Introduction
Neurohypopheseal diabetes insipidus is a disorder
caused by increased urine production and volume depletion due to
vasopressin (AVP) deficiency. Most cases are linked with structural
damage of the hypothalamus, pituitary stalk or posterior pituitary
gland (1). Familial neurohypophyseal
diabetes insipidus (FNDI) is a rare inherited disease that accounts
for only 1% of central diabetes insipidus cases (2). The disorder usually presents in early
childhood with polyuria and polydipsia, and follow-up evaluation
frequently reveals loss of the posterior pituitary bright spot on
T1-weighted magnetic resonance imaging (MRI) (3–5). FNDI is
caused by mutations of the arginine vasopressin-neurophysin II
(AVP-NPII) gene (GenBank ID, NM_000490.4), which is located on
chromosome 20 and encodes a preproprotein that is proteolytically
processed to generate multiple products, including AVP, neurophysin
II and copeptin (6). Biosynthesis of
AVP is tightly regulated by translation and post-translational
processes, including enzymatic cleavage of the AVP-neurophysin II
prohormone and appropriate secretion of AVP into the circulation
upon various stimuli (7,8). Since the publication of the first
genetic study on FNDI (9), ~70
mutations have been described and all are located within the 2.5
kb-long AVP-NPII gene. The proposed pathogenic mechanisms include
aberrant preprohormone processing leading to the gradual
destruction of AVP-secreting cells (10) and mutations that alter the amino acid
structure of the mature AVP hormone (11).
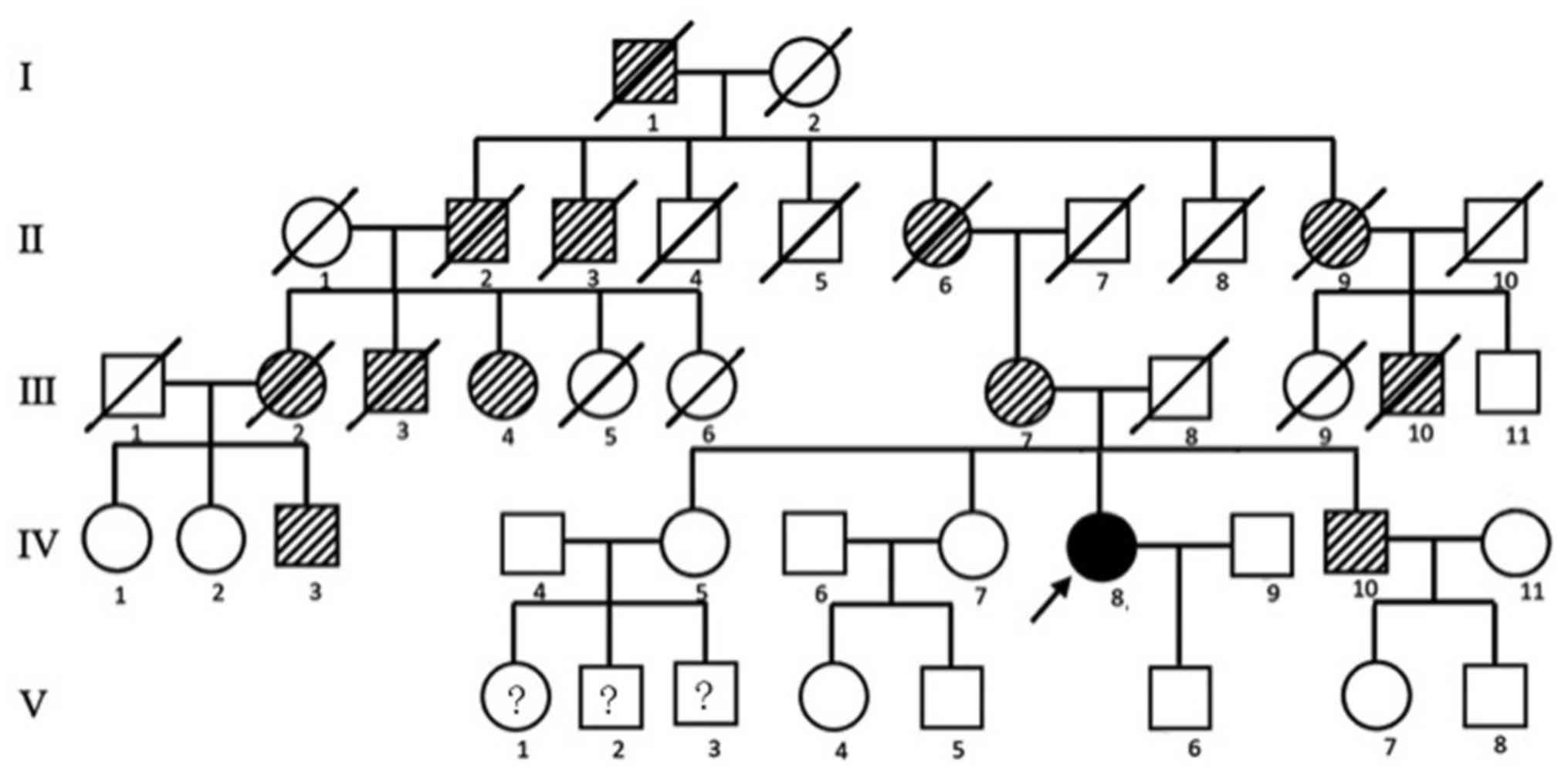
The present study reports on a female patient with
autosomal dominant FNDI linked to a novel nonsense mutation of
AVP-NPII gene and her extended family. The clinical, biochemical
and radiological characteristics of the patient were investigated.
The novel nonsense mutation was also identified in another affected
family member.
Patients and methods
Patients
A 46-year-old Chinese woman was referred to Peking
Union Medical College Hospital (Beijing, China) for evaluation of
acute urinary retention. A urinary catheter was placed to alleviate
severe bladder distension. She reported having polyuria and
polydipsia since age 13, but had not sought any medical care, as
she always had access to water. The patient was born at term
without any complications and had a normal pubescence. A total of
12 additional family members spanning four generations, including
the patient's mother and younger brother, also had teenage-onset of
polyuria and polydipsia. Daily urine volumes varied from 6.0 to
15.0 liters. No mental retardation or delay in puberty had been
identified. The average final body height was 160.1 cm for females
and 175.2 cm for male members of the family. The pedigree of the
family is presented in Fig. 1.
Clinical, laboratory and radiological
assessments
Clinical data were collected in accordance with the
Declaration of Helsinki. Written informed consent was obtained from
the patient (IV-8), the patient's mother (III-7) and an
asymptomatic sister of the patient (IV-7). A water deprivation test
followed by the administration of vasopressin was performed to
diagnose central diabetes insipidus as described previously
(12). Enhanced MRI of the
hypothalamo-hypophyseal area and pelvis was performed. The residual
urine volume was measured by performing an ultrasound scan of the
bladder.
Genomic DNA extraction, amplification
and sequencing of the AVP-NPII gene
The AVP-NPII gene was analyzed by direct sequencing
from genomic DNA extracted from leucocytes of peripheral blood with
the QIAamp DNA Mini kit (Qiagen GmbH). In brief, PCR was performed
in a 40-µl reaction volume containing 100 ng genomic DNA, 20 µl 2X
GC PCR buffer, 0.1 µM of each dNTP, 0.1 µM of each primer and 2.5
units of rTaq polymerase (Takara Bio, Inc.) in a thermocycler (ABI
9700; Applied Biosystems; Thermo Fisher Scientific, Inc.). All
genomic DNA was denatured at 94°C for 10 min and amplified with 35
cycles. For exon 1, the cycles were programmed as 94°C for 30 sec,
60°C for 30 sec and 72°C for 40 sec. The cycling conditions for
exons 2 and 3 were programmed as follows: Module 1 was 94°C for 5
min, 65°C for 5 min and 74°C for 5 min (1 cycle); module 2 was 94°C
for 1 min, 65°C for 1 min and 74°C for 4 min (35 cycles); module 3
was 74°C for 10 min; module 4 was a hold at 4°C. Primers for
amplification and sequencing of the AVP-NPII gene are provided in
Table I. Amplified products were
detected by agarose gel electrophoresis and sequenced using an ABI
3730 DNA analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.).
 | Table I.Primer sequences for PCR of the
arginine vasopressin-neurophysin II gene. |
Table I.
Primer sequences for PCR of the
arginine vasopressin-neurophysin II gene.
| Exon/direction | Sequences
(5′-3′) |
|---|
| 1 |
|
|
Forward |
ATGATCCCCTGCACAGACAG |
|
Reverse |
CTGCCCAGCCATGCCATG |
| 2 |
|
|
Forward |
TCGCTGCGTTCCCCTCCAACCCCTCGACTC |
|
Reverse |
CGCCCCCCCCCAGGCCCGCCCCCGCCGCGC |
| 3 |
|
|
Forward |
CCCAGGCGCCCGTGCTCACACGTCCTCCCG |
|
Reverse |
CCTCTCTCCCCTTCCCTCTTCCCGCCAGAG |
Three-dimensional structural models of the predicted
wild-type and mutant proteins were generated and comparisons
between the proteins were performed using the PHYRE2 Protein Fold
Recognition Server database (http://wwwsbg.bio.ic.ac.uk/phyre2) and the SWISS-MODEL
protein structure homology-modelling server (http://swissmodel.expasy.org) as reported previously
(5).
Results
Clinical and MRI assessments
Diabetes insipidus was confirmed via a standard
water deprivation test. After 8 h of water deprivation, the plasma
osmolality rose from 301 to 315 mOsm/kg and the urine osmolality
rose from 50 to 65 mOsm/kg. 4 U vasopressin was subsequently
injected intramuscularly. Following 2 h, the plasma osmolality
decreased from 315 to 302 mOsm/kg and the urine osmolality
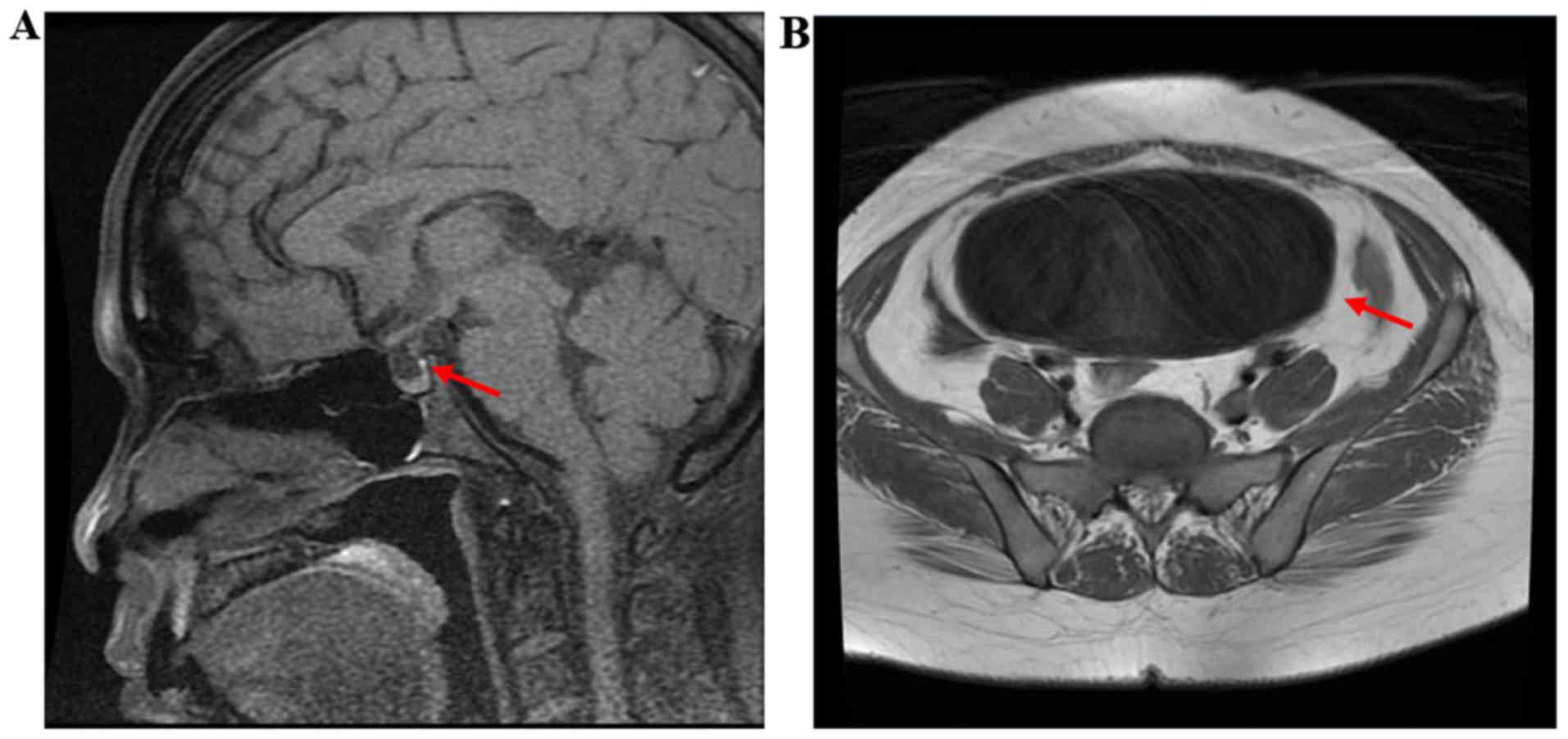
increased from 65 to 587 mOsm/kg. As presented in Fig. 2A, the cranial MRI revealed a high
signal region in the posterior lobe of the pituitary gland. Serum
cortisol, thyroid function, growth hormone levels, insulin-like
growth factor 1 and estrogen were all within normal ranges. Pelvic
MRI revealed a significantly enlarged urinary bladder (Fig. 2B). The daily urine output decreased
from 15 l to 2 l after 1 week of treatment with 100 µg
1-deamino-8-D-arginine-vasopressin three times per day. The dosage
was then tapered to 50 µg, which was administered 3 times per day
and maintained thereafter. The residual urine volume decreased from
1,250 to 4 ml and the urinary catheter was removed.
PCR and sequencing
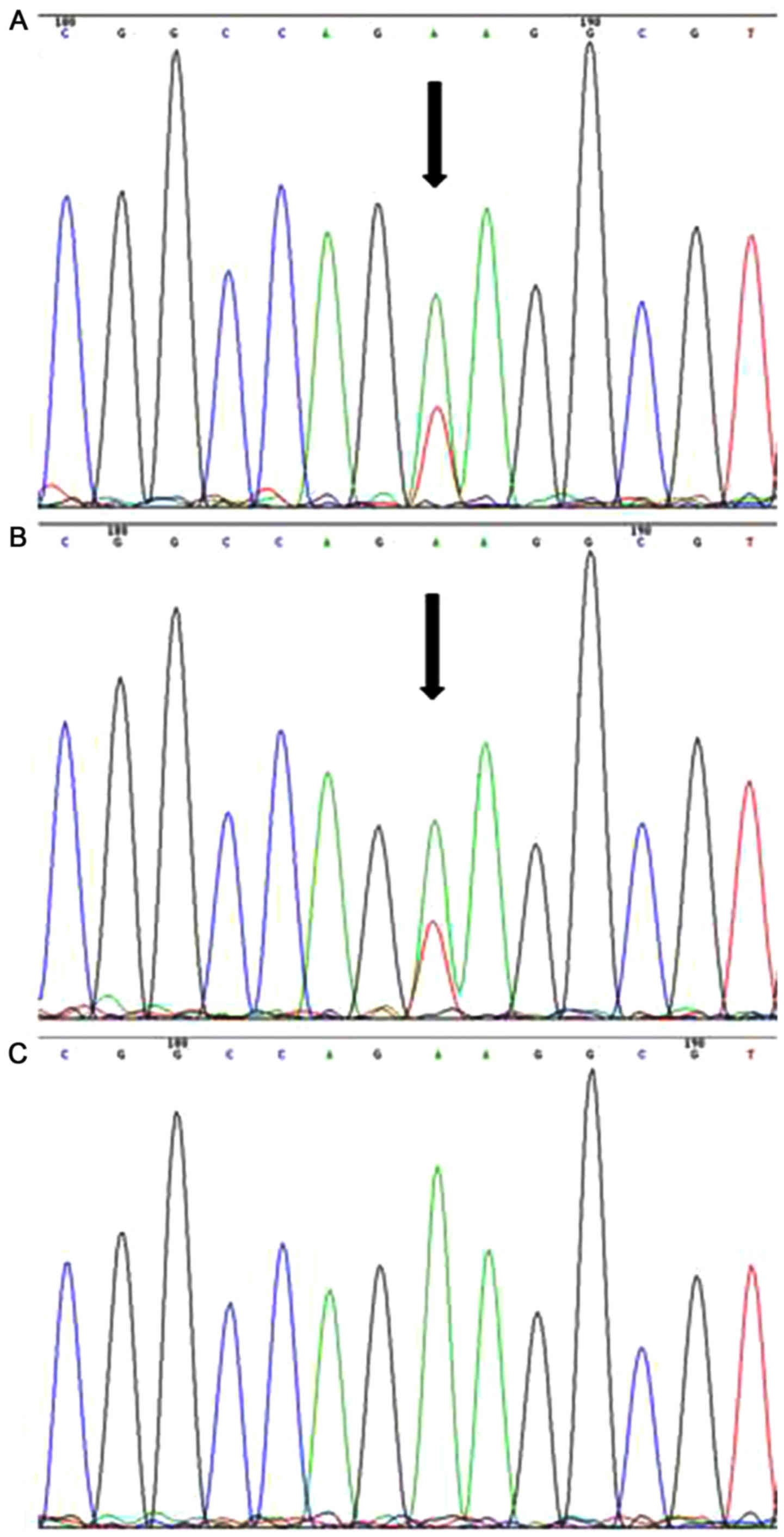
The AVP-NPII genes were sequenced from the patient,
the patient's mother and a sister of the patient. Sequence analyses
of the patient's entire AVP-NPII coding region revealed a novel
heterogeneous nonsense mutation at codon 268 (c.268A>T), which
results in a substitution in exon 2 of Lys (AAG) with a stop codon
(TAG) and corresponds to lysine 90 of the NPII moiety (p.Lys90Ter)
(Fig. 3A). The same mutation was
identified in the patient's mother (Fig.
3B), but not in the patient's sister, who had a normal
phenotype (Fig. 3C). The 3-D
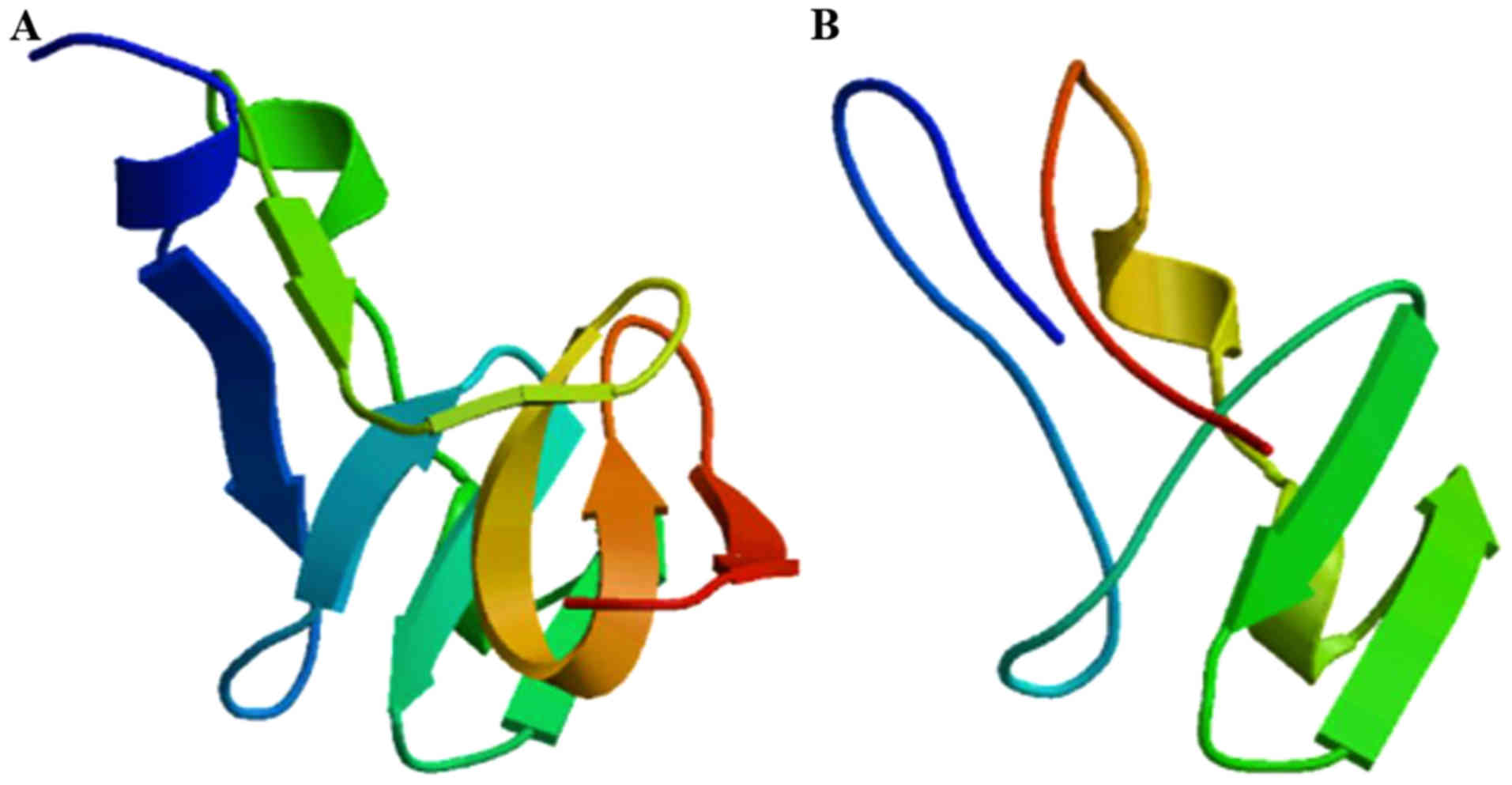
structures of the wild-type and mutant (p.Lys90Ter) AVP-NPII
proteins were modeled using the above-mentioned computational
servers. As presented in Fig. 4, the
Lys90Ter mutation is located in the middle of NPII and leads to the
loss of 74 amino acids, including 6 cysteine residues. The loss of
cysteine residues results in aberrant disulfide bonds, which is
expected to alter the protein structure.
Discussion
The present study reports on a novel p.Lys90Ter
mutation identified in exon 2 of the AVP-NPII gene that is linked
with FNDI. The pedigree examined included cases spanning four
generations, with no apparent gender preponderance among affected
individuals. Similar to the observations of previously published
studies on FNDI, the affected individuals in the present study were
normal at birth and gradually developed symptoms of vasopressin
deficiency beginning in childhood or adolescence (13). None of the subjects had any mental
deficits or impaired pubescent development.
FNDI is a rare, single-gene disorder caused by
mutations in the 2.5-kb AVP-NPII gene located in chromosome region
20p13 (14,15). The AVP-NPII gene has three exons:
Exon 1 encodes a signal peptide, AVP, and the
NH2-terminal portion of neurophysin II, exon 2 encodes
the central region of neurophysin II and exon 3 encodes the
COOH-terminal region of neurophysin II and copeptin. AVP is
synthesized in the supraoptic nucleus and paraventricular nucleus
of the hypothalamus and then packaged into neurosecretory vesicles
along with its carrier protein neurophysin II. It is then
transported axonally to the neurohypophysis. Most of the mutations
reported to be associated with FNDI are located in the region
encoding the NPII moiety, which is the intracellular binding
protein for AVP. Several mutations are localized to the coding
region for signaling peptide or AVP per se. The mutant NPII is
assumed to interfere with axonal transport of intracellular AVP
proteolysis (16,17).
A total of 7 mutations spanning all 3 exons of the
AVP-NPII gene have been previously described in Chinese populations
(Table II) (18–20),
including 3 missense mutations in exon 2. To the best of our
knowledge, the present study is the first to report on a nonsense
mutation in exon 2 of the AVP-NPII gene in Chinese individuals. The
mutation results in a preterm stop codon and thus a truncated NPII
moiety. Wild-type pro-vasopressin has 164 amino acids and 7
disulfide bonds between all 14 cysteine residues (21). The mutant NPII loses 6 of these
cysteine residues and the resultant aberrant disulfide bonds cause
misfolding of the prohormone. As a result, the prohormone is likely
retained within the endoplasmic reticulum and exceeds its capacity
to tolerate the ‘unfolded protein response’, causing stress that
may trigger apoptosis (22,23). However, further investigations are
required to verify the molecular mechanisms underlying FNDI. It may
be hypothesized that FNDI follows a pattern of autosomal dominant
inheritance, which is consistent in the present pedigree.
| Table II.Arginine vasopressin-neurophysin II
gene mutations in familial neurohypophyseal diabetes insipidus
reported in the Chinese population. |
Table II.
Arginine vasopressin-neurophysin II
gene mutations in familial neurohypophyseal diabetes insipidus
reported in the Chinese population.
| First author
(year) | Coding region | cDNA mutation | Amino acid
change | Mutation type | Mode of
inheritance | Family history | (Refs.) |
|---|
| Tian (2016) | Exon 1 | c.2delT | p.M1_T4del | Frameshift
mutation | Autosomal
dominant | Yes | (18) |
| Tian (2016) | Exon 1 | c.50C>A | p.S17Y | Missense
mutation | Autosomal
dominant | Yes | (18) |
| Tian (2016) | Exon 1 | c.52-54delTCC | p.S18del | Frameshift
mutation | Autosomal
dominant | No | (18) |
| Tian (2016) | Exon 2 | c.127C>G | p.P43A | Missense
mutation | Autosomal
dominant | No | (18) |
| Tian (2016) | Exon 3 | c.329C>A | p.C110Y | Missense
mutation | Autosomal
dominant | No | (18) |
| Ye (2013) | Exon 2 | c.1516G>T | p.G17V | Missense
mutation | Autosomal
dominant | Yes | (19) |
| Luo (2012) | Exon 2 | c.193T>A | p.C65S | Missense
mutation | Autosomal
dominant | Yes | (20) |
| Present study | Exon 2 | c.268A>T | p.Lys90Ter | Nonsense
mutation | Autosomal
dominant | Yes |
|
Previous studies have described the MRI presentation
of subjects with FNDI. In one study, the presence of a bright spot
in the posterior lobe appeared to vary between members of the same
family (5). In another case series,
such a spot was entirely absent from all 13 patients (24). The MRI of the patient in the current
study revealed a bright spot on the posterior lobe of pituitary
gland. More serious urological complications are uncommon in these
patients, as they retain a limited capacity to secrete AVP during
severe dehydration.
In conclusion, the present study identified a novel
nonsense mutation in the AVP-NPII gene linked with NDI in a Chinese
patient whose family members shared a history of polyuria and
polydipsia across four generations. This heterogeneous Lys90Ter
mutation is located in the middle of NPII and leads to loss of 6
cysteine residues and aberrant disulfide bonds, which is expected
to alter the mature protein structure. Although a number of studies
have described variant mutations of NPII in FNDI, further
elucidation of the molecular mechanisms underlying specific mutant
interference with the axonal transport of intracellular AVP
proteolysis are required.
Acknowledgements
Not applicable.
Funding
This work was supported by the Special Research Fund
for Central Universities, Peking Union Medical College (grant no.
2017PT31004) and the CAMS Innovation Fund for Medical Science
(grant no. CAMS-2016-I2M-1-002□2016-I2M-1-008).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HY analyzed and interpreted the patient data and
wrote the primary manuscript. KY performed the PCR experiments. LW
collected the blood samples of the patient and the patient's
relatives and analyzed the gene sequencing data. FG supervised the
experiments and data analysis. ZJ helped to interpret the patient's
clinical data. HZ revised the primary manuscript and was a major
contributor in writing the manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of Peking Union Medical College Hospital (Beijing,
China).
Patient consent for publication
Written informed consent was obtained from the
patient, the patient's mother and an asymptomatic sister prior to
genetic testing and the publication of data and images in the
present study.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AVP-NPII
|
arginine vasopressin-neurophysin
II
|
|
FNDI
|
familial neurohypophyseal diabetes
insipidus
|
References
|
1
|
Baylis PH and Robertson GL: Vasopressin
function in familial cranial diabetes insipidus. Postgrad Med J.
57:36–40. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deniz F, Acar C, Saglar E, Erdem B,
Karaduman T, Yonem A, Cagiltay E, Ay SA and Mergen H:
Identification of a novel deletion in AVP-NPII gene in a patient
with central diabetes insipidus. Ann Clin Lab Sci. 45:588–592.
2015.PubMed/NCBI
|
|
3
|
Saglar E, Karaduman T, Ozcan M, Erdem B,
Oflaz O, Sahin D, Deniz F, Ay AS and Mergen H: Identification of
novel mutations in AVP-NPII gene. FEBS J. 283:1312016.
|
|
4
|
Elias PC, Elias LL, Torres N, Moreira AC,
Antunes-Rodrigues J and Castro M: Progressive decline of
vasopressin secretion in familial autosomal dominant
neurohypophyseal diabetes insipidus presenting a novel mutation in
the vasopressin-neurophysin II gene. Clin Endocrinol (Oxf).
59:511–518. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Turkkahraman D, Saglar E, Karaduman T and
Mergen H: AVP-NPII gene mutations and clinical characteristics of
the patients with autosomal dominant familial central diabetes
insipidus. Pituitary. 18:898–904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gainer H, Yamashita M, Fields RL, House SB
and Rusnak M: The magnocellular neuronal phenotype: Cell-specific
gene expression in the hypothalamo-neurohypophysial system. Prog
Brain Res. 139:1–14. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Murphy D and Wells S: In vivo gene
transfer studies on the regulation and function of the vasopressin
and oxytocin genes. J Neuroendocrinol. 15:109–125. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Repaske DR, Phillips JA III, Kirby LT, Tze
WJ, D'Ercole AJ and Battey J: Molecular analysis of autosomal
dominant neurohypophyseal diabetes insipidus. J Clin Endocrinol
Metab. 70:752–757. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ito M, Mori Y, Oiso Y and Saito H: A
single base substitution in the coding region for neurophysin II
associated with familial central diabetes insipidus. J Clin Invest.
87:725–728. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Christensen JH and Rittig S: Familial
neurohypophyseal diabetes insipidus-an update. Semin Nephrol.
26:209–223. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Willcutts MD, Felner E and White PC:
Autosomal recessive familial neurohypophyseal diabetes insipidus
with continued secretion of mutant weakly active vasopressin. Hum
Mol Genet. 8:1303–1307. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Trimpou P, Olsson DS, Ehn O and Ragnarsson
O: Diagnostic value of the water deprivation test in the
polyuria-polydipsia syndrome. Hormones. 16:414–422. 2017.PubMed/NCBI
|
|
13
|
Rittig S, Robertson GL, Siggaard C, Kovács
L, Gregersen N, Nyborg J and Pedersen EB: Identification of 13 new
mutations in the vasopressin-neurophysin II gene in 17 kindreds
with familial autosomal dominant neurohypophyseal diabetes
insipidus. Am J Hum Genet. 58:107–17. 1996.PubMed/NCBI
|
|
14
|
Sausville E, Carney D and Battey J: The
human vasopressin gene is linked to the oxytocin gene and is
selectively expressed in a cultured lung cancer cell line. J Biol
Chem. 260:10236–10241. 1985.PubMed/NCBI
|
|
15
|
Rao VV, Löffler C, Battey J and Hansmann
I: The human gene for oxytocin-neurophysin I (OXT) is physically
mapped to chromosome 20p13 by in situ hybridization. Cytogenet Cell
Genet. 61:271–273. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Christensen JH, Siggaard C, Corydon TJ,
Robertson GL, Gregersen N, Bolund L and Rittig S: Impaired
trafficking of mutated AVP prohormone in cells expressing rare
disease genes causing autosomal dominant familial neurohypophyseal
diabetes insipidus. Clin Endocrinol (Oxf). 60:125–136. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kobayashi H, Fujisawa I, Ikeda K, Son C,
Iwakura T, Yoshimoto A, Kasahara M, Ishihara T and Ogawa Y: A novel
heterozygous missense mutation in the vasopressin moiety is
identified in a Japanese person with neurohypophyseal diabetes
insipidus. J Endocrinol Invest. 29:252–256. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tian D, Cen J, Nie M and Gu F:
Identification of five novel arginine vasopressin gene mutations in
patients with familial neurohypophyseal diabetes insipidus. Int J
Mol Med. 38:1243–1249. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye D, Dong F, Lu W, Zhang Z, Lu X, Li C
and Liu Y: A missense mutation in the arginine-vasopressin
neurophysin-II gene causes autosomal dominant neurohypophyseal
diabetes insipidus in a Chinese family. Clin Endocrinol (Oxf).
78:920–925. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luo Y, Wang B, Qiu Y, Zhang C, Jin C, Zhao
Y, Zhu Q and Ma X: Clinical and molecular analysis of a Chinese
family with autosomal dominant neurohypophyseal diabetes insipidus
associated with a novel missense mutation in the
vasopressin-neurophysin II gene. Endocrine. 42:208–213. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Baglioni S, Corona G, Maggi M, Serio M and
Peri A: Identification of a novel mutation in the arginine
vasopressin-neurophysin II gene affecting the sixth intrachain
disulfide bridge of the neurophysin II moiety. Eur J Endocrinol.
151:605–611. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Arima H, Morishita Y, Hagiwara D, Hayashi
M and Oiso Y: Endoplasmic reticulum stress in vasopressin neurons
of familial diabetes insipidus model mice: Aggregate formation and
mRNA poly(A) tail shortening. Exp Physiol. 99:66–71. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ahner A and Brodsky JL: Checkpoints in
ER-associated degradation: Excuse me, which way to the proteasome?
Trends Cell Biol. 14:474–478. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gudinchet F, Brunelle F, Barth MO, Taviere
V, Brauner R, Rappaport R and Lallemand D: MR imaging of the
posterior hypophysis in children. AJR Am J Roentgenol. 153:351–354.
1989. View Article : Google Scholar : PubMed/NCBI
|