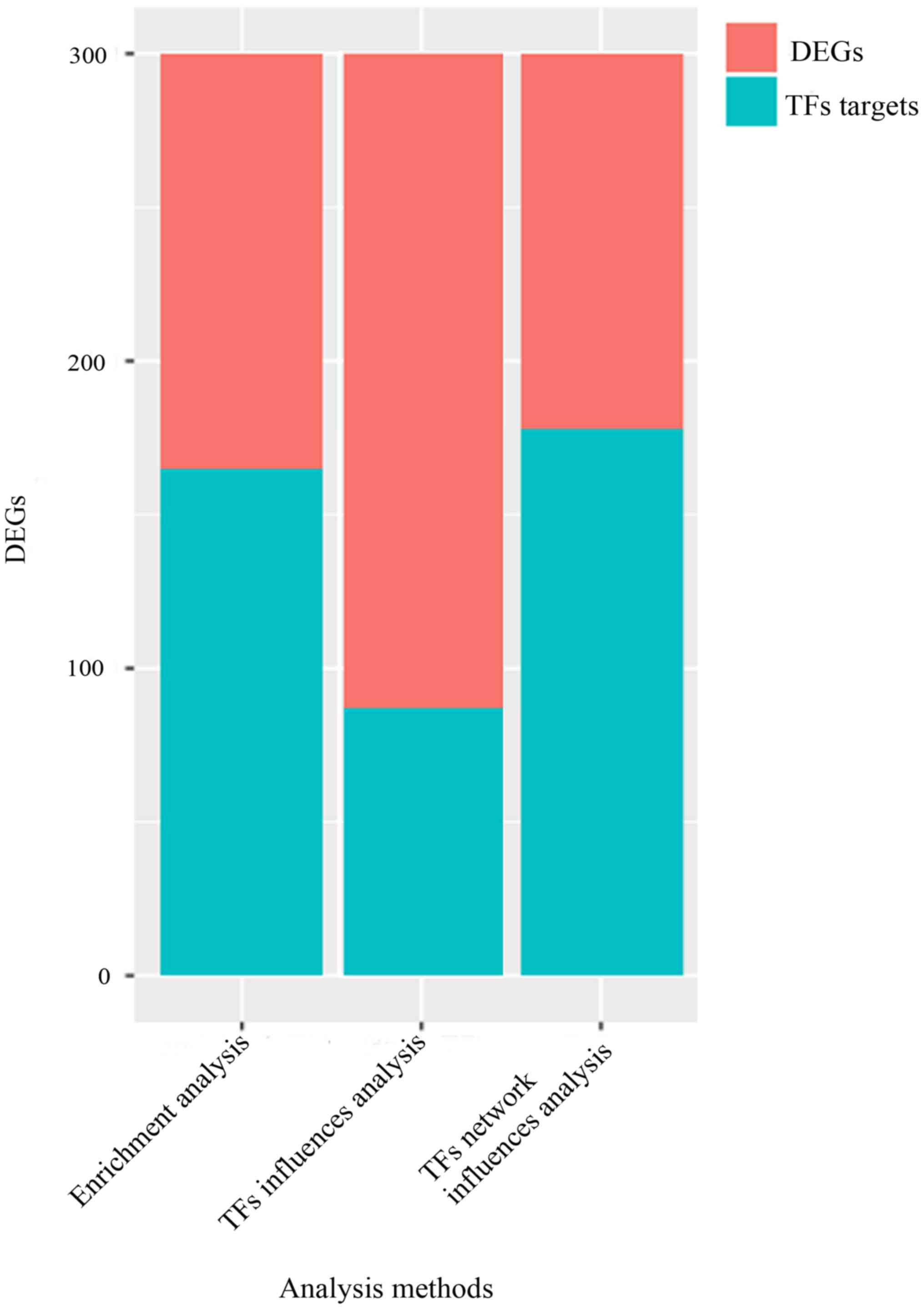
165 or 87 TF targets, as shown in Fig.
1. Thus, the optimal set of TFs were FOXO1, KLF16, RXRA,
RARA, HNF4A, CEBPB, ESR1, SOX8, ZNF219, and SP1.
Discussion
Bioinformatics methods were utilized to identify
crucial TFs for clinical OP treatment. The results suggested that
300 DEGs such as upregulated LYG2, PPEF1, TTC16 were gained in OP
group compared with control group. A total of 165 TF targets from
DEGs were enriched based on TF genes enrichment analysis, 87 TF
targets from DEGs were attained by their IF analysis and 178 TF
targets from DEGs were achieved by TF network influence analysis.
According to the optimal TF set with TFs having maximum coverage of
DEGs, 178 TF targets were the most. Thus, the optimal sets of TFs
were FOXO1, KLF16, RXRA, RARA, HNF4A, CEBPB, ESR1, SOX8,
ZNF219, and SP1. These TFs identified from third
bioinformatics methods are possibly capable of aiding in a better
understanding of the underlying molecular mechanisms, implying
these crucial TFs are likely to be the potential therapy target for
OP.
It is well-established that TF, specific to binding
to its target gene, thus exerts inhibitory or facilitative role in
gene expression, showing a significant part in the multitude of
biological processes involved in diseases (15–17).
Particularly, it is well known that runt-related TF-2 (Runx-2),
served as a master member of osteogenic differentiation specific
TFs during the early stage of osteogenesis, upregulates the
transcription of various mineralized related protein genes in
osteoblasts and chondrocytes, and thus promotes these cells to
differentiate into osteoblasts (18,19).
Hence, Runx-2 is also proved to play a key role in bone metabolism
regulation and bone development, indicating it is of great
significance to prevent and treat OP. Osterix, a
zinc-finger-containing TF, is an essential osteogenic marker for
the differentiation of preosteoblasts into mature osteoblasts
during the late stage of osteoblast differentiation, which is
required for bone formation (20,21). It
is reported that nuclear receptors, emerged as a family of TFs, are
fundamental regulators of maintaining bone development and
remodeling, besides, several drugs targeting it are widely applied
in treating bone diseases such OP via regulating rates of bone
formation and resorption (22).
Although some TFs are proved to play an essential role in bone
metabolism, global prediction of TFs in OP are still relatively
scarce.
Thus, in the present study, comprehensive
bioinformatics methods were introduced to obtain key TFs from a
global point of view. First, 300 DEGs were obtained including top
10 DEGs such as LYG2, PPEF1, TTC16 all upregulated in OP according
to 5 OP samples compared with 5 normal samples. Following, based on
third bioinformatics methods, 165 TF targets, 87 TF targets and 178
TF targets were identified, respectively. The 178 TF targets had
the most coverage of TFs, thus, the optimal TF set of these TF
targets were FOXO1, KLF16, RXRA, RARA, HNF4A, CEBPB, ESR1, SOX8,
ZNF219, and SP1. It is suggested that deletion of
forkhead box O1 (FoxO1), one of members of the Forkhead box O
(FOXO) family of TFs, could lead to a bone formation increase,
which was maintained up to 24 months in mice while a lower number
of adipocytes in the bone marrow of Foxo-deleted mice at this late
age was presented (23), indicating
FoxO1 possibly plays a crucial regulatory role in bone metabolism.
It has also been demonstrated that Retinoid-X receptor-α (RXRA),
one of nuclear hormone superfamily, is an essential cofactor in the
action of 1,25-dihydroxyvitamin D (1,25[OH]2-vitamin D) and
epigenetically regulates activation of vitamin D, indicating it may
influence bone mineral accrual (24–26). It
is shown that CCAAT/enhancer-binding protein β (CEBPB), a TF,
reduced bone mass in knockout mice (27). It has been found that estrogen
receptor α (ESR1) is associated with low mineral osseous
densitometry in women after menopause (28). Schmidt et al have shown that
Sox8-deficient mouse exhibit a severely impaired bone formation,
which is modulated by a strongly reduced expression of runt-related
TF 2 (29). Taken together, the
above evidence indicates that the comprehensive bioinformatics
analysis of TFs from a point of view provided by our study may aid
in a better understanding of the molecular mechanism of OP. Albeit
a novel insight of comprehending OP molecular pathogenesis emerged,
many further studies are required due to the presence of some
limitations in this study. Relatively small sample number and
datasets, verification of in vitro and in vivo
experiments are needed in further investigations.
In conclusion, our study provided novel insight into
the mechanism by which crucial TFs were identified by comprehensive
bioinformatics analysis of OP microarray data. These selected TFs
are possible potential targets in the management of OP.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
CSW conceived the study and drafted the manuscript.
YLiu, YLi and XL performed the experiments, analyzed the data and
revised the manuscript. All authors read and approved the final
study.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pavone V, Testa G, Giardina SMC, Vescio A,
Restivo DA and Sessa G: Pharmacological therapy of osteoporosis: A
systematic current review of literature. Front Pharmacol.
8:8032017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dhruv HD, McDonough Winslow WS, Armstrong
B, Tuncali S, Eschbacher J, Kislin K, Loftus JC, Tran NL and Berens
ME: Reciprocal activation of transcription factors underlies the
dichotomy between proliferation and invasion of glioma cells. PLoS
One. 8:e721342013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheng D, Wang S, Jia W, Zhao Y, Zhang F,
Kang J and Zhu J: Regulation of human and mouse telomerase genes by
genomic contexts and transcription factors during embryonic stem
cell differentiation. Sci Rep. 7:164442017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Silva LM, Hirai KE, de Sousa JR, de Souza
J, Dias LB Jr, Carneiro FRO, Aarão TLS, Fuzii HT and Quaresma JAS:
NFκB transcription factor (p65) immunohistochemistry in leprosy
dermal microvasculature. Microb Pathog. 113:427–431. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He X, Lindsay-Mosher N, Li Y, Molinaro AM,
Pellettieri J and Pearson BJ: FOX and ETS family transcription
factors regulate the pigment cell lineage in planarians.
Development. 144:4540–4551. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heppler LN and Frank DA: Targeting
oncogenic transcription factors: Therapeutic implications of
endogenous STAT inhibitors. Trends Cancer. 3:816–827. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ruetz T, Pfisterer U, Di Stefano B,
Ashmore J, Beniazza M, Tian TV, Kaemena DF, Tosti L, Tan W, Manning
JR, et al: Constitutively active SMAD2/3 are broad-scope
potentiators of transcription-factor-mediated cellular
reprogramming. Cell Stem Cell. 21:791–805.e9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garg P, Mazur MM, Buck AC, Wandtke ME, Liu
J and Ebraheim NA: Prospective review of mesenchymal stem cells
differentiation into osteoblasts. Orthop Surg. 9:13–19. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Benisch P, Schilling T, Klein-Hitpass L,
Frey SP, Seefried L, Raaijmakers N, Krug M, Regensburger M, Zeck S,
Schinke T, et al: The transcriptional profile of mesenchymal stem
cell populations in primary osteoporosis is distinct and shows
overexpression of osteogenic inhibitors. PLoS One. 7:e451422012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng G, Tu K, Yang Q, Xiong Y, Wei C, Xie
L, Zhu Y and Li Y: ITFP: An integrated platform of mammalian
transcription factors. Bioinformatics. 24:2416–2417. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marbach D, Lamparter D, Quon G, Kellis M,
Kutalik Z and Bergmann S: Tissue-specific regulatory circuits
reveal variable modular perturbations across complex diseases. Nat
Methods. 13:366–370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han H, Shim H, Shin D, Shim JE, Ko Y, Shin
J, Kim H, Cho A, Kim E, Lee T, et al: TRRUST: A reference database
of human transcriptional regulatory interactions. Sci Rep.
5:114322015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Szklarczyk D, Franceschini A, Kuhn M,
Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork
P, et al: The STRING database in 2011: Functional interaction
networks of proteins, globally integrated and scored. Nucleic Acids
Res. 39:(Database). D561–D568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wilkinson AC, Nakauchi H and Göttgens B:
Mammalian transcription factor networks: Recent advances in
interrogating biological complexity. Cell Syst. 5:319–331. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cohen-Solal KA, Kaufman HL and Lasfar A:
Transcription factors as critical players in melanoma invasiveness,
drug resistance, and opportunities for therapeutic drug
development. Pigment Cell Melanoma Res. 31:241–252. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thu HE, Mohamed IN, Hussain Z and Shuid
AN: Exploring molecular mechanism of bone-forming capacity of
Eurycoma longifolia: Evidence of enhanced expression of
bone-related biomarkers. J Ayurveda Integr Med.
S0975-9476(17)30007-4. 2017.PubMed/NCBI
|
|
19
|
Zhao XL, Chen JJ, Zhang GN, Wang YC, Si
SY, Chen LF and Wang Z: Small molecule T63 suppresses osteoporosis
by modulating osteoblast differentiation via BMP and WNT signaling
pathways. Sci Rep. 7:103972017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang C: Transcriptional regulation of
bone formation by the osteoblast-specific transcription factor Osx.
J Orthop Surg Res. 5:372010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Choi YH, Han Y, Jin SW, Lee GH, Kim GS,
Lee DY, Chung YC, Lee KY and Jeong HG: Pseudoshikonin I enhances
osteoblast differentiation by stimulating Runx2 and Osterix. J Cell
Biochem. 119:748–757. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zuo H and Wan Y: Nuclear receptors in
skeletal homeostasis. Curr Top Dev Biol. 125:71–107. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Iyer S, Ambrogini E, Bartell SM, Han L,
Roberson PK, de Cabo R, Jilka RL, Weinstein RS, O'Brien CA,
Manolagas SC, et al: FOXOs attenuate bone formation by suppressing
Wnt signaling. J Clin Invest. 123:3409–3419. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takeyama K and Kato S: The vitamin D3
1alpha-hydroxylase gene and its regulation by active vitamin D3.
Biosci Biotechnol Biochem. 75:208–213. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li G, Yin W, Chamberlain R, Hewett-Emmett
D, Roberts JN, Yang X, Lippman SM and Clifford JL: Identification
and characterization of the human retinoid X receptor alpha gene
promoter. Gene. 372:118–127. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang M, Zhao LJ, Zhou Y, Badr R, Watson
P, Ye A, Zhou B, Zhang J, Deng HW, Recker RR, et al: SNP rs11185644
of RXRA gene is identified for dose-response variability to vitamin
D3 supplementation: A randomized clinical trial. Sci Rep.
7:405932017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brommage R, Liu J, Hansen GM, Kirkpatrick
LL, Potter DG, Sands AT, Zambrowicz B, Powell DR and Vogel P:
High-throughput screening of mouse gene knockouts identifies
established and novel skeletal phenotypes. Bone Res. 2:140342014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gonçalves CG, Almeida BC, Camargo-Kosugi
CM, Costa AM, Silva ID and Haidar MA: Polymorphisms in CYP17, COMT,
and ESR1 genes in women after menopause and association with bone
mineral density. Genet Mol Res. 14:15802–15810. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schmidt K, Schinke T, Haberland M, Priemel
M, Schilling AF, Mueldner C, Rueger JM, Sock E, Wegner M and Amling
M: The high mobility group transcription factor Sox8 is a negative
regulator of osteoblast differentiation. J Cell Biol. 168:899–910.
2005. View Article : Google Scholar : PubMed/NCBI
|