Introduction
Trisomy 16 is recognized as the most common type of
trisomy in first-trimester spontaneous abortions, occurring in 1%
of all clinically recognized pregnancies, while being rarer in the
second and third trimesters (1–3). Early
lethality and incompatibility with life have been described as its
major outcomes (1). Trisomy 16 may
be classified into three major types: Full trisomy, mosaics and
partial trisomy of 16p or 16q. Since Schmickel (4) reported the first case of 16q trisomy as
identified using a chromosome banding technique in 1975, >30
cases of partial trisomy 16q have been described. The common
clinical features of trisomy 16q include low birthweight,
hypotonia, failure to thrive, psychomotor retardation, periorbital
edema, high prominent forehead, microcephaly, low-set ears, flat
nasal bridge, small and down-slanting palpebral fissures,
micrognathia, hypertelorism, long philtrum and posterior cleft
palate (3). In the literature, few
cases of 16q trisomy among liveborn infants have been reported. The
unbalanced segregation of a parental balanced translocation
frequently leads to partial trisomy 16, which is common in newborns
and is associated with a wide range of clinical congenital
abnormalities (5).
Pure terminal deletions of 2p are rare in a clinical
context. To date, ~30 such patients have been reported, who share
the following common clinical features: Early-onset
obesity/overweight associated with intellectual disabilities and
behavioral difficulties (6–8).
The present case study reports on a male newborn
with multi-organ malformations exhibiting partial trisomy
16q21→qter and monosomy 2p25.3→pter. The proband exhibited the
clinical manifestations of congenital muscular torticollis and
congenital laryngomalacia, which have not been reported previously
in either trisomy 16q or monosomy 2p. The clinical features of
cases reported in the published literature were also described.
Case report
The proband was admitted to the neonatal department
at The First Hospital of Jilin University (Changchun, Jilin) due to
wet lung and small stature for his gestational age directly after
birth in March 2017. The patient was born at 40 weeks 3 days of
gestation, with a body weight of 2,350 g, body height of 46 cm and
Apgar scores of 6 at 1 min and 8 at 5 min. The infant was the first
child of the non-consanguineous and healthy couple: A 32-year-old
father and a 28-year-old mother, who had taken a tocolytic agent
due to suspected risk of spontaneous abortion in the first
trimester. Physical examination indicated that the proband had
small anterior fontanelles, prominent forehead, low hairline,
telecanthus, flat nasal bridge, choanal atresia, clinodactyly of
the fifth fingers, small penis and right cryptorchidism. Brain
magnetic resonance imaging indicated no obvious brain
abnormalities, but regular follow-up to dynamically observe the
proband's neurological condition remained necessary. At the age of
2 days, abnormal findings from chest X-rays revealed pneumonia in
the patient, with the ultrasonic cardiogram exhibiting patent
ductus arteriosus. Given an intolerance for normal feeding of the
patient, feeding was accomplished via a nasogastric tube. The
patient also presented with poor psychomotor development.
Considering that the subject failed a hearing screening test and
was positive for cytomegalovirus pp65 antigen, ganciclovir therapy
was performed. Subsequently, congenital muscular torticollis (at 19
days) and congenital laryngomalacia (at 24 days) were diagnosed.
Finally, the proband developed stable vital signs and was
discharged at the age of one month old. At 19 months after the
proband's birth, a follow-up was performed, during which the
presence of low-set ears, torticollis, hypotonia and an inability
to walk were noted, and laryngeal wheezing during sleep was
reported. The parents refused surgical treatments for the right
cryptorchidism and laryngomalacia. The protocol of the present
study was approved by the Ethics Committee of the First Hospital of
Jilin University (Changchun, China) and written informed consent
was obtained from the parents of the patient.
From the proband, 5 ml of peripheral blood was
collected using a standard vacuum extraction blood-collecting
system containing EDTA and heparin. Genomic DNA was isolated from
whole blood using a QIAamp DNA Mini kit (Qiagen GmbH) in accordance
with the manufacturer's protocol.
Chromosome microarray (CMA) was performed using
Affymetrix CytoScan HD arrays, in accordance with the
manufacturer's protocol. The procedure included genomic DNA
extraction, digestion and ligation, PCR amplification, PCR product
purification, quantification and fragmentation, labeling, array
hybridization, washing and scanning. The array was designed
specifically for cytogenetic research, including ~1,950,000 copy
number variation markers and 750,000 single-nucleotide polymorphism
markers. Data were analyzed using Affymetrix Chromosome Analysis
Suite v3.3 Software. Thresholds for genome-wide screening were set
at ≥100 kb for gains and ≥50 kb for losses. The detected copy
number gains or losses were systematically evaluated for clinical
significance by comparing them with values reported in the
scientific literature and the following databases: i) Database of
Genomic Variants (http://projects.tcag.ca/variation/), ii) DECIPHER
(http://decipher.sanger.ac.uk/), iii)
ISCA (https://www.iscaconsortium.org/), iv)
ECARUCA (http://www.ecaruca.net), v) Online
Mendelian Inheritance in Man (OMIM; http://www.ncbi.nlm.nih.gov/omim) and vi) Clinical
Genome Resource (https://www.clinicalgenome.org/; (9). The CMA results for the proband revealed
a 1.4 Mb deletion of 2p25.3 (12,770-1,393,107; Fig. 1A) and a 30.2 Mb duplication of
16q21q24.3 (59,939,853-90,155,062; Fig.
1B).
To further identify the chromosomal anomalies, blood
from the proband and the proband's parents was obtained for
karyotyping after obtaining written informed consent, in accordance
with conventional G-banding techniques. The ISCN 2013 nomenclature
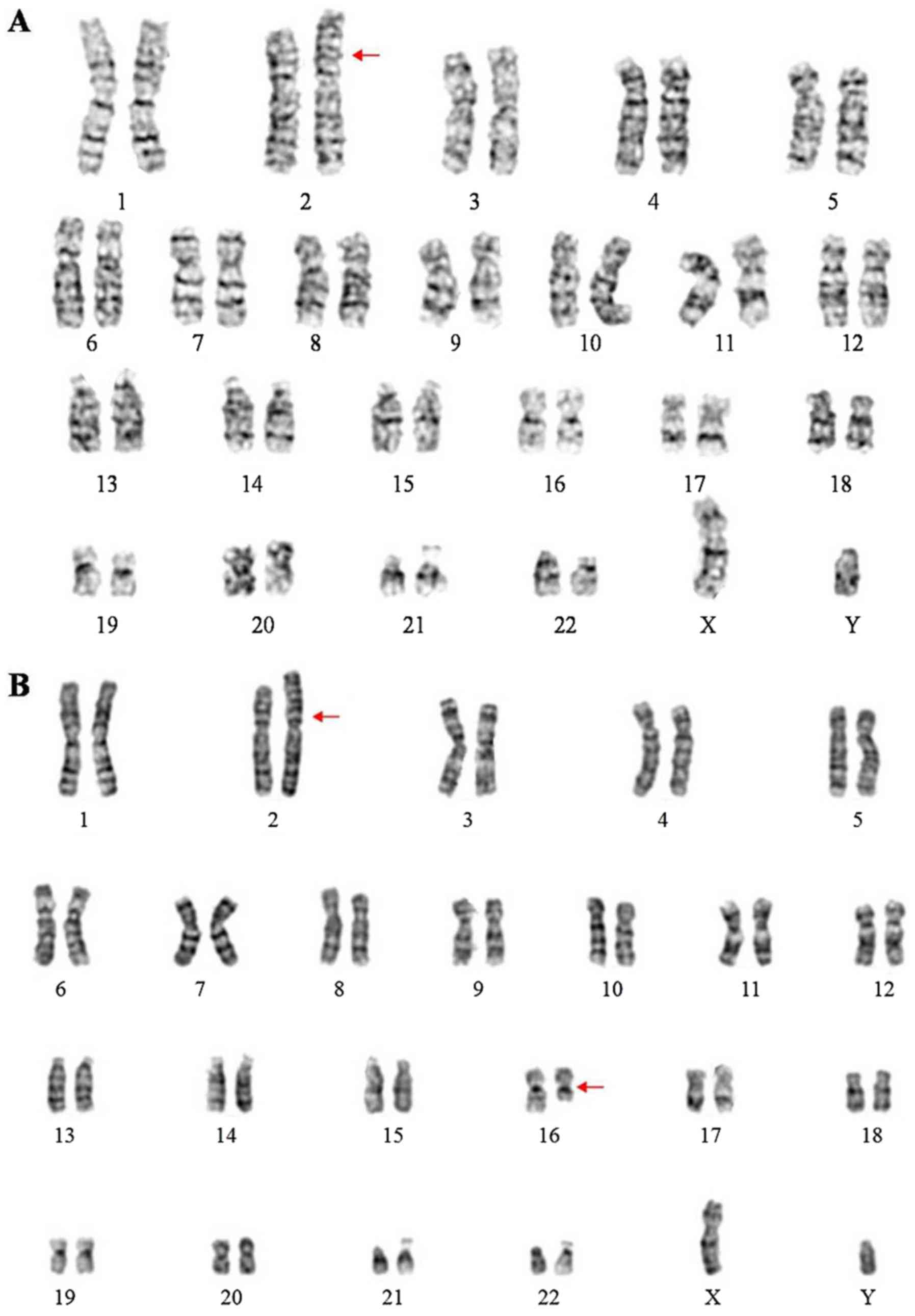
was used to describe the karyotype (10). The proband's karyotype was
46,XY,der(2)t(2;16)(p25;q21)pat (Fig.
2A). The proband's father's karyotype was
46,XY,t(2;16)(p25;q21) (Fig. 2B) and
the proband's mother's karyotype was 46,XX. Thus, the proband was
trisomic for 16q21→qter due to unbalanced segregation of a
paternally inherited balanced translocation. According to the
follow-up, the patient remained alive and stable although he did
not accept any further surgical treatments.
Discussion
The present study reports on a male infant with an
unbalanced der(2)t(2;16)(p25;q21) chromosome resulting in a 2p25.3
deletion and 16q24.3 duplication, presenting with psychomotor
retardation, small anterior fontanelles, prominent forehead, low
hairline, telecanthus, flat nasal bridge, choanal atresia, finger
and genital deformities and multiple other malformations.
Trisomy 16q21→qter is a rare chromosomal abnormality
among the different types of trisomy 16q. Brisset et al
(1) established a series of
phenotype-genotype correlations for trisomy16q: High/prominent
forehead, bitemporal narrowing, periorbital edema in the neonatal
period; severe mental retardation; vertebral, genital and anal
abnormalities associated with 16q24; distal joint contractures and
camptodactyly with 16q23; cleft palate and renal anomalies with
16q22; beaked nose and gall bladder agenesis with 16q21; gut
malrotation, and lung and liver anomalies with 16q13; and
behavioral abnormalities with band 16q11-q13. Barber et al
(11) concluded that duplications of
proximal 16q were not associated with dysmorphic features, but were
linked to speech delay, learning difficulties and behavioral
problems. Laus et al (5)
suggested that the involved region ranging from 16q11 to 16q22 was
necessary for the characteristic phenotypes of trisomy 16q.
To establish phenotype-karyotype correlations for
trisomy16q21→qter, the present study mainly compared the clinical
features of the present case with those from the literature on
partial trisomy 16q21→qter cases, as reviewed in Table I (3,12–16). The
rates of the different clinical characteristics are as follows:
Psychomotor retardation (7/7), prominent forehead (6/7), hypotonia
(6/7), low-set ears (6/7), respiratory distress (5/7), urogenital
anomalies (5/7), flat nasal bridge (5/7), hypertelorism (4/7), weak
sucking (4/7), clinodactyly of the fifth fingers (3/7) and small
anterior fontanelles (2/7). To the best of our knowledge, the
present study was the first to report on congenital muscular
torticollis and congenital laryngomalacia in a case of partial
trisomy 16q21→qter. It is generally accepted that unbalanced
segregation of a parental balanced translocation usually leads to
chromosome partial trisomy and monosomy (16). All cases of trisomy 16q21→qter in the
reviewed literature were derived from a balanced translocation of
parental origin, involving monosomy of the other chromosomes
(2p25.3, 4q35.2, 9p24, 10q26.3, 15p13, 18p11.2 and 22p12) at the
same time. In the present case, the proband's karyotype was
46,XY,der(2)t(2;16)(p25;q21)pat. The proband's mother had a normal
karyotype, while the proband's father's karyotype was
46,XY,t(2;16)(p25;q21). The proband had thus clearly inherited a
paternal unbalanced translocation between chromosomes 2 and 16. It
is difficult to distinguish whether the clinical phenotypes are
associated with trisomy 16q and monosomy 2p separately or are a
consequence of the genome rearrangements. The major limitation of
the present study is that published studies on partial trisomy
16q21→qter are limited, most of which attempt to establish the
phenotype- karyotype correlation through the clinical
manifestations. Part of the characteristics of the present case are
different from those of cases reported in the previous literature,
while partial phenotypes overlap with them, which enhances the
current knowledge on the phenotype-karyotype correlation to a
certain extent. Furthermore, as presented in Table I, poor postnatal survival has been
reported, ranging from 18 days to 7 years. Although it is
impossible to make an accurate prediction regarding the proband's
survival, it is difficult to be optimistic about his longevity.
 | Table I.Clinical features of patients with
partial trisomy 16q21→qter. |
Table I.
Clinical features of patients with
partial trisomy 16q21→qter.
|
Phenotype/presentation | Balestrazzi et
al, 1979 (12) | Garau et al,
1980 (13) | Lessick et al,
1989 (14) | Maher et al,
1991 (3) | De Carvalho et
al, 2010 (15) | Mishra et al,
2018 (16) | Present case |
|---|
| Parental
translocation | t(16;22) | t(16;18) | t(9;16) | t(10;16) | t(4;16) | t(15;16) | t(2;16) |
|
| (q21;p12) mat | (q21;p11.2) pat | (p24;q21)mat | (q26.3;q21)pat | (q35.2;q21)mat | (p13;q21) mat | (p25;q21)pat |
| Sex/gestational
age | M/40 w | F/at term | F/at term | M/32 w | F/41 w | M/33 w | M/40+3 d |
| Birth weight (g) | 2,600 | 2,470 | 2,325 | IGR(<3rd
centile) | 2,400 | 1,230 | 2,350 |
| Birth head
circumference (cm) | N.R. | N.R. | 32.5 | 29 | 35 | 28 | N.R. |
| Body length (cm) | N.R. | 40 | 46 | 42 | 48 | 42 | 46 |
| Psychomotor
retardation | + | + | + | + | + | + | + |
| Hypotonia | + | + | + | N.R. | + | + | + |
| High/prominent
forehead | + | + | + | + | + | N.R. | + |
| Small anterior
fontanelles | + | + | N.R. | N.R. | + | N.R. | + |
| Small palpebral
fissures | N.R. | N.R. | + | N.R. | + | + | N.R. |
| Epicanthus | + | + | + | N.R. | + | N.R. | N.R. |
| Hypertelorism | + | − | N.R. | N.R. | + | N.R. | + |
| Strabismus | + | + | + | N.R. | − | N.R. | N.R. |
| Broad flat nasal
bridge | + | N.R. | − | + | + | N.R. | + |
| Long philtrum | + | + | N.R. | + | + | N.R. | N.R. |
| Thin upper lip | + | N.R. | + | N.R. | + | N.R. | N.R. |
| Micrognathia | + | N.R. | + | + | + | N.R. | N.R. |
| Dysplastic/low-set
ears | + | N.R. | + | + | + | + | + |
| Abnormal palmar
creases | + | + | + | + | + | N.R. | N.R. |
| Abnormalities in
palate | + | N.R. | High palate | N.R. | High palate | Cleft palate | High palate |
| Disease | − | − | − | Congenital heart
disease | Congenital heart
disease | Congenital heart
disease | Congenital muscular
torticollis and congenital laryngomalacia |
| Urogenital
anomalies | + | + | − | + | + | N.R. | + |
| Weaksucking | + | + | N.R. | N.R. | + | N.R. | + |
| Clinodactyly of the
fifth fingers | N.R. | N.R. | + | N.R. | + | N.R. | + |
| Respiratory
distress | + | + | + | N.R. | + | N.R. | + |
| Survival time | 3 y6 m | 22 d | 6 m | 18 d | 7 y | 10 m | 19 ma |
The selection of plausible candidate genes
exhibiting a ‘dose effect’ may partly explain the clinically
observed phenotypes. The 2p25.3 region involved in the present case
contains the genes family with sequence similarity 110 member C
(FAM110C), SH3 and SYLF domain containing 1(SH3YL1),
acid phosphatase 1 (ACP1), FAM150B, transmembrane protein 18
(TMEM18) and syntrophin gamma 2(SNTG2). Doco-Fenzy
et al (7) reported on five
patients with a 2p25 deletion presenting with early-onset obesity,
hyperphagia, intellectual deficiency and behavioral difficulties.
They speculated that the ACP1 and TMEM18 genes may be
involved in early-onset obesity. Heterozygous loss of the
SNTG2 gene has been reported to be associated with
intellectual deficiency and behavioral difficulties. Zou et
al (17) reported on a
7-year-old female patient with a de novo unbalanced
der(2)t(2;16)(p25.3;q24.3) chromosome resulting in 2p25.3 deletion
and 16q24.3 duplication. However, the distinct trisomy 16q causes
various clinical manifestations, which explains that monosomy 2p
may have mild effects when associated with trisomy 16q. However,
the haploinsufficiency of genes in the region of 2p25.3 has yet to
be evaluated.
The genes in the region of 16q21-16q24.3 and the
associated diseases are summarized in Table II. The region contains >250 genes
including 61 morbidity-associated genes. The gene chromatin
licensing and DNA replication factor 1(CDT1), located at
16q24.3, which is required for DNA replication at multiple stages
of development and mitosis, is expressed only in the G1 and S
phases (18). The homozygous or
compound heterozygous mutation of the gene CDT1 has been
indicated to be associated with Meier-Gorlin syndrome 4. Patients
with this syndrome usually have short stature, distinctive facial
features, low-set/rotated ears, micrognathia, full lips, a narrow
nose with a high nasal bridge, patellar aplasia/hypoplasia and
abnormalities in sexual development. Small testes and
cryptorchidism are also involved in this condition, along with
difficulty in feeding and breathing problems (19). Ankyrin repeat domain
11(ANKRD11), located in 16q24.3, is a member of a family of
ankyrin repeat-containing cofactors that interacts with p160
nuclear receptor coactivators and inhibits ligand-dependent
transcriptional activation (20).
Heterozygous mutation in the ANKRD11 gene has been reported
to lead to KBG syndrome, characterized by short stature, global
developmental delay, intellectual disability, distinctive
craniofacial features, seizures, hypertelorism and skeletal
abnormality (brachydactyly or clinodactyly) (21,22).
However, the triplosensitivity of these genes caused by partial
trisomy 16q has not been clearly described.
| Table II.Genes in the region of 16q21-16q24.3
and the associated diseases. |
Table II.
Genes in the region of 16q21-16q24.3
and the associated diseases.
| Gene | Location | OMIM | Description | Disease |
|---|
| BEAN1 | 16q21 | 612051 | Brain expressed
associated with NEDD4 1 | Spinocerebellar
ataxia 31 |
| TK2 | 16q21 | 188250 | Thymidine kinase
2 | Mitochondrial DNA
depletion syndrome 2 (myopathic type); progressive external
ophthalmoplegia with mitochondrial DNA deletions, autosomal
recessive 3 |
| CBFB | 16q22.1 | 121360 | Core-binding factor
subunit beta | Leukemia, Acute
Myeloid; AML |
| HSF4 | 16q22.1 | 602438 | Heat shock
transcription factor 4 | Cataract 5,
multiple types |
| NOL3 | 16q22.1 | 605235 | Nucleolar protein
3 | Myoclonus, familial
cortical |
| HSD11B2 | 16q22.1 | 614232 | Hydroxysteroid
11-beta dehydrogenase 2 | Apparent
mineralocorticoid excess |
| CTCF | 16q22.1 | 604167 | CCCTC-binding
factor | Mental retardation,
autosomal dominant 21 |
| ACD | 16q22.1 | 609377 | ACD, shelterin
complex subunit and telomerase recruitment factor | Dyskeratosis
Congenita, Autosomal Dominant 6; DKCA6 |
| LCAT | 16q22.1 | 606967 |
Lecithin-cholesterol acyltransferase | Fish-eye disease;
Norum disease |
| AGRP | 16q22.1 | 602311 | Agouti-related
neuropeptide | Obesity |
| CDH3 | 16q22.1 | 114021 | Cadherin 3 | Ectodermal
dysplasia, ectrodactyly and macular dystrophy; hypotrichosis,
congenital, with juvenile macular dystrophy |
| PRMT7 | 16q22.1 | 610087 |
Proteinargininemethyltransferase 7 | Short stature,
brachydactyly, intellectual developmental disability and
seizures |
| CDH1 | 16q22.1 | 192090 | Cadherin 1 | Breast cancer;
Blepharocheilodontic syndrome 1; Gastric Cancer, Hereditary
Diffuse; HDGC; Ovarian Cancer; Prostate cancer; Endometrial
Cancer |
| COG8 | 16q22.1 | 606979 | Component of
oligomericgolgi complex 8 | Congenital disorder
of glycosylation, type IIh |
| COG4 | 16q22.1 | 606976 | Component of
oligomericgolgi complex 4 | Congenital disorder
of glycosylation, type IIj |
| AARS | 16q22.1 | 601065 |
Alanyl-tRNAsynthetase | Charcot-Marie-tooth
disease, axonal, type 2N; epileptic encephalopathy, early
infantile, 29 |
| NQO1 | 16q22.1 | 125860 | NAD(P)H quinone
dehydrogenase 1 | Nad(P)H
Dehydrogenase, Quinone 1; NQO1 |
| VAC14 | 16q22.1-q22.2 | 604632 | Vac14, PIKFYVE
complex component | Striatonigral
degeneration, childhood-onset |
| HYDIN | 16q22.2 | 610812 | HYDIN, axonemal
central pair apparatus protein | Ciliary dyskinesia,
primary, 5 |
| DHODH | 16q22.2 | 126064 | Dihydroorotate
dehydrogenase (quinone) | Miller
syndrome |
| TAT | 16q22.2 | 613018 | Tyrosine
aminotransferase | Tyrosinemia, type
II |
| HP | 16q22.2 | 140100 | Haptoglobin |
Anhaptoglobinemia |
| ZFHX3 | 16q22.2-q22.3 | 104155 | Zinc finger
homeobox 3 | Prostate
cancer |
| RFWD3 | 16q23.1 | 614151 | Ring finger and WD
repeat domain 3 | Fanconi anemia;
complementationgroup W |
| FA2H | 16q23.1 | 611026 | Fatty acid
2-hydroxylase | Spastic paraplegia
35; autosomal recessive |
| CHST6 | 16q23.1 | 605294 | Carbohydrate
sulfotransferase 6 | Macular corneal
dystrophy |
| TMEM231 | 16q23.1 | 614949 | Transmembrane
protein 231 | Joubert syndrome
20; Meckel syndrome 11 |
| KARS | 16q23.1 | 601421 |
Lysyl-tRNAsynthetase | Charcot-Marie-Tooth
disease; recessive intermediate; B; deafness, autosomal recessive
89 |
| ADAMTS18 | 16q23.1 | 607512 | ADAM
metallopeptidase with thrombospondin type 1 motif 18 | Microcornea; myopic
chorioretinal atrophy; telecanthus |
| WWOX | 16q23.1-q23.2 | 605131 | WW domain
containing oxidoreductase | Esophageal cancer;
spinocerebellar ataxia, autosomal recessive 12; epileptic
encephalopathy, early infantile, 28 |
| MAF | 16q23.2 | 177075 | MAF bZIP
transcription factor types | Ayme-Gripp
syndrome; cataract 21, multiple |
| GCSH | 16q23.2 | 238330 | Glycine cleavage
system protein H | Glycine
encephalopathy |
| BCMO1 | 16q23.2 | 605748 | Beta-carotene
oxygenase 1 | Hypercarotenemia
and vitamin A deficiency, autosomal dominant |
| GAN | 16q23.2 | 605379 | Gigaxonin | Giant axonal
neuropathy-1 |
| PLCG2 | 16q24.1 | 600220 | Phospholipase C
gamma 2 | Familial cold
autoinflammatory syndrome 3; autoinflammation, antibody deficiency
and immune dysregulation syndrome |
| MLYCD | 16q23.3 | 606761 | Malonyl-CoA
decarboxylase | Malonyl-CoA
decarboxylase deficiency |
| SLC38A8 | 16q23.3 | 615585 | Solute carrier
family 38 member 8 | Foveal hypoplasia
2, with or without optic nerve misrouting and/or anterior segment
dysgenesis |
| DNAAF1 | 16q24.1 | 613190 | Dynein axonemal
assembly factor 1 | Ciliary dyskinesia,
primary, 13 |
| RF8 | 16q24.1 | 601565 | Interferon
regulatory factor 8 | Immunodeficiency
32A, mycobacteriosis, autosomal dominant; immunodeficiency 32B,
monocyte and dendritic cell deficiency, autosomal recessive |
| FOXF1 | 16q24.1 | 601089 | Forkhead box
F1 | Alveolar capillary
dysplasia with misalignment of pulmonary veins |
| FOXC2 | 16q24.1 | 602402 | Forkhead box
C2 |
Lymphedema-distichiasis syndrome |
| FBXO31 | 16q24.2 | 609102 | F-box protein
31 | Mental retardation,
autosomal recessive 45 |
| JPH3 | 16q24.2 | 605268 | Junctophilin 3 | Huntington
disease-like 2 |
| CA5A | 16q24.2 | 114761 | Carbonic anhydrase
5A | Hyperammonemia due
to carbonicanhydrase VA deficiency |
| ZNF469 | 16q24.2 | 612078 | Zinc finger protein
469 | Brittle cornea
syndrome 1 |
| CDT1 | 16q24.3 | 605525 | Chromatin licensing
and DNA replication factor 1 | Meier-Gorlin
syndrome 4 |
| CYBA | 16q24.2 | 608508 | Cytochrome b-245
alpha chain | Chronic
granulomatous disease, autosomal, due to deficiency of CYBA |
| MVD | 16q24.2 | 603236 |
Mevalonatediphosphate decarboxylase | Porokeratosis 7,
multiple types |
| GALNS | 16q24.3 | 612222 | Galactosamine
(N-acetyl)-6-sulfatase |
Mucopolysaccharidosis IVA |
| ACSF3 | 16q24.3 | 614245 | Acyl-CoA synthetase
family member 3 | Combined malonic
and methylmalonicaciduria |
| CDH15 | 16q24.3 | 114019 | Cadherin 15 | Mental retardation,
autosomal dominant 3 |
| ANKRD11 | 16q24.3 | 611192 | Ankyrin repeat
domain 11 | KBG syndrome |
| SPG7 | 16q24.3 | 602783 | SPG7, paraplegin
matrix AAA peptidase subunit | Spastic paraplegia
7, autosomal recessive |
| CHMP1A | 16q24.3 | 164010 | Charged
multivesicular body protein 1A | Pontocerebellar
hypoplasia, type 8 |
| CDK10 | 16q24.3 | 603464 | Cyclin-dependent
kinase 10 | Al Kaissi
syndrome |
| FANCA | 16q24.3 | 607139 | FA complementation
group A | Fanconianemia,
complementation group A |
| MC1R | 16q24.3 | 155555 | Melanocortin 1
receptor | Albinism,
oculocutaneous, type II; Skin/hair/eye pigmentation, variation in,
2; analgesia from kappa-opioid receptor agonist, female-specific;
melanoma, cutaneous malignant, 5 |
| TUBB3 | 16q24.3 | 602661 | Tubulin beta 3
class III | Fibrosis of
extraocular muscles, congenital, 3A; cortical dysplasia, complex,
with other brain malformations 1 |
| GAS8 | 16q24.3 | 605178 | Growth
arrest-specific 8 | Ciliary dyskinesia,
primary, 33 |
| PIEZO1 | 16q24.3 | 611184 | Piezo type
mechanosensitive ion channel component 1 | Dehydrated
hereditary stomatocytosis with or without pseudohyperkalemia and/or
perinatal edema; Lymphatic malformation 6 |
| APRT | 16q24.3 | 102600 | Adenine
phosphoribosyltransferase | Adenine
phosphoribosyltransferase deficiency |
In conclusion, the present case study reports on a
newborn suffering from multiple organ malformations, with 2p25.3
deletion and 16q21q24.3 duplication. Concomitant partial monosomy
2p and partial trisomy 16q is unusual in a clinical context. The
present results not only underline the importance of trisomy
16q21→qter, but also uncover certain unprecedented clinical
features, which expand on the current knowledge of the clinical
phenotypes of trisomy 16q21→qter syndrome. The limited number of
relevant previous studies put a restriction on the clear
delineation of the phenotype-karyotype correlation of trisomy
16q21→qter. Considering the observed clinical manifestations, the
SNTG2, CDT1 and ANKRD11 genes are plausible
candidates contributing to the proband's phenotype. As more and
more clinical and molecular cytogenetic evaluations of cases with
partial trisomy 16q emerge, it is anticipated that more accurate
information on genotype-phenotype correlations will be established.
Taking all of the information obtained into consideration,
pre-implantation genetic diagnosis and prenatal diagnosis would be
the most suitable choice if the proband's parents intend to
conceive again.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Special Funds
for Projects of Independent Innovation and New and Hi-tech Industry
Development of Jilin Province (grant no. 2017C025).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FY obtained the clinical information, collected data
from the literature and wrote the manuscript. YJ and YP critically
reviewed the manuscript. LeL and LiL performed the cytogenetic
study and CMA analysis on the proband and the proband's parents. RL
acquired funding and designed the study. RW conceived and designed
the study, and performed the final review and editing of the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by the Medical Ethics
Committee of the First Hospital of Jilin University (Changchun,
China). The parents of the patient provided written informed
consent for their child to participate in this study, and they also
consented to participate themselves.
Patient consent for publication
The present case report was published with the
informed consent of the patient's parents, whose anonymities were
preserved.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brisset S, Joly G, Ozilou C, Lapierre JM,
Gosset P, LeLorc'h M, Raoul O, Turleau C, Vekemans M and Romana SP:
Molecular characterization of partial trisomy 16q24.1-qter:
Clinical report and review of the literature. Am J Med Genet.
113:339–345. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hassold T, Merrill M, Adkins K, Freeman S
and Sherman S: Recombination and maternal age-dependent
nondisjunction: Molecular studies of trisomy 16. Am J Hum Genet.
57:867–874. 1995.PubMed/NCBI
|
|
3
|
Maher ER, Willatt L, Cuthbert G, Chapman C
and Hodgson SV: Three cases of 16q duplication. J Med Genet.
28:801–802. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schmickel R, Poznanski A and Himebaugh J:
16q trisomy in a family with a balanced 15/16 translocation. Birth
Defects Orig Artic Ser. 11:229–236. 1975.PubMed/NCBI
|
|
5
|
Laus AC, Baratela WA, Laureano LA, Santos
SA, Huber J, Ramos ES, Rebelo CC, Squire JA and Martelli L:
Karyotype/phenotype correlation in partial trisomies of the long
arm of chromosome 16: Case report and review of literature. Am J
Med Genet A 158A. 821–827. 2012. View Article : Google Scholar
|
|
6
|
De Rocker N, Vergult S, Koolen D, Jacobs
E, Hoischen A, Zeesman S, Bang B, Béna F, Bockaert N, Bongers EM,
et al: Refinement of the critical 2p25.3 deletion region: The role
of MYT1L in intellectual disability and obesity. Genet Med.
17:460–466. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Doco-Fenzy M, Leroy C, Schneider A, Petit
F, Delrue MA, Andrieux J, Perrin-Sabourin L, Landais E, Aboura A,
Puechberty J, et al: Early-onset obesity and paternal 2pter
deletion encompassing the ACP1, TMEM18, and MYT1L genes. Eur J Hum
Genet. 22:471–479. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stevens SJ, van Ravenswaaij-Arts CM,
Janssen JW, Klein Wassink-Ruiter JS, van Essen AJ, Dijkhuizen T,
van Rheenen J, Heuts-Vijgen R, Stegmann AP, Smeets EE and Engelen
JJ: MYT1L is a candidate gene for intellectual disability in
patients with 2p25.3 (2pter) deletions. Am J Med Genet A 155A.
2739–2745. 2011. View Article : Google Scholar
|
|
9
|
Wu XL, Li R, Fu F, Pan M, Han J, Yang X,
Zhang YL, Li FT and Liao C: Chromosome microarray analysis in the
investigation of children with congenital heart disease. BMC
Pediatr. 17:1172017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shaffer LG, Slovak ML and Campbell LJ:
ISCN 2013: An international system for human cytogenetic
nomenclature. Basel, Switzerland: Karger S.; pp. 1382013,
PubMed/NCBI
|
|
11
|
Barber JC, Zhang S, Friend N, Collins AL,
Maloney VK, Hastings R, Farren B, Barnicoat A, Polityko AD,
Rumyantseva NV, et al: Duplications of proximal 16q flanked by
heterochromatin are not euchromatic variants and show no evidence
of heterochromatic position effect. Cytogenet Genome Res.
114:351–358. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Balestrazzi P, Giovannelli G, Landucci
Rubini L and Dallapiccola B: Partial trisomy 16q resulting from
maternal translocation. Hum Genet. 49:229–235. 1979.PubMed/NCBI
|
|
13
|
Garau A, Crisponi G, Peretti D, Peretti D,
Vanni R and Zuffardi O: Trisomy 16q21=to qter. Hum Genet.
53:165–167. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lessick ML, Israel J, Wong PW and Szego K:
Partialtrisomy 16q secondary to a maternal 9;16 translocation. J
Med Genet. 26:63–64. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
de Carvalho AF, da Silva Bellucco FT, dos
Santos NP, Pellegrino R, de Azevedo Moreira LM, Toralles MB,
Kulikowski LD and Melaragno MI: Trisomy 16q21→qter: Seven-year
follow-up of a girl with unusually long survival. Am J Med Genet A
152A. 2074–2078. 2010. View Article : Google Scholar
|
|
16
|
Mishra R, Paththinige CS, Sirisena ND,
Nanayakkara S, Kariyawasam UGIU and Dissanayake VHW: Partial
trisomy 16q21→qter due to an unbalanced segregation of a maternally
inherited balanced translocation 46,XX,t(15;16)(p13;q21): A case
report and review of literature. BMC Pediatr. 18:42018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zou YS, Van Dyke DL and Ellison JW:
Microarray comparative genomic hybridization and FISH studies of an
unbalanced cryptic telomeric 2p deletion/16q duplication in a
patient with mental retardation and behavioral problems. Am J Med
Genet A 143A. 746–751. 2007. View Article : Google Scholar
|
|
18
|
Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic
C, Walter JC and Dutta A: Inhibition of eukaryotic DNA replication
by geminin binding to Cdt1. Science. 290:2309–2312. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
de Munnik SA, Bicknell LS, Aftimos S,
Al-Aama JY, van Bever Y, Bober MB, Clayton-Smith J, Edrees AY,
Feingold M, Fryer A, et al: Meier-Gorlin syndrome
genotype-phenotype studies: 35 individuals with pre-replication
complex gene mutations and 10 without molecular diagnosis. Eur J
Hum Genet. 20:598–606. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang A, Yeung PL, Li CW, Tsai SC, Dinh
GK, Wu X, Li H and Chen JD: Identification of a novel family of
ankyrin repeats containing cofactors for p160 nuclear receptor
coactivators. J Biol Chem. 279:33799–33805. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sirmaci A, Spiliopoulos M, Brancati F,
Powell E, Duman D, Abrams A, Bademci G, Agolini E, Guo S, Konuk B,
et al: Mutations in ANKRD11 cause KBG syndrome, characterized by
intellectual disability, skeletal malformations, and macrodontia.
Am J Hum Genet. 89:289–294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kleyner R, Malcolmson J, Tegay D, Ward K,
Maughan A, Maughan G, Nelson L, Wang K, Robison R and Lyon GJ: KBG
syndrome involving a single-nucleotide duplication in ANKRD11. Cold
Spring Harb Mol Case Stud. 2:a0011312016. View Article : Google Scholar : PubMed/NCBI
|