Introduction
Hemorrhagic shock and resuscitation (HSR) is known
to induce a pulmonary inflammatory response that leads to acute
lung injury, which is referred to as acute respiratory distress
syndrome (ARDS) (1–4). Although lung protective ventilation has
been proposed for the treatment of ARDS in clinical settings
(5,6), there is no definitive pharmacological
therapy to prevent pulmonary inflammation in ARDS (6–8).
Carbon monoxide (CO) is widely known to be a toxic
gaseous molecule that produces carboxyhemoglobin (COHb) due to its
higher affinity to hemoglobin (9,10).
However, small amounts of CO are endogenously produced by the
enzymatic reaction of heme oxygenase (HO) that degrades heme to
biliverdin, iron (II) ion, and CO. In addition, heme oxygenase-1
(HO-1), the inducible isoform of HO that is rapidly induced by a
vast array of stress stimuli, plays an important role in organ
protection by degrading free heme, a prooxidant, and producing CO
(11,12). Recent findings have indicated that
exogenous inhalation of low-dose concentrations of CO conferred
cytoprotective effects against insults in various animal models of
inflammatory disorders (13–17). Our previous study also demonstrated
that inhalation of CO at 250 ppm significantly ameliorated the
HSR-induced acute lung injury via its anti-inflammatory and
anti-apoptotic properties (18).
However, even this low dose of CO inhalation therapy elevated
arterial COHb levels by approximately 20% (19), which may be toxic to humans (20). To avoid the adverse effect of CO
inhalation, carbon monoxide-releasing molecules (CORMs) have been
developed by coordinating CO with transition metal carbonyl
complexes, which spontaneously liberate and deliver CO to organ
tissues under physiological conditions through all possible routes
of administration (21–24). CORMs have also been reported to exert
cytoprotective effects against various animal models of oxidative
damage without altering the blood COHb levels (22–28).
However, to our knowledge, few studies have focused on the effect
of CORMs on HSR-induced acute lung injury, although a previous
report showed the beneficial effect of CORM-3
[tricarbonylchloro-glycinate ruthenium(II) (Ru(CO)3Cl-glycinate); a
water-soluble form of CORMs] on HSR-induced liver injury (29). Moreover, the influence of CORMs on
arterial oxygenation and hemodynamics during the acute phase of HSR
remains to be elucidated.
Thus, in the present study, we investigated whether
CORM-3 administration immediately after resuscitation exerted a
therapeutic effect on HSR-induced lung injury in a rat HSR model
without showing any detrimental influence on oxygenation and
hemodynamics.
Materials and methods
Animals
This study was approved by the Animal Use and Care
Committee of the Okayama University Medical School (OKU-2015429) on
September 2, 2015 and conformed to the guidelines for the care and
use of laboratory animals that followed the ARRIVE (Animal
Research: Reporting of In Vivo Experiments) guidelines
(30) and the 2013 AVMA euthanasia
guidelines (31). Male
Sprague-Dawley rats weighing 370 to 430 g were purchased from Clea
Japan, Inc. (Tokyo, Japan). The rats were housed in
temperature-controlled rooms at 25°C with 12-h light/dark cycles
and allowed free access to water and chew until the start of
experiments. The total number of rats was 117 in this study; sham
group (n=29), HSR group (n=27), HSR/CORM-3 group (n=38), and
HSR/inactive CORM-3 (iCORM-3) group (n=23).
Preparation of drugs
Water-soluble CORM-3 was purchased from
Sigma-Aldrich Japan Inc. (Tokyo, Japan). CORM-3 was solubilized in
distilled water (20 mg/ml stock) and stored at −20°C until the
experiments. iCORM-3, an inactive counterpart of CORM-3, was
prepared by incubating CORM-3 in a phosphate-buffered solution (pH
7.4) at room temperature for 2 days to liberate all of the CO gas
from the molecule before the experiment (32). At the time of administration, the
stock solution was diluted four-fold with sterile saline to make a
5 mg/ml final concentration of CORM-3 and iCORM3. Then, 4 mg/kg of
CORM-3 or iCORM-3 (0.8 ml/kg of CORM-3 or iCORM-3 solution) was
intravenously administered through the left femoral vein.
Protocol for HSR
Rats were anesthetized with intraperitoneal sodium
pentobarbital (50 mg/kg) injection and subjected to the HSR
procedure as previously described (33,34). In
brief, the left inguinal artery and vein were dissected using
aseptic techniques, and heparinized polyethylene catheters were
inserted into the left femoral vessels. The left femoral artery
catheter was used to monitor the arterial blood pressure during the
HSR procedure while the left femoral vein catheter was used to
withdraw or return the shed blood. Hemorrhagic shock was induced by
collecting blood into a heparinized syringe (10 units/ml) from the
left femoral vein over 15 min until a mean arterial blood pressure
of 30 mmHg was achieved. This pressure level was maintained for an
additional 45 min by further blood withdrawal, after which the rats
were resuscitated by reinfusing all the shed blood over 15 min.
After a 45-min post-reinfusion monitoring period, the surgical
incision was closed. The rats in the sham group underwent all
surgical procedures except bleeding. The animals were allowed to
breathe spontaneously without tracheal intubation throughout the
experiment. All procedures were performed on a heating pad that
could perform automatic regulation of rectal body temperature
within the physiological range.
Experimental design
To examine the effects of CORM-3 treatment on
HSR-induced lung injury, the rats subjected to HSR were randomly
assigned to the following groups based on the treatment at the end
of resuscitation: The HSR/CORM-3 group was administered 4 mg/kg of
CORM-3; the HSR/iCORM-3 group was administered 4 mg/kg of iCORM-3;
and the HSR group was vehicle-treated (same amount of normal saline
as CORM-3 or iCORM-3). Drugs were intravenously administered
through the left femoral vein immediately after returning all the
shed blood. At specific time points following resuscitation, rats
were euthanized by exsanguination from the left femoral artery
under inhaled anesthesia by ethyl ether with spontaneous breathing.
The lungs were excised and frozen immediately in liquid nitrogen
and stored at −80°C until further use. For histological
examinations, the right upper lobe of the lung was fixed in 10%
neutral buffered formalin and embedded in paraffin.
Arterial blood gas analysis
Arterial blood samples were drawn from the left
femoral artery catheter at the following time points during HSR: 0
min, at the beginning of the hemorrhagic shock; 60 min, at the end
of the hemorrhagic shock; 80 min, at 5 min after the completion of
the blood reinfusion and the subsequent drug administration; and
120 min, at the end of the monitoring phase after resuscitation.
Arterial blood gas (ABG) analysis was performed with ABL80 CO-OX
(Radiometer Medical ApS, Copenhagen, Denmark).
Histopathological examination
For histological examinations, the right upper lobe
of the lungs was excised 12 h after resuscitation and fixed in 10%
neutral buffered formalin, embedded in paraffin, and sectioned at a
thickness of 5 µm. After deparaffinization and dehydration, the
sections were stained with hematoxylin and eosin for microscopic
examination. Lung histological changes were assessed on a light
microscope by three observers in a blinded fashion, in accordance
with previously described methods (35–37) with
modifications for five independent experiments. In brief, a total
of 10 randomly selected areas from each lung section were graded
separately as 0 (no findings or normal), 1 (mild), 2 (moderate), or
3 (severe) for each of the following four parameters: Congestion,
pulmonary edema, cellular infiltration, and hemorrhage. The sum of
the individual scores for each parameter was calculated as the lung
injury score.
Terminal deoxynucleotidyl
transferase-mediated dUTP-fluorescein isothiocyanate nick-end
labeling staining
Transferase-mediated dUTP-fluorescein isothiocyanate
(FITC) nick-end labeling (TUNEL) staining was performed using the
MEBSTAIN Apoptosis TUNEL Kit Direct (No. 8445; MBL, Nagano, Japan)
according to the manufacturer's instructions. Sections were
incubated with terminal deoxynucleotidyl transferase and
FITC-conjugated dUTP, followed by counterstaining with 0.5 µg/ml
propidium iodide. The number of TUNEL-positive cells was counted in
five nonconsecutive sections per rat at a magnification of ×400 in
a blinded manner with a Zeiss confocal laser scanning microscope
model LSM510 (Zeiss, Jena, Germany).
Lung wet weight to dry weight
(wet/dry) ratio
Left lung tissue samples obtained 12 h after
resuscitation were weighed (wet weight) and dried at 110°C for 24
h. Then, the dry tissue weight was measured, and the wet/dry weight
ratio was determined as an indicator of pulmonary edema by dividing
the lung wet weight by the lung dried weight (19,34,38).
Measurement of protein concentration
in the bronchoalveolar lavage fluid
Protein concentration in the bronchoalveolar lavage
(BAL) fluid was measured at 6 h after HSR. Rats were subjected to
euthanasia and tracheotomy at 6 h after HSR, followed by lavage via
the tracheal tube with 5 ml of saline (39). The collected BAL fluids were
centrifuged at 240 × g for 15 min at 4°C, and the supernatants were
stored at −30°C until measurement. Protein concentrations in the
BAL fluids were determined using the Pierce™ BCA Protein Assay Kit
(Pierce, Rockford, IL, USA) according to the manufacturer's
instructions. The absorbance was measured with a microplate reader,
Multiskan JX (Thermo Fisher Scientific, Inc., Yokohama, Japan).
RNA isolation and northern blot
analysis
Total RNA was isolated from lung tissues at 3 h
after HSR by using TRI REAGENT® (Molecular Research
Center, Inc., Cincinnati, OH, USA) according to the manufacturer's
instructions. Northern blot analysis was performed as described
previously (40,41). In brief, 20 µg of total RNA was
subjected to electrophoresis in 1.2% (w/v) agarose gel containing
6.5% (v/v) formaldehyde and transferred to a Bio-Rad Zeta-Probe GT
Blotting Membrane (Bio-Rad Laboratories, Richmond, CA, USA). The
blotted membrane was hybridized with [α-32P] dCTP-labeled cDNA
probes for tumor necrosis factor (TNF)-α and inducible nitric oxide
synthase (iNOS), respectively, followed by rigorous washing. The
membrane was exposed to a sheet of Fuji Medical X-ray film
(Fujifilm Co., Tokyo, Japan) with an intensifying screen at −70°C.
Autoradiographs were quantified with an image scanner (ChemiDoc™
XRS plus; Bio-Rad Laboratories, Tokyo, Japan) and computerized
image analysis software (Image Lab™ version 5.0; Bio-Rad
Laboratories, Tokyo, Japan). The relative amounts of radiolabeled
cDNA that hybridized to the blots were normalized to the levels of
18S ribosomal RNA for correction of loading errors.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from lung tissues as
described above. Purification of total RNA was performed using the
RNeasy® Mini kit (Qiagen Sciences, Germantown, MD, USA).
After removal of potentially contaminating DNA with DNase I
(RNase-Free DNase set; Qiagen GmbH, Hilden, Germany), reverse
transcription of total RNA was performed by using a
QuantiTect® Reverse Transcription Kit (Qiagen GmbH,
Hilden, Germany) to generate first-strand cDNA. The PCR reaction
mixture was prepared by using SYBR® Premix Ex Taq™
(Takara Bio Inc., Shiga, Japan). The PCR was performed using a
LightCycler (Roche Diagnostics GmbH, Mannheim, Germany), as
previously described (42). The
sequences of the upstream and downstream primers for interleukin-10
(IL-10), interleukin-1β (IL-1β), and β-actin were as follows:
5′-ACGCTGTCATCGATTTCTC-3′ and 5′-GGCCTTGTAGACACCTTTG-3′ for IL-10;
5′-AGCTATGGCAACTGTCCCTGAA-3′ and 5′-CATCTGGACAGCCCAAGTCAAfor IL-1β;
5′-AACCCTAAGGCCAACCGTGAA-3′ and 5′-CAGGGACAACACAGCCTGGA-3′ for
β-actin, respectively. The mRNA levels of IL-10 and IL-1β were
normalized to the mRNA level of β-actin.
Protein extraction and measurement of
protein expression by enzyme-linked immunosorbent assay
(ELISA)
To extract protein from lung tissue, lung
homogenates at 12 h after HSR were prepared as previously described
with minor modifications (43). In
brief, 0.4 g of lung samples were mixed with 1 ml of a
phosphate-buffered saline −0.1% Triton-X100 solution containing a
protease inhibitor cocktail (cOmplete; Roche Diagnostics
Indianapolis, IN, USA), homogenized, and centrifuged at 23,800 × g
for 15 min at 4°C. The supernatants were collected and stored for
subsequent analysis. The protein levels of IL-1β and macrophage
inflammatory protein-2 (MIP-2) in the lung homogenates were
determined by an ELISA kit (R&D Systems®,
Minneapolis, MN, USA) according to the manufacturer's instructions.
All assays were performed in duplicate. Data are presented as the
amount per gram of fresh weight of lung tissue.
Western blot analysis
Western blot analysis was performed as described
previously (44). The protein
concentrations of lung homogenates were determined using the Pierce
BCA™ Protein Assay Kit (Pierce, Rockford, IL, USA). Samples
equivalent to 50 µg were applied to 12.5 or 15% (w/v)
polyacrylamide-SDS gels. After electrophoretic separation, the
proteins were transferred to ‘Amersham’ Hybond-polyvinylidene
fluoride membranes (GE Healthcare Japan Co., Tokyo, Japan). The
membrane was blocked with Tris-buffered saline containing 4% (w/v)
BlockAce™ (DS Pharma Biomedical Co., Ltd., Osaka, Japan) for 1 h at
room temperature, followed by incubation with the primary antibody
against cleaved caspase-3 (rabbit anti-cleaved caspase-3 [Asp175]
polyclonal antibody: 9661S, Cell Signaling, 1:500 dilution) or
GAPDH (rabbit anti-GAPDH [FL-335] polyclonal antibody: sc-25778,
SANTA CRUZ, 1:5,000 dilution) for 20 h at 4°C. After washing the
membranes with buffer, the membranes were incubated with the
secondary antibody (goat anti-rabbit IgG-HRP: sc-2004; Santa Cruz,
1:20,000 dilution) for 30 min at room temperature. The membranes
were stained with Clarity Western ECL Substrate (Bio-Rad, USA)
according to the manufacturer's instructions. Antigen-antibody
complexes were visualized with an image scanner (ChemiDoc XRS Plus
Imaging System, Bio-Rad) and the intensity of signals was
quantified by using an analysis software (Image Lab Version 5.0,
Bio-Rad).
Statistical analysis
Data are expressed as mean ± standard error of the
mean (SEM). Statistical analyses were performed using analysis of
variance followed by the Tukey-Kramer multiple comparisons method,
as appropriate. A two-sided P<0.05 was considered to indicate a
statistically significant difference. Comparisons were examined by
using JMP Pro 12® software (SAS Institute Inc., Cary,
NC, USA).
Results
Effect of CORM-3 on HSR-induced
histological damage in the lung
First, we assessed the effect of CORM-3 on
HSR-induced histological damage in the lungs 12 h after
resuscitation by performing microscopic examinations of lung
sections stained with hematoxylin and eosin (magnification, ×400).
Lung sections of the HSR group revealed prominent histopathological
changes, including inflammatory cell infiltration, alveolar septal
thickening, interstitial edema, and hyaline membrane formation,
while those of the sham group appeared almost normal. In contrast,
CORM-3 treatment following HSR markedly improved these
histopathological changes, although iCORM-3 administration did not
affect the pathological findings caused by HSR (Fig. 1A). These findings were confirmed by
the lung injury score, which was used to evaluate histopathological
changes related to congestion, edema, inflammation, and hemorrhage
in a blinded fashion. The lung injury scores in rats of the HSR
group were significantly higher than those in the sham group, but
CORM-3 treatment significantly reduced the score to levels almost
identical to those in sham-operated rats (Fig. 1B).
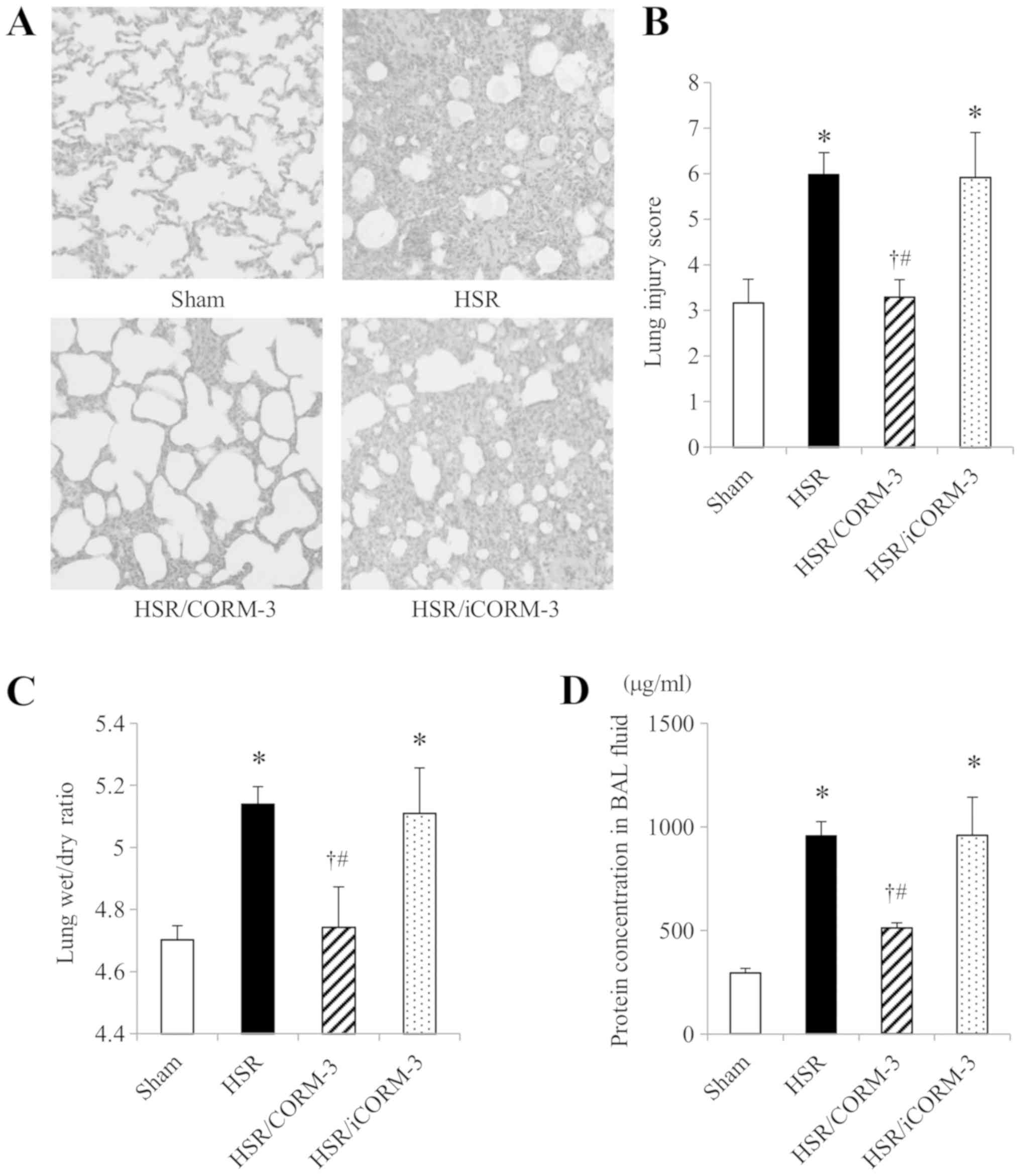 | Figure 1.Effect of CORM-3 on HSR-induced lung
histological damage, lung wet/dry ratio, and the protein
concentration in the BAL fluid. Lungs were excised 12 h following
resuscitation and stained with hematoxylin and eosin for
microscopic examination. Lung wet/dry ratio and protein
concentration in the BAL fluid were evaluated at 6 h following HSR.
(A) Representative images from five independent experiments
(hematoxylin-eosin staining; original magnification, ×400). (B)
Lung injury score; the severity of histopathological changes in the
lungs 12 h following HSR was assessed by three independent
observers. Ten areas randomly selected from each lung section were
graded separately as 0 (no findings or normal), 1 (mild), 2
(moderate), or 3 (severe) for each of the following four
parameters: Congestion, pulmonary edema, cellular infiltration, and
hemorrhage. The sum of these four scores was calculated as the lung
injury score. (C) Lung wet/dry ratio. Excised left lungs at 12 h
following HSR were weighed (wet weight) and dried at 110°C for 24 h
(dry weight). The wet/dry ratio was calculated by dividing the lung
wet weight by the dry weight. (D) Protein concentration in the BAL
fluid. BAL fluid was collected by lavage with 5 ml of saline via a
tracheotomy at 6 h after HSR. Statistical analysis was performed
using analysis of variance followed by Tukey-Kramer multiple
comparisons method. Data in each analysis are presented as the mean
± SEM (n=5 for each group). *P<0.05 (sham vs. HSR, sham vs.
HSR/iCORM-3), †P<0.05 (HSR vs. HSR/CORM-3), and
#P<0.05 (HSR/CORM-3 vs. HSR/iCORM-3). BAL,
bronchoalveolar lavage; CORM-3, carbon monoxide-releasing
molecule-3; HSR, hemorrhagic shock and resuscitation; iCORM-3,
inactive carbon monoxide-releasing molecule-3; SEM, standard error
of the mean. |
Effect of CORM-3 on lung wet/dry ratio
and the protein concentration in the BAL fluid
Since the lung histological examination showed
HSR-induced pulmonary edema and its improvement by CORM-3, we
further evaluated the lung wet/dry ratio and the protein
concentration in the BAL fluid as indicators of lung edema and
vascular permeability, respectively. Consistent with the
histological findings, the lung wet/dry ratio at 12 h after HSR in
the HSR group was markedly higher than that in the sham group
(Fig. 1C). In contrast, CORM-3
treatment following HSR significantly suppressed the HSR-induced
increase in the lung wet/dry ratio. However, the ratio in the
HSR/iCORM-3 group was as high as that in the HSR group.
Furthermore, we also found that the protein concentration in the
BAL fluid in HSR animals was significantly higher (approximately
3-fold higher) than that in sham-operated animals (Fig. 1D). Consistent with the observations
for the wet/dry ratio, only CORM-3 treatment significantly reduced
the protein concentration. These results, together with the
histopathological findings, indicate that CORM-3 treatment
ameliorated lung edema and the HSR-induced increase in pulmonary
vascular permeability.
Effect of CORM-3 on gene expressions
of proinflammatory mediators in the lung
CORM-3 treatment significantly suppressed the
HSR-induced infiltration of inflammatory cells to the lung in
histological examinations. Therefore, in order to clarify the
anti-inflammatory effect of CORM-3, we examined the effect of
CORM-3 on the gene expressions of inflammatory mediators in the
lung. We measured the gene expression levels of the proinflammatory
mediators TNF-α and iNOS by northern blot analysis and that of
IL-1β by RT-qPCR using lung samples obtained 3 h after HSR. While
the mRNA levels in the sham group were relatively low, those in the
HSR group had significantly increased (Fig. 2A-C). In contrast, CORM-3 treatment
markedly reduced the increased expression levels observed in the
HSR group to less than half of the mRNA levels in the HSR group,
although the HSR/iCORM-3 group showed no change in those mRNA
levels in comparison with the levels in the HSR group (Fig. 2A-C).
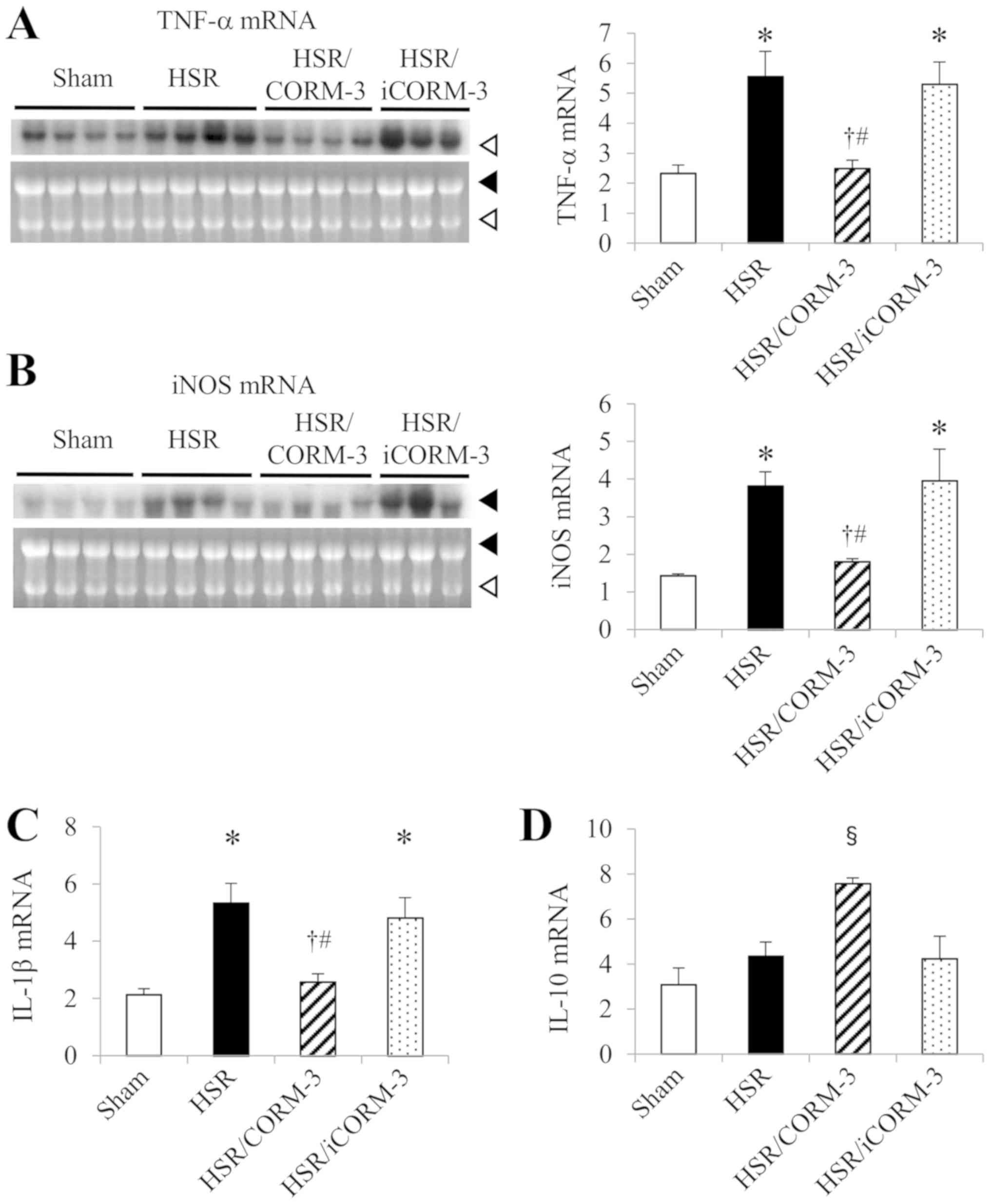 | Figure 2.Effect of CORM-3 on gene expressions
of pro- and anti-inflammatory mediators in the lung following HSR.
Lungs from HSR rats were excised at 3 h following resuscitation.
Then, the mRNA levels of TNF-a and iNOS were measured by northern
blot analysis. (A and B) The closed arrowhead indicates 28S
ribosomal RNA, while the open arrowhead indicates 18S ribosomal
RNA. The relative amounts of hybridized radiolabeled cDNAs were
normalized to 18S ribosomal RNA levels; mRNA levels of IL-1β (C)
and IL-10 (D) were measured by RT-qPCR. Data from RT-qPCR were
normalized to β-actin levels. Statistical analysis was performed
using analysis of variance followed by the Tukey-Kramer multiple
comparisons method among groups. Data in each analysis are
presented as the mean ± SEM in arbitrary units (n=3–5 for each
group). *P<0.05 (sham vs. HSR, sham vs. HSR/iCORM-3),
†P<0.05 (HSR vs. HSR/CORM-3),
#P<0.05 (HSR/CORM-3 vs. HSR/iCORM-3), and
§P<0.05 (HSR/CORM-3 vs. sham, HSR, and HSR/iCORM-3).
CORM-3, carbon monoxide-releasing molecule-3; HSR, hemorrhagic
shock and resuscitation; iCORM-3, inactive carbon
monoxide-releasing molecule-3; IL-1β, interleukin-1β; IL-10,
interleukin-10; iNOS, inducible nitric oxide synthase; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction; SEM,
standard error of the mean; TNF-α, tumor necrosis factor-α. |
Effect of CORM-3 on the expressions of
proinflammatory mediators in the lung
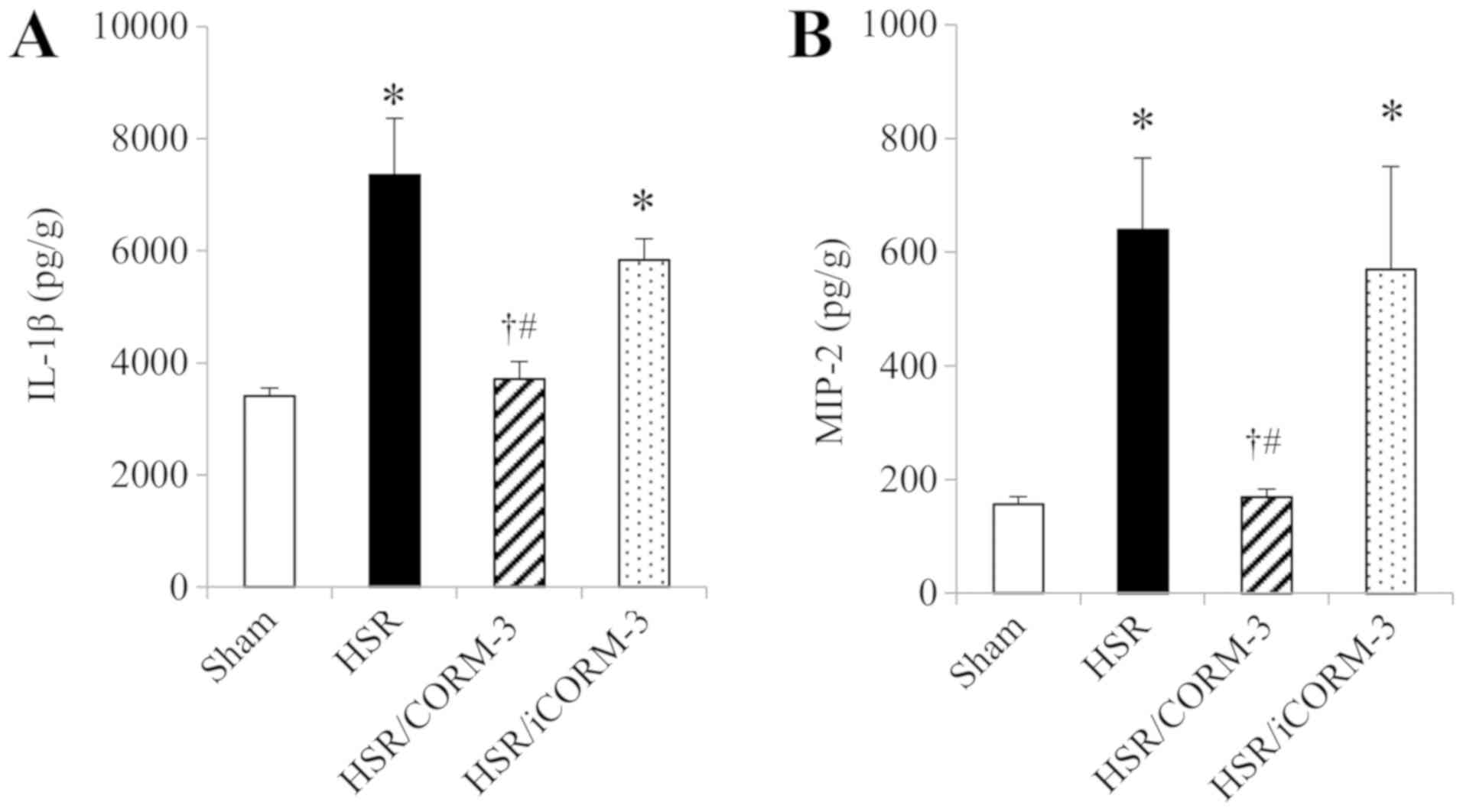
Next, we measured the protein expression levels of
inflammatory cytokines such as IL-1β and MIP-2 in the lung
homogenates at 12 h following HSR by ELISA. The expression levels
of these cytokines were markedly higher in the HSR group than in
the sham group (Fig. 3A and B). In
contrast, CORM-3 treatment significantly reduced the elevated
expression levels of these cytokines to almost the same levels as
those observed in the sham group, while iCORM-3 did not show any
influence on the HSR-induced upregulation of these inflammatory
cytokines (Fig 3A and B).
Effect of CORM-3 on the gene
expression of IL-10, an anti-inflammatory cytokine, in the
lung
To further investigate the anti-inflammatory effects
of CORM-3, we examined the gene expression of IL-10, an
anti-inflammatory cytokine, by RT-qPCR in the lungs at 3 h after
HSR. Although pulmonary gene expression of IL-10 mRNA was barely
detectable in the sham group, it was increased by the HSR
procedure. Notably, the CORM-3 treatment group showed drastically
upregulated IL-10 mRNA levels compared to those in the other three
groups (Fig. 2D).
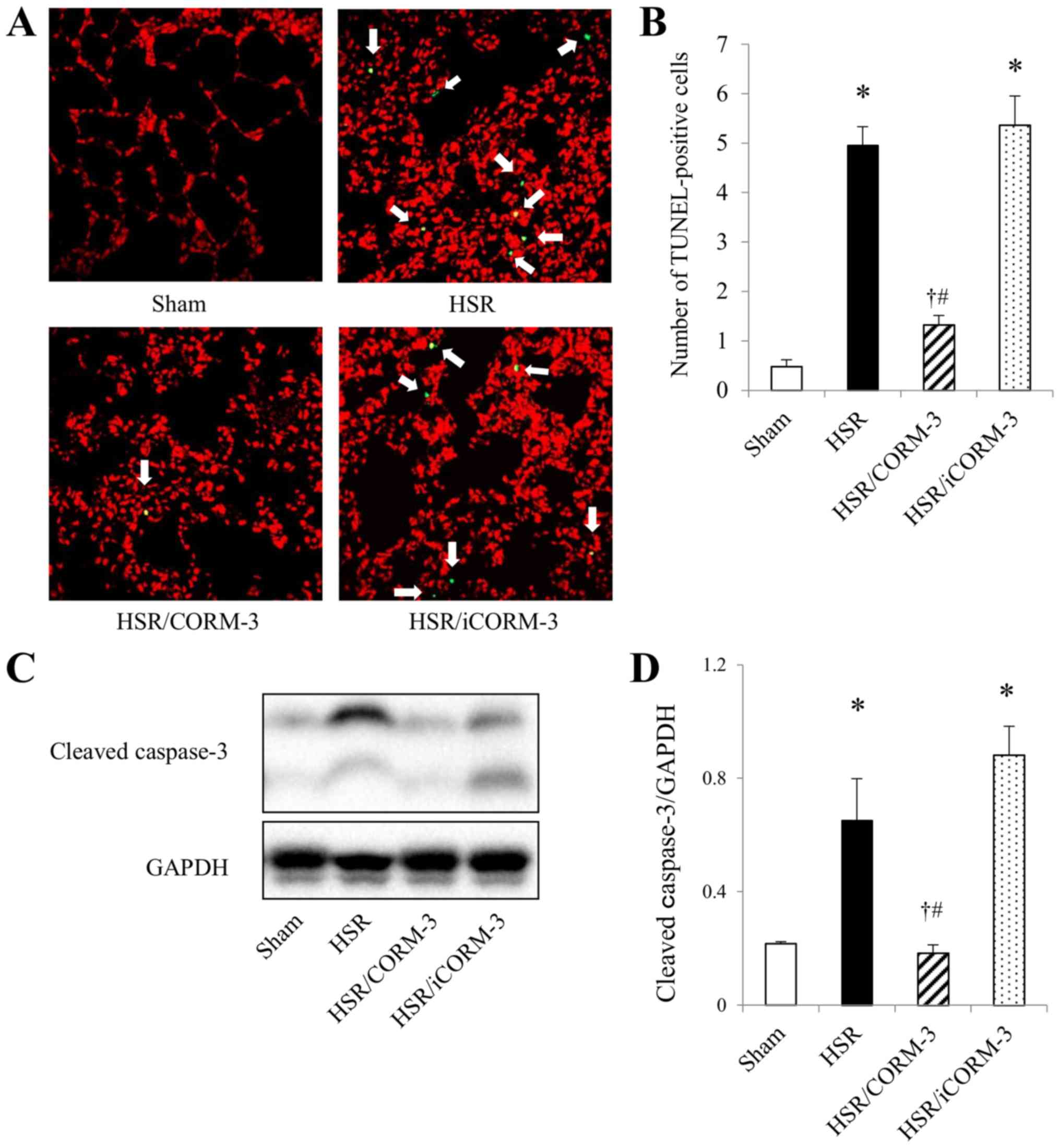
Effect of CORM-3 on apoptotic cell
death in the lungs following HSR
Pulmonary ischemia reperfusion induces tissue
inflammation, which causes oxidative stress and ultimately leads to
the apoptosis of alveolar endothelial cells (45). Therefore, we examined the effect of
CORM-3 on apoptotic cell death by performing TUNEL staining, which
specifically stains the 3′-OH DNA ends generated by DNA
fragmentation of apoptosis, and assessed the protein expressions of
cleaved caspase-3, an executive molecule of apoptosis, in the lungs
at 12 h after HSR. While the lung sections from the sham group
showed a few TUNEL-positive cells, the HSR procedure resulted in a
significant increase in the number of TUNEL-positive cells
(Fig. 4A and B). In contrast, CORM-3
treatment significantly reduced the number of TUNEL-positive cells
to approximately one-fourth of the HSR group, although the
HSR/iCORM-3 group showed an increase in the number of
TUNEL-positive cells to the same extent as that noted in the HSR
group. Consistent with the changes observed in TUNEL staining, the
level of cleaved caspase-3 expression was significantly elevated in
the HSR group, while it was barely detectable in the sham group
(Fig 4C and D). Conversely, CORM-3
treatment significantly suppressed the upregulation of cleaved
caspase-3 to one-third of that in the HSR group, whereas iCORM-3
administration did not exert any effect on the expression of
cleaved caspase-3 (Fig. 4C and D).
These findings indicate that CORM-3 treatment significantly
attenuated HSR-induced lung injury by its anti-apoptotic
effect.
CORM-3 administration has no influence
on the hemodynamic status during HSR
Since CORM-3 has been reported to have a
vasodilatory effect through the activation of soluble guanylate
cyclase and large conductance calcium-activated potassium channels
(BKCa channels) (21,32,46), we
examined the effect of CORM-3 treatment on the hemodynamic status.
Although only the shed blood was returned at the time of
resuscitation without any additional fluid requirement in the rats
subjected to HSR, the time course changes in mean arterial blood
pressure and heart rate in all experimental groups were almost
identical throughout the HSR procedure (Fig. 5), indicating that CORM-3 at this dose
did not affect the animals' hemodynamic status during HSR.
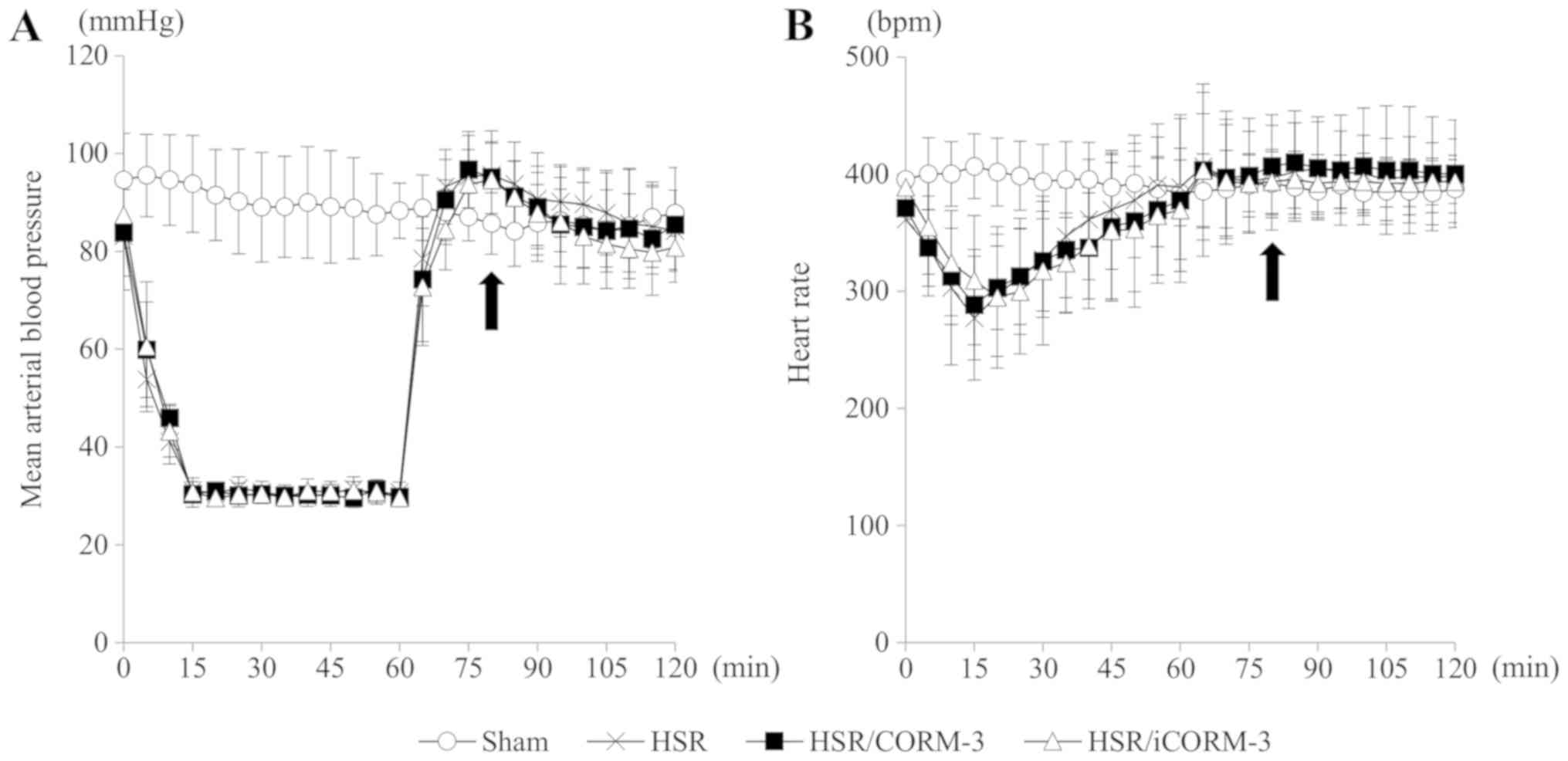 | Figure 5.CORM-3 administration has no
influence on vital signs during HSR. Rats subjected to HSR were
administered CORM-3, iCORM-3, or a vehicle (normal saline) through
the left femoral vein immediately following the completion of
resuscitation. Mean arterial blood pressure and heart rate were
measured at 5 min intervals during the HSR procedure. (A) Mean
arterial blood pressure at 5-min intervals during HSR. The
horizontal axis indicates the time after the beginning of the
hemorrhagic shock, and the vertical axis shows the mean arterial
blood pressure. The closed arrow indicates the time of the drug or
vehicle administration. (B) Heart rate at 5-min intervals during
HSR. The horizontal axis indicates the time after the beginning of
the hemorrhagic shock, and the vertical axis shows the heart rate.
The closed arrow indicates the time of the drug or vehicle
administration. Statistical analysis was performed using analysis
of variance followed by Tukey-Kramer multiple comparisons method
for the mean arterial blood pressure and heart rate at each time
point in the HSR, HSR/CORM-3, and HSR/iCORM-3 groups. Data are
presented as the mean ± SEM (n=10 for each group). CORM-3, carbon
monoxide-releasing molecule-3; HSR, hemorrhagic shock and
resuscitation; iCORM-3, inactive carbon monoxide-releasing
molecule-3; SEM, standard error of the mean. |
CORM-3 administration following HSR
has no influence on blood COHb levels and preserves oxygenation as
measured by ABG analysis during HSR
Since CORM-3 releases trace amounts of CO after
intravenous administration, its treatment may cause an increase in
blood COHb levels that could lead to derangement of the physiologic
homeostasis. Thus, we performed ABG analysis at four specific time
points during HSR, as indicated in the Materials and methods
section. Notably, there were no significant differences on the
blood COHb levels among all experimental groups at all time points,
and CORM-3 treatment did not affect arterial oxygenation, as
indicated by the maintenance of the PaO2 and
SaO2 levels (Fig. 6).
Although the levels of hemoglobin (Hb), pH, and base excess (BE) at
the end of the hemorrhagic shock in all groups subjected to HSR
were significantly lower than those in the sham group, probably
because of the systemic ischemia and metabolic acidosis induced by
the hemorrhagic shock, there were no significant differences in
these indices among the groups subjected to HSR. These results
indicate that CORM-3 treatment influenced neither blood COHb levels
nor the physiologic homeostasis in the arterial blood.
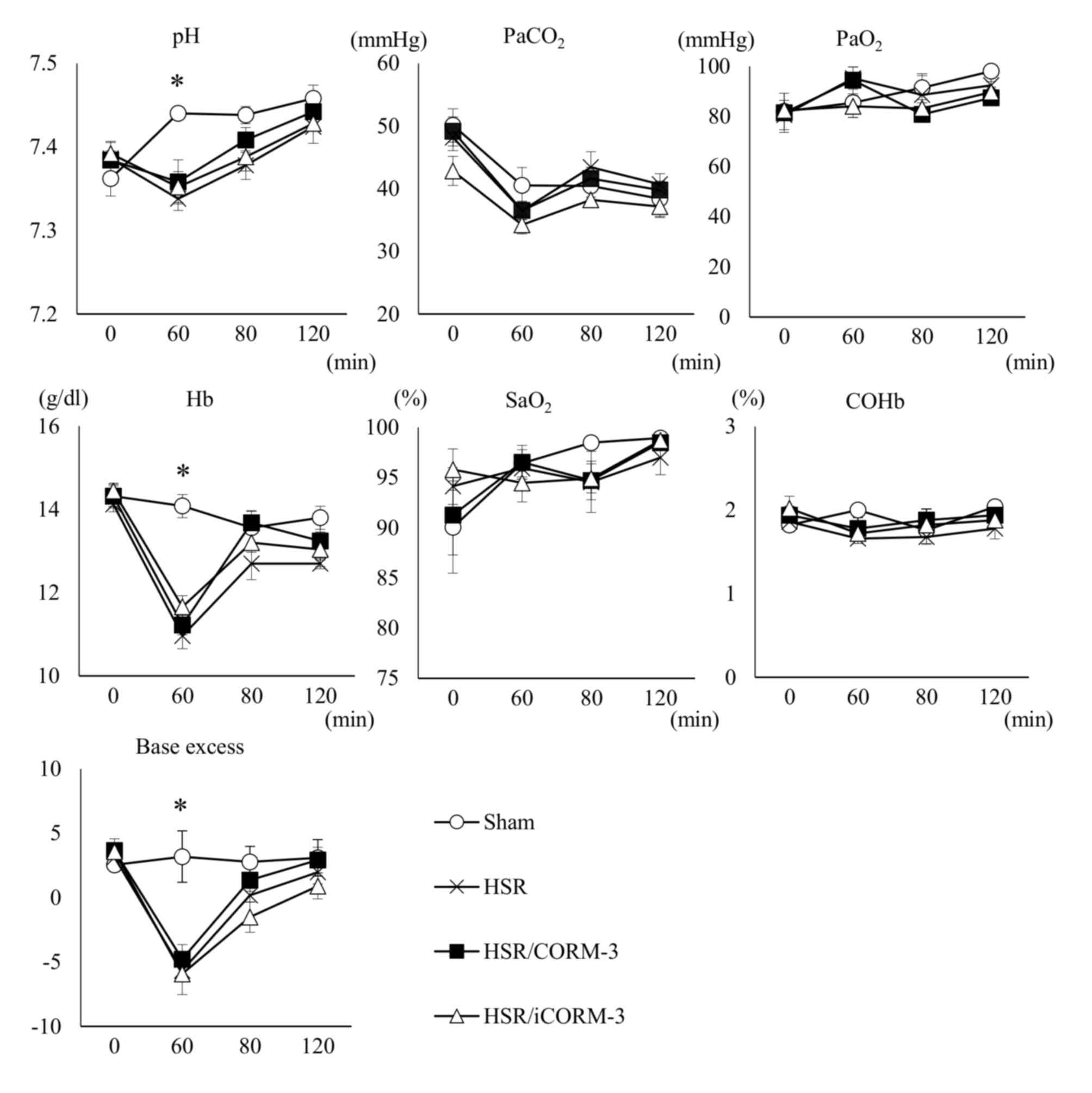 | Figure 6.CORM-3 administration following HSR
has no influence on blood carboxyhemoglobin levels and preserves
oxygenation, as measured by ABG analysis, during HSR. Arterial
blood gas (ABG) analysis was performed at the following time points
during HSR: 0 min, at the beginning of the hemorrhagic shock; 60
min, at the end of the hemorrhagic shock; 80 min, at 5 min after
the completion of blood reinfusion and the subsequent drug
administration; 120 min, at the end of the resuscitation. The
horizontal axis in each graph indicates the time after the
beginning of the hemorrhagic shock, and the vertical axis shows
each parameter of the ABG analysis. Statistical analysis was
performed using analysis of variance followed by the Tukey-Kramer
multiple comparisons method among groups. Data are presented as the
mean ± SEM (n=5 for each group). *P<0.05 (sham vs. HSR,
HSR/CORM-3, and HSR/iCORM-3 at 60 min). ABG, arterial blood gas;
COHb, carboxyhemoglobin; CORM-3, carbon monoxide-releasing
molecule-3; iCORM-3, inactive carbon monoxide-releasing molecule-3;
Hb, hemoglobin; HSR, hemorrhagic shock and resuscitation;
PaCO2, partial pressure of carbon dioxide;
PaO2, partial pressure of oxygen; SaO2,
saturation of arterial blood oxygen; SEM, standard error of the
mean. |
Discussion
The present study demonstrated that administration
of CORM-3 immediately after the completion of resuscitation
following hemorrhagic shock ameliorated the HSR-induced acute lung
injury. This fact was ascertained by the improvements in the
histological damage and lung edema caused by the increased
pulmonary vascular permeability. We also found that CORM-3
treatment significantly decreased the expressions of inflammatory
mediators, such as TNF-α, iNOS, IL-1β, and MIP-2, and increased the
expression of IL-10, an anti-inflammatory cytokine. Moreover,
CORM-3 administration decreased apoptotic cell death, evidenced by
the decrease in TUNEL-positive cells and cleaved caspase-3
expression. In contrast, CORM-3 administration did not influence
arterial COHb levels and oxygenation or exert any detrimental
effects on hemodynamic status in HSR animals. These findings
indicate that CORM-3 exerts potent therapeutic effects on
HSR-induced acute lung injury, which are at least in part mediated
through its anti-inflammatory and anti-apoptotic properties.
Hemorrhagic shock and resuscitation causes oxidative
stress and produces excess amounts of free heme by the
destabilization of hemoproteins, leading to various tissue injuries
involving the lung, liver, kidney, and intestine (12). To protect against the insult, the
enzymatic reaction of HO-1 is rapidly induced to catalyze the
degradation of free heme into biliverdin, iron (II) ion, and CO
(11,12). Our group and others have reported
that induction of HO-1 ameliorated oxidative tissue injuries
induced by HSR, and the deletion or inhibition of HO-1 conversely
exacerbated the injuries (34,47,48).
Thus, the HO-1 system is thought to constitute an essential
cytoprotective component against HSR-induced oxidative organ
damage. Although CO is a byproduct of heme breakdown, the molecule
itself has been reported to possess anti-inflammatory and
anti-apoptotic properties mediated through several signaling
pathways, including p38 mitogen-activated protein kinases (MAPK),
nuclear factor κB (NF-κB), the nucleotide-binding domain,
leucine-rich-containing family, pyrin domain-containing-3
(NLRP3)-dependent inflammasome, and modulation of mitochondrial
biogenesis, among others (49–56).
CORMs, which release CO, also have been reported to exert a
cytoprotective effect against organ injury through the same
signaling pathways as CO (26–28,54,57,58).
Previous studies for acute lung injury induced by thermal injury or
sepsis indicated that CORMs-released CO suppressed NF-κB
activation, thereby decreasing the expression of proinflammatory
mediators, including TNF-α, IL-1β, and NO (57,58). The
beneficial effects of CORMs have also been reported to be
associated with the upregulation of IL-10, mediated through
induction of HO-1, in a p38MAPK-dependent manner (27). CORMs-induced IL-10 expression
attenuates activation of the NLRP-3-dependent inflammasome, which
exacerbates the inflammatory response by producing IL-1β in
sepsis-induced lung injury (54).
These findings are consistent with our results in the present
study. As for the model of HSR-induced lung injury, our previous
studies demonstrated that CO inhalation therapy at 250 ppm exerted
anti-inflammatory and anti-apoptotic effects against HSR-induced
lung injury, mediated through the activation of peroxisome
proliferator-activated receptor (PPAR)-γ, or the suppression of
NF-κB and activator protein-1 (18,19). The
anti-inflammatory and anti-apoptotic effect of CORM-3 demonstrated
in the present study might be involved in those upstream signaling
pathways. Although further investigation is needed, the present
study is valuable in demonstrating the beneficial effect of CORM-3
against HSR-induced lung injury. As far as we know, few studies
have investigated the effect of CORM-3 on acute lung injury induced
by HSR. Although Nassour et al demonstrated that CORM-3
reduces systemic inflammation and hepatic sinusoidal endothelial
injury after HSR (29), they did not
focus on lung injury. Moreover, they administered CORM-3 during the
resuscitation period, whereas we administered CORM-3 immediately
after completion of resuscitation. Thus, the present study is, to
the best of our knowledge, the first study to demonstrate the
usefulness of CORM-3 administration post-resuscitation against
HSR-induced lung injury.
We previously demonstrated that inhalation of CO at
250 ppm for 3 h after completion of resuscitation significantly
attenuated HSR-induced acute lung injury via the anti-inflammatory
and anti-apoptotic properties of CO (18). However, even 1 h of CO inhalation at
250 ppm significantly increased blood COHb levels by up to
approximately 20% in rats (13,19,49),
which may exert toxic effects in humans (20). Notably, in the present study, CORM-3
treatment markedly ameliorated HSR-induced acute lung injury
without influencing the blood COHb levels. CORMs are transition
metal carbonyl complexes coordinated with CO that can liberate and
deliver CO to tissues once in contact with suitable physiological
conditions (21–24). CORM-3, a water-soluble form of CORMs,
rapidly releases CO immediately after administration into lysis
buffer (t1/2<1 min) (23). Thus,
our findings indicated that intravenous administration of CORM-3
rapidly releases CO following its incorporation into the lung,
which leads to the attenuation of HSR-induced lung injury and
inflammation.
CORMs have been reported to have a vasodilatory
effect that involves the activation of soluble guanylyl cyclase and
BKCa channels (46). According to
previous studies, 20–30 µmol/kg of CORM-3 administration reduced
the mean arterial blood pressure by approximately 10 mmHg in a
normal rat model, and its vasodilatory effect was dose-dependent
(21,32). However, in the present study, time
course changes in mean arterial pressure after resuscitation were
not different among the HSR/CORM-3, HSR/iCORM-3, and HSR groups
(Fig. 5). The absence of the
vasodilatory effect of CORM-3 may be attributable to two factors.
First, the dosage of CORM-3 in our study was 4 mg/kg (13.58
µmol/kg), which was relatively lower than the dosages in previous
studies (21,32). Second, as shown in Fig. 2B, the HSR procedure increased the
pulmonary expression of iNOS that led to the production of NO, a
potent vasodilator. The iNOS gene is expressed in nearly every
organ and is associated with refractory hypotension after
hemorrhagic shock (45,59). In contrast, CORM-3 treatment markedly
decreased the expression of iNOS in the lung in the present study
(Fig. 2B). Thus, we speculate that
the reduction in iNOS expression with CORM-3 administration may
compensate for the vasodilatory effect of CORM-3, although we
examined the modulation of iNOS expression by CORM-3 only in the
lung. In any case, our findings underscore the safety of CORM-3
treatment with respect to hemodynamic stability, since CORM-3
administration did not show any apparent detrimental influence on
vital signs even in the acute, hemodynamically instable state after
HSR.
There are several limitations to the present study.
First, we did not directly measure the CO concentration in the
pulmonary tissue of our rat HSR model. However, in a pilot study
prior to the presented experiments, we confirmed increases in CO
concentrations in the blood and various tissues after CORM-3
administration in healthy control rats (unpublished data). CORM-3
administration significantly increased in a dose-dependent fashion
the CO concentration in lung tissue of these animals. In addition,
we administered in the present study iCORM-3 that still contained
ruthenium as the transition metal but lost the capability to
release CO. This drug did not ameliorate HSR-induced lung injury,
did not exert any effects on the expression of either pro- or
anti-inflammatory cytokines, and did not influence lung vascular
permeability and apoptosis parameters. Thus, it is considered that
the therapeutic effect of CORM-3 demonstrated in the present study
is dependent on the CO released by CORM-3. Second, we did not
investigate the effect of CORM-3 in organ tissues other than the
lung. Previous studies indicated therapeutic effects of CORM-3 on
the liver, the kidneys, and the heart, among others (26,29,60).
However, our previous study revealed that the established HSR
procedure mainly injured the lung, and only to a lesser extent the
liver and the kidneys (34). Thus,
the beneficial effects of CORM-3 in the present study are less
likely to be based on an indirect modification of the lung by
changes in other organs. Third, we showed that CORM-3 did not have
any detrimental effect on the ABG analysis and the vital signs
during HSR. However, long-term effects or unwanted effects in other
organs including the brain, liver, and kidneys were not
investigated. Since CORM-3 is a ruthenium-based complex, a possible
accumulation of ruthenium after CO liberation could result in
undesired effects. Further studies are needed to investigate the
long-term safety of CORM-3 in various organs.
In conclusion, the HSR procedure caused acute lung
injury in a rat model, as demonstrated by the upregulation of
proinflammatory mediators, neutrophil migration into the lung, and
pulmonary edema, which led to apoptotic cell death. CORM-3
administration immediately after HSR ameliorated the HSR-induced
lung injury via suppression of proinflammatory cytokines, induction
of IL-10 gene expression, and reduction of apoptotic cell death. In
addition, we did not observe any detrimental effect on the ABG
analysis and vital signs during HSR. These findings suggest that
CORM-3 treatment ameliorated HSR-induced lung injury, at least in
part, through anti-inflammatory and anti-apoptotic effects.
Although further studies are needed to clarify the precise
mechanism and pharmacological features of CORM-3, CORM-3
administration may be a promising therapeutic approach for acute
lung injury after HSR.
Acknowledgements
The authors would like to thank Dr Reiko Akagi
(Yasuda Women's University, Hiroshima, Japan) for providing cDNAs
of TNF and iNOS. The authors would also like to thank Dr Akihiko
Taniguchi (Okayama University Graduate School of Medicine,
Dentistry, and Pharmaceutical Sciences, Okayama, Japan) for
technical support of BAL analysis.
Funding
This study was supported by the Japan Society for
the Promotion of Science (JSPS) Grant-in-Aid for Scientific
Research (KAKENHI) Grant no. JP16K10972.
Availability of data and materials
The dataset used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YK, TT, HS, KI, and HM contributed to the conception
and design of the study. YK, RN, EO, and HS performed experiments
and collected data. YK, HS, KI, and TT analyzed and interpreted the
data. YK drafted and wrote the manuscript. TT and HM revised the
manuscript critically for important intellectual content. HM
supervised the study and gave final approval of the version to be
published. All authors approved the final version of the
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Use and
Care Committee of the Okayama University Medical School
(OKU-2015429) on September 2, 2015. The care and handling of the
animals were conducted in accordance with the National Institutes
of Health guidelines for Animal Research. The present study also
conformed to guidelines for the care and use of laboratory animals
that followed the ARRIVE (Animal Research: Reporting of In
Vivo Experiments) guidelines and the 2013 AVMA euthanasia
guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ABG
|
arterial blood gas
|
|
ARDS
|
acute respiratory distress
syndrome
|
|
BAL
|
bronchoalveolar lavage
|
|
BE
|
base excess
|
|
BKCa channels
|
large conductance calcium-activated
potassium channels
|
|
CO
|
carbon monoxide
|
|
COHb
|
carboxyhemoglobin
|
|
CORMs
|
carbon monoxide-releasing
molecules
|
|
CORM-3
|
carbon monoxide-releasing
molecule-3
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
Hb
|
hemoglobin
|
|
HO
|
heme oxygenase
|
|
HO-1
|
heme oxygenase-1
|
|
HSR
|
hemorrhagic shock and
resuscitation
|
|
iCORM-3
|
inactive carbon monoxide-releasing
molecule-3
|
|
IL-10
|
interleukin-10
|
|
IL-1β
|
interleukin-1β
|
|
iNOS
|
inducible nitric oxide synthase
|
|
MAPK
|
mitogen-activated protein kinases
|
|
MIP-2
|
macrophage inflammatory protein-2
|
|
NF-κB
|
nuclear factor κB
|
|
NLRP3
|
nucleotide-binding domain,
leucine-rich-containing family, pyrin domain-containing-3
|
|
PaCO2
|
partial pressure of carbon dioxide
|
|
PaO2
|
partial pressure of oxygen
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
SaO2
|
saturation of arterial blood
oxygen
|
|
SEM
|
standard error of the mean
|
|
TNF-α
|
tumor necrosis factor-α
|
|
TUNEL
|
transferase-mediated dUTP-fluorescein
isothiocyanate (FITC) nick-end labeling
|
References
|
1
|
Dewar D, Moore FA, Moore EE and Balogh Z:
Postinjury multiple organ failure. Injury. 40:912–918. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ciesla DJ, Moore EE, Johnson JL, Cothren
CC, Banerjee A, Burch JM and Sauaia A: Decreased progression of
postinjury lung dysfunction to the acute respiratory distress
syndrome and multiple organ failure. Surgery. 140:640–648. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ware LB: Pathophysiology of acute lung
injury and the acute respiratory distress syndrome. Semin Respir
Crit Care Med. 27:337–349. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
ARDS Definition Task Force, ; Ranieri VM,
Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E,
Camporota L and Slutsky AS: Acute respiratory distress syndrome:
The berlin definition. JAMA. 307:2526–2533. 2012.PubMed/NCBI
|
|
5
|
Acute Respiratory Distress Syndrome
Network, ; Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson
BT and Wheeler A: Ventilation with lower tidal volumes as compared
with traditional tidal volumes for acute lung injury and the acute
respiratory distress syndrome. N Engl J Med. 342:1301–1308. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tonelli AR, Zein J, Adams J and Ioannidis
JP: Effects of interventions on survival in acute respiratory
distress syndrome: An umbrella review of 159 published randomized
trials and 29 meta-analyses. Intensive Care Med. 40:769–787. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adhikari N, Burns KE and Meade MO:
Pharmacologic therapies for adults with acute lung injury and acute
respiratory distress syndrome. Cochrane Database Syst Rev.
CD0044772004.PubMed/NCBI
|
|
8
|
Peter JV, John P, Graham PL, Moran JL,
George IA and Bersten A: Corticosteroids in the prevention and
treatment of acute respiratory distress syndrome (ARDS) in adults:
Meta-analysis. BMJ. 336:1006–1009. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rose JJ, Wang L, Xu Q, McTiernan CF, Shiva
S, Tejero J and Gladwin MT: Carbon monoxide poisoning:
Pathogenesis, management, and future directions of therapy. Am J
Respir Crit Care Med. 195:596–606. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gullotta F, di Masi A, Coletta M and
Ascenzi P: CO metabolism, sensing, and signaling. Biofactors.
38:1–13. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bilban M, Haschemi A, Wegiel B, Chin BY,
Wagner O and Otterbein LE: Heme oxygenase and carbon monoxide
initiate homeostatic signaling. J Mol Med (Berl). 86:267–279. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takahashi T, Shimizu H, Morimatsu H,
Maeshima K, Inoue K, Akagi R, Matsumi M, Katayama H and Morita K:
Heme oxygenase-1 is an essential cytoprotective component in
oxidative tissue injury induced by hemorrhagic shock. J Clin
Biochem Nutr. 44:28–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Otterbein LE, Mantell LL and Choi AM:
Carbon monoxide provides protection against hyperoxic lung injury.
Am J Physiol. 276:L688–L694. 1999.PubMed/NCBI
|
|
14
|
Sarady JK, Zuckerbraun BS, Bilban M,
Wagner O, Usheva A, Liu F, Ifedigbo E, Zamora R, Choi AM and
Otterbein LE: Carbon monoxide protection against endotoxic shock
involves reciprocal effects on iNOS in the lung and liver. FASEB J.
18:854–856. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song R, Kubo M, Morse D, Zhou Z, Zhang X,
Dauber JH, Fabisiak J, Alber SM, Watkins SC, Zuckerbraun BS, et al:
Carbon monoxide induces cytoprotection in rat orthotopic lung
transplantation via anti-inflammatory and anti-apoptotic effects.
Am J Pathol. 163:231–242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang X, Shan P, Otterbein LE, Alam J,
Flavell RA, Davis RJ, Choi AM and Lee PJ: Carbon monoxide
inhibition of apoptosis during ischemia-reperfusion lung injury is
dependent on the p38 mitogen-activated protein kinase pathway and
involves caspase 3. J Biol Chem. 278:1248–1258. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zuckerbraun BS, Mccloskey CA, Gallo D, Liu
F, Ifedigbo E, Otterbein LE and Billiar TR: Carbon monoxide
prevents multiple organ injury in a model of hemorrhagic shock and
resuscitation. Shock. 23:527–532. 2005.PubMed/NCBI
|
|
18
|
Kawanishi S, Takahashi T, Morimatsu H,
Shimizu H, Omori E, Sato K, Matsumi M, Maeda S, Nakao A and Morita
K: Inhalation of carbon monoxide following resuscitation
ameliorates hemorrhagic shock-induced lung injury. Mol Med Rep.
7:3–10. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kanagawa F, Takahashi T, Inoue K, Shimizu
H, Omori E, Morimatsu H, Maeda S, Katayama H, Nakao A and Morita K:
Protective effect of carbon monoxide inhalation on lung injury
after hemorrhagic shock/resuscitation in rats. J Trauma.
69:185–194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gorman D, Drewry A, Huang YL and Sames C:
The clinical toxicology of carbon monoxide. Toxicology. 187:25–38.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Motterlini R, Clark JE, Foresti R,
Sarathchandra P, Mann BE and Green CJ: Carbon monoxide-releasing
molecules: Characterization of biochemical and vascular activities.
Circ Res. 90:e17–e24. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Motterlini R, Mann BE, Johnson TR, Clark
JE, Foresti R and Green CJ: Bioactivity and pharmacological actions
of carbon monoxide-releasing molecules. Curr Pharm Des.
9:2525–2539. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Motterlini R and Otterbein LE: The
therapeutic potential of carbon monoxide. Nat Rev Drug Discov.
9:728–743. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Motterlini R: Carbon monoxide-releasing
molecules (CO-RMs): Vasodilatory, anti-ischaemic and
anti-inflammatory activities. Biochem Soc Trans. 35:1142–1146.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Foresti R, Bani-Hani MG and Motterlini R:
Use of carbon monoxide as a therapeutic agent: Promises and
challenges. Intensive Care Med. 34:649–658. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo Y, Stein AB, Wu WJ, Tan W, Zhu X, Li
QH, Dawn B, Motterlini R and Bolli R: Administration of a
CO-releasing molecule at the time of reperfusion reduces infarct
size in vivo. Am J Physiol Heart Circ Physiol. 286:H1649–H1653.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
De Backer O, Elinck E, Blanckaert B,
Leybaert L, Motterlini R and Lefebvre RA: Water-soluble
CO-releasing molecules reduce the development of postoperative
ileus via modulation of MAPK/HO-1 signalling and reduction of
oxidative stress. Gut. 58:347–356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fredenburgh LE, Kraft BD, Hess DR, Harris
RS, Wolf MA, Suliman HB, Roggli VL, Davies JD, Winkler T, Stenzler
A, et al: Effects of inhaled CO administration on acute lung injury
in baboons with pneumococcal pneumonia. Am J Physiol Lung Cell Mol
Physiol. 309:L834–L846. 2015.PubMed/NCBI
|
|
29
|
Nassour I, Kautza B, Rubin M, Escobar D,
Luciano J, Loughran P, Gomez H, Scott J, Gallo D, Brumfield J, et
al: Carbon monoxide protects against hemorrhagic shock and
resuscitation-induced microcirculatory injury and tissue injury.
Shock. 43:166–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kilkenny C, Browne WJ, Cuthill IC, Emerson
M and Altman DG: Improving bioscience research reporting: The
ARRIVE guidelines for reporting animal research. Osteoarthritis
Cartilage. 20:256–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
American Veterinary Medical Association:
AVMA guidelines for the euthanasia of animals: 2013 edition.
https://www.avma.org/KB/Policies/Documents/euthanasia.pdfAugust
4–2018
|
|
32
|
Foresti R, Hammad J, Clark JE, Johnson TR,
Mann BE, Friebe A, Green CJ and Motterlini R: Vasoactive properties
of CORM-3, a novel water-soluble carbon monoxide-releasing
molecule. Br J Pharmacol. 142:453–460. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Inoue K, Takahashi T, Uehara K, Shimuzu H,
Ido K, Morimatsu H, Omori E, Katayama H, Akagi R and Morita K:
Protective role of heme oxygenase 1 in the intestinal tissue injury
in hemorrhagic shock in rats. Shock. 29:252–261. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Maeshima K, Takahashi T, Uehara K, Shimizu
H, Omori E, Yokoyama M, Tani T, Akagi R and Morita K: Prevention of
hemorrhagic shock-induced lung injury by heme arginate treatment in
rats. Biochem Pharmacol. 69:1667–1680. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Murakami K, McGuire R, Cox RA, Jodoin JM,
Bjertnaes LJ, Katahira J, Traber LD, Schmalstieg FC, Hawkins HK,
Herndon DN and Traber DL: Heparin nebulization attenuates acute
lung injury in sepsis following smoke inhalation in sheep. Shock.
18:236–241. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zegdi R, Fabre O, Cambillau M, Fornès P,
Tazi KA, Shen M, Hervé P, Carpentier A and Fabiani JN: Exhaled
nitric oxide and acute lung injury in a rat model of extracorporeal
circulation. Shock. 20:569–574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang H, Meng F, Li W, Tong L, Qiao H and
Sun X: Splenectomy ameliorates acute multiple organ damage induced
by liver warm ischemia reperfusion in rats. Surgery. 141:32–40.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stephens KE, Ishizaka A, Larrick JW and
Raffin TA: Tumor necrosis factor causes increased pulmonary
permeability and edema. Comparison to septic acute lung injury. Am
Rev Respir Dis. 137:1364–1370. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wohlauer M, Moore EE, Silliman CC, Fragoso
M, Gamboni F, Harr J, Accurso F, Wright F, Haenel J, Fullerton D
and Banerjee A: Nebulized hypertonic saline attenuates acute lung
injury following trauma and hemorrhagic shock via inhibition of
matrix metalloproteinase-13. Crit Care Med. 40:2647–2653. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kosaka J, Morimatsu H, Takahashi T,
Shimizu H, Kawanishi S, Omori E, Endo Y, Tamaki N, Morita M and
Morita K: Effects of biliverdin administration on acute lung injury
induced by hemorrhagic shock and resuscitation in rats. PLoS One.
8:e636062013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nakahira K, Takahashi T, Shimizu H,
Maeshima K, Uehara K, Fujii H, Nakatsuka H, Yokoyama M, Akagi R and
Morita K: Protective role of heme oxygenase-1 induction in carbon
tetrachloride-induced hepatotoxicity. Biochem Pharmacol.
66:1091–1105. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Umeda K, Takahashi T, Inoue K, Shimizu H,
Maeda S, Morimatsu H, Omori E, Akagi R, Katayama H and Morita K:
Prevention of hemorrhagic shock-induced intestinal tissue injury by
glutamine via heme oxygenase-1 induction. Shock. 31:40–49. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kurimoto E, Miyahara N, Kanehiro A, Waseda
K, Taniguchi A, Ikeda G, Koga H, Nishimori H, Tanimoto Y, Kataoka
M, et al: IL-17A is essential to the development of
elastase-induced pulmonary inflammation and emphysema in mice.
Respir Res. 14:52013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yamaoka M, Shimizu H, Takahashi T, Omori E
and Morimatsu H: Dynamic changes in Bach1 expression in the kidney
of rhabdomyolysis-associated acute kidney injury. PLoS One.
12:e01809342017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rushing GD and Britt LD: Reperfusion
injury after hemorrhage: A collective review. Ann Surg.
247:929–937. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Alshehri A, Bourguignon MP, Clavreul N,
Badier-Commander C, Gosgnach W, Simonet S, Vayssettes-Courchay C,
Cordi A, Fabiani JN, Verbeuren TJ and Félétou M: Mechanisms of the
vasorelaxing effects of CORM-3, a water-soluble carbon
monoxide-releasing molecule: Interactions with eNOS. Naunyn
Schmiedebergs Arch Pharmacol. 386:185–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shimzu K, Takahashi T, Iwasaki T, Shimizu
H, Inoue K, Morimatsu H, Omori E, Matsumi M, Akagi R and Morita K:
Hemin treatment abrogates monocrotaline-induced pulmonary
hypertension. Med Chem. 4:572–576. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tamion F, Richard V, Bonmarchand G, Leroy
J, Lebreton J and Thuillez C: Induction of heme-oxygenase-1
prevents the systemic responses to hemorrhagic shock. Am J Respir
Crit Care Med. 164:1933–1938. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Otterbein LE, Otterbein SL, Ifedigbo E,
Liu F, Morse DE, Fearns C, Ulevitch RJ, Knickelbein R, Flavell RA
and Choi AM: MKK3 mitogen-activated protein kinase pathway mediates
carbon monoxide-induced protection against oxidant-induced lung
injury. Am J Pathol. 163:2555–2563. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang X, Shan P, Alam J, Fu X and Lee PJ:
Carbon monoxide differentially modulates STAT1 and STAT3 and
inhibits apoptosis via a phosphatidylinositol 3-kinase/Akt and p38
kinase-dependent STAT3 pathway during anoxia-reoxygenation injury.
J Biol Chem. 280:8714–8721. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dolinay T, Szilasi M, Liu M and Choi AM:
Inhaled carbon monoxide confers antiinflammatory effects against
ventilator-induced lung injury. Am J Respir Crit Care Med.
170:613–620. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Koulouras VP, Li R, Chen L and
Hedenstierna GG: Effects of inhaled carbon monoxide and
glucocorticoids in porcine endotoxin sepsis. Int J Clin Exp Med.
4:53–66. 2011.PubMed/NCBI
|
|
53
|
Jung SS, Moon JS, Xu JF, Ifedigbo E, Ryter
SW, Choi AM and Nakahira K: Carbon monoxide negatively regulates
NLRP3 inflammasome activation in macrophages. Am J Physiol Lung
Cell Mol Physiol. 308:L1058–L1067. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kim SK, Joe Y, Chen Y, Ryu J, Lee JH, Cho
GJ, Ryter SW and Chung HT: Carbon monoxide decreases interleukin-1β
levels in the lung through the induction of pyrin. Cell Mol
Immunol. 14:349–359. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Queiroga CS, Almeida AS and Vieira HL:
Carbon monoxide targeting mitochondria. Biochem Res Int.
2012:7498452012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gomez H, Kautza B, Escobar D, Nassour I,
Luciano J, Botero AM, Gordon L, Martinez S, Holder A, Ogundele O,
et al: Inhaled carbon monoxide protects against the development of
shock and mitochondrial injury following hemorrhage and
resuscitation. PLoS One. 10:e01350322015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sun B, Sun H, Liu C, Shen J, Chen Z and
Chen X: Role of CO-releasing molecules liberated CO in attenuating
leukocytes sequestration and inflammatory responses in the lung of
thermally injured mice. J Surg Res. 139:128–135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Qin W, Zhang J, Lv W, Wang X and Sun B:
Effect of carbon monoxide-releasing molecules II-liberated CO on
suppressing inflammatory response in sepsis by interfering with
nuclear factor kappa B activation. PLoS One. 8:e758402013.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Taylor BS and Geller DA: Molecular
regulation of the human inducible nitric oxide synthase (iNOS)
gene. Shock. 13:413–424. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tayem Y, Johnson TR, Mann BE, Green CJ and
Motterlini R: Protection against cisplatin-induced nephrotoxicity
by a carbon monoxide-releasing molecule. Am J Physiol Renal
Physiol. 290:F789–F794. 2006. View Article : Google Scholar : PubMed/NCBI
|