Introduction
Ischemic stroke, a major cause of mortality and
long-term disability among adults worldwide, often occurs when a
cerebral blood vessel is ruptured or occluded, resulting in various
acute and chronic diseases of the brain (1). Advances in medicine have led to the
development of thrombolytic agents and intravascular techniques,
which markedly decrease functional deficits (2,3). Early
reperfusion is crucial for the rapid recovery of cerebral blood
flow in ischemic tissue, mainly involved in neuroprotection and
thrombolysis. However, the re-establishment of blood flow following
ischemia is always associated with certain side effects, which is
termed ischemia/reperfusion (IR) injury (4). Continually, cerebral IR promotes the
necrosis and apoptosis of nerve cells (5). Therefore, innovative treatment
strategies are vital for defending the brain against IR injury to
obtain a favorable prognosis.
Ischemic preconditioning (IPreC) was first described
in 1986 in a canine myocardial ischemia model (6). IPreC means a brief ischemic event,
which can mobilize intrinsic protective mechanisms that improve
tolerance to severe ischemic insult (7). IPreC stands for an important adaptation
of the central nervous system to sublethal ischemia, which is an
effective method against IR injury through transient repeated
ischemia (8,9). Numerous studies have confirmed that
IPreC improves ischemic tolerance in animal organs and tissues
(10,11). In previous years, orthopedic studies
have focused on the protective effects of IPreC in IR injury of
limb skeletal muscles (12,13). According to the report of Dong et
al (14), IPreC protected
against IR injury in the rat sciatic nerve. However, the underlying
mechanisms of IPreC resulting in neuronal protective effects
remains to be fully elucidated.
Accumulated evidence has confirmed that oxidative
stress is implicated in the pathogenesis of ischemic and
reperfusion injury in the brain (15,16).
Reports have indicated that oxidative burst lasted for several
minutes upon the onset of reperfusion and persistently increased
the production of oxygen radicals (17,18). The
overproduction of reactive oxygen species (ROS) results in
oxidative damage, including lipid peroxidation, protein oxidation
and DNA damage, which can lead to cell death (19). The brain is susceptible to the damage
caused by oxidative stress and data support the hypothesis that
oxidative stress is a potent mediator of cerebral IR injury
(20,21). Therefore, alleviating oxidative
stress may be a major target for the treatment of cerebral IR.
Cerebral IR injury and its associated inflammation
are vital in the evolution of brain injury (22). The activation of transcriptional
regulatory factors can result in inflammatory responses, leading to
the release of various pro-inflammatory factors (23). It has been reported that nuclear
factor-κB (NF-κB) may be the ‘main switch’ of the neurovascular
unit inflammatory reactions (24).
It was found that the activation of NF-κB induced by transient
ischemia occurs prior to DNA fragmentation (25). Several neuroprotective reagents
exhibit the effect of suppressing the expression and/or activity of
NF-κB, suggesting that NF-κB is important in regulating transient
ischemia-induced neuronal death (26,27).
Therefore, the present study investigated the involvement of NF-κB
in the effect of IPreC on cerebral IR injury.
In the present study, the effect of IPreC on
cerebral IR injury and its underlying mechanisms were detected. The
result demonstrated that IPreC mitigated cerebral IR injury via
alleviating free radicals and the inflammatory response.
Materials and methods
Experimental animals
In total, 24 male Sprague-Dawley rats (age, 10–12
weeks; weight, 200–250 g) were obtained from the Experimental
Animal Center of Binzhou Medical College (Binzhou, China). All
experimental protocols were approved by the Animal Ethics Committee
of Binzhou Medical College. All rats were fed in ventilated cages
at 24°C and 30–50% humidity in a 12/12 h light/dark cycle, with
free access to food and water for 1 week.
Induction of transient cerebral
ischemia
Experiments were performed according to previous
reports (28,29). Briefly, each rat was placed in the
supine position and its neck was incised in the middle ventral site
to expose the left carotid artery. The left carotid artery was then
isolated from the vagus nerve and clamped via small vascular clips
to induce hypotension for 1 h by occlusion. A cerebral animal model
was thus established. Subsequently, the rats were randomly divided
into three groups (n=8); i) control group, healthy rats; ii) IR
group, cerebral animal model treated with another 24 h reperfusion;
iii) IR + IPreC group, rats were anesthetized with xylazine (1.5
mg/100 g) and ketamine (4 mg/100 g) by intramuscular injection, and
then fixed on the operating table in a supine position. Following
shaving and sterilization, a 3–4-cm-long cervical median incision
was made. The left common carotid artery and the left external
carotid artery were exposed, followed by three cycles of 10-min
occlusion (left internal carotid artery was occluded using a
microclamp) and 10 min of reperfusion (microclamp was removed to
restore blood flow). Following IPreC for 72 h, the rats were
treated by the induction of transient cerebral IR (30). The rats were then sacrificed with an
overdose of urethane for subsequent experiments.
Determination of cerebral infarction
area
Following craniotomy, the brain was harvested and
then stored at −80°C for 15 min. The brain tissues were cut into
2-mm sections, and then stained with 2% 2,3,5-triphenyl-tetrazolium
chloride (TTC) solution at 37°C for 30 min and soaked in 10%
paraformaldehyde solution overnight. The areas of infarction were
observed in gray under an inverted microscope. Image-Pro Plus 6.0
(Media Cybernetics Inc., Rockville, MD, USA) was used to measure
the area of cerebral infarction.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) staining
Cryosections were cut (6-µm-thick) and placed on
slides. The measurement of apoptotic nuclei was accomplished by
TUNEL in brain sections using an In-Situ Cell Death Detection kit
(Roche Diagnostics GmbH, Mannheim, Germany). Briefly, the sections
were washed with PBS containing 1% Triton-100 for 2 min on ice, and
then incubated with 50 µl TUNEL reaction mixture for 60 min at
37°C. Subsequently, the sections were stained with DAPI. The
TUNEL-positive nuclei were detected using a fluorescence microscope
(Olympus Corporation, Tokyo, Japan).
Western blot assay
Tissue proteins were extracted from the brains and
were homogenized in RIPA lysis buffer containing 1% PMSF on ice.
The lysates were centrifuged at 10,000 × g for 15 min at 4°C, and
supernatant was collected. Total protein was quantified using a
bicinchoninic acid assay (Solarbio Biotechnology Co., Ltd.,
Beijing, China) and 20 µg protein/lane was separated via SDS-PAGE
on a 10% gel. The separated proteins were transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA).
Subsequently, the membranes were blocked with 5% non-fat skim milk
for 2 h at room temperature, followed by incubation at 4°C
overnight with the following primary antibodies: B-cell lymphoma 2
(Bcl-2) (1:1,000; cat. no. 15071), Bcl-2-associated X protein (Bax)
(1:1,000; cat. no. 5023), interleukin (IL)-1β (1:1,000; cat. no.
12703), IL-1 receptor antagonist (IL-1Ra) (1:1,000; cat. no. 3865),
transforming growth factor-β-activated kinase 1 (TAK1; 1:1,000;
cat. no. 5206), P65 (1:1,000; cat. no. 8242), phosphorylated
(p-)P65 (1:1,000; cat. no. 3033), tumor necrosis factor (TNF)-α
(1:1,000; cat. no. 6945) and GAPDH (1:1,000; cat. no. 5174; all
Cell Signaling Technology, Inc., Danvers, MA, USA). Following
washing with TBST twice, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibody anti-rabbit
IgG (1:1,000; cat. no. 14708; CST) in TBST solution for 1 h at room
temperature. Following washing, the signals of detected proteins
were assessed using an enhanced chemiluminescence reaction system
(EMD Millipore). ImageJ software (version 1.48; National Institutes
of Health, Bethesda, MD, USA) was used to evaluate the protein
levels.
Measurement of oxidative and
anti-oxidative parameters in the brain
The brain tissues were homogenized in nine volumes
of ice-cold 0.9% NaCl solution and centrifuged at 10,000 × g for 10
min at 4°C. The supernatant was collected for the measurement of
oxidative stress parameters. Malondialdehyde (MDA), lactate
dehydrogenase (LDH), nitric oxide (NO) and superoxide dismutase
(SOD) levels in the brain were detected using corresponding
biochemical methods according to the instructions of the reagent
kits (Nanjing Jiancheng Bioengineering Institute, Nanjing,
China).
Flow cytometry
Venous blood from the antecubital veins was
collected in an EDTA-coated tube. Flow cytometry was performed
within 4 h of blood sampling. Briefly, the red blood cells were
lysed using 1% red blood cell lysis buffer (Nordic Biosite AB,
Täby, Sweden). White blood cells (5×105) were collected
in each test tube. The cells were then washed with an isotonic
buffer (BD FACS Flow; BD Biosciences, Franklin Lakes, NJ, USA) and
centrifuged at 600 × g for 5 min at 4°C. This process was repeated
three times. Subsequently, cells were resuspended in the isotonic
buffer, and monoclonal antibodies CD11b (1:1,000; ab8878) and CD18
(1:1,000; ab119830; both Abcam, Cambridge, UK) were added to each
sample and incubated in the dark at 4°C for 20 min. The cells were
then washed and resuspended in the isotonic FACS Flow buffer. The
stained cells were detected using flow cytometry (BD FACSCanto II;
BD Biosciences).
ELISA assay
The release of serum IL-6 and IL-10 was measured
using ELISA kits (Dakewe Biotech Co., Ltd., Shenzhen, China)
according to the manufacturer's protocol. The reaction product was
determined at 450 nm wavelength and the optical density values were
detected and analyzed.
Statistical analysis
The results were analyzed using SPSS 17.0 software
(SPSS, Inc., Chicago, IL, USA). The differences of the indicators
between the three groups were compared by one-way analysis of
variance followed by Boferroni's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
IPreC alleviates cell apoptosis
To determine whether IPreC can mitigate cerebral IR
injury, the area of cerebral infarction was detected using TTC
staining. As illustrated in Fig. 1A,
the increased area of cerebral infarction induced by IR was
markedly suppressed by IPreC. In addition, IR-induced cell
apoptosis was significantly inhibited by IPreC (Fig. 1B). The increased expression of
Bcl-2/Bax in the IR + IPreC group compared with the IR group
further validated this (Fig. 1C).
These results indicated that IPreC ameliorated IR-induced cell
apoptosis in the brain.
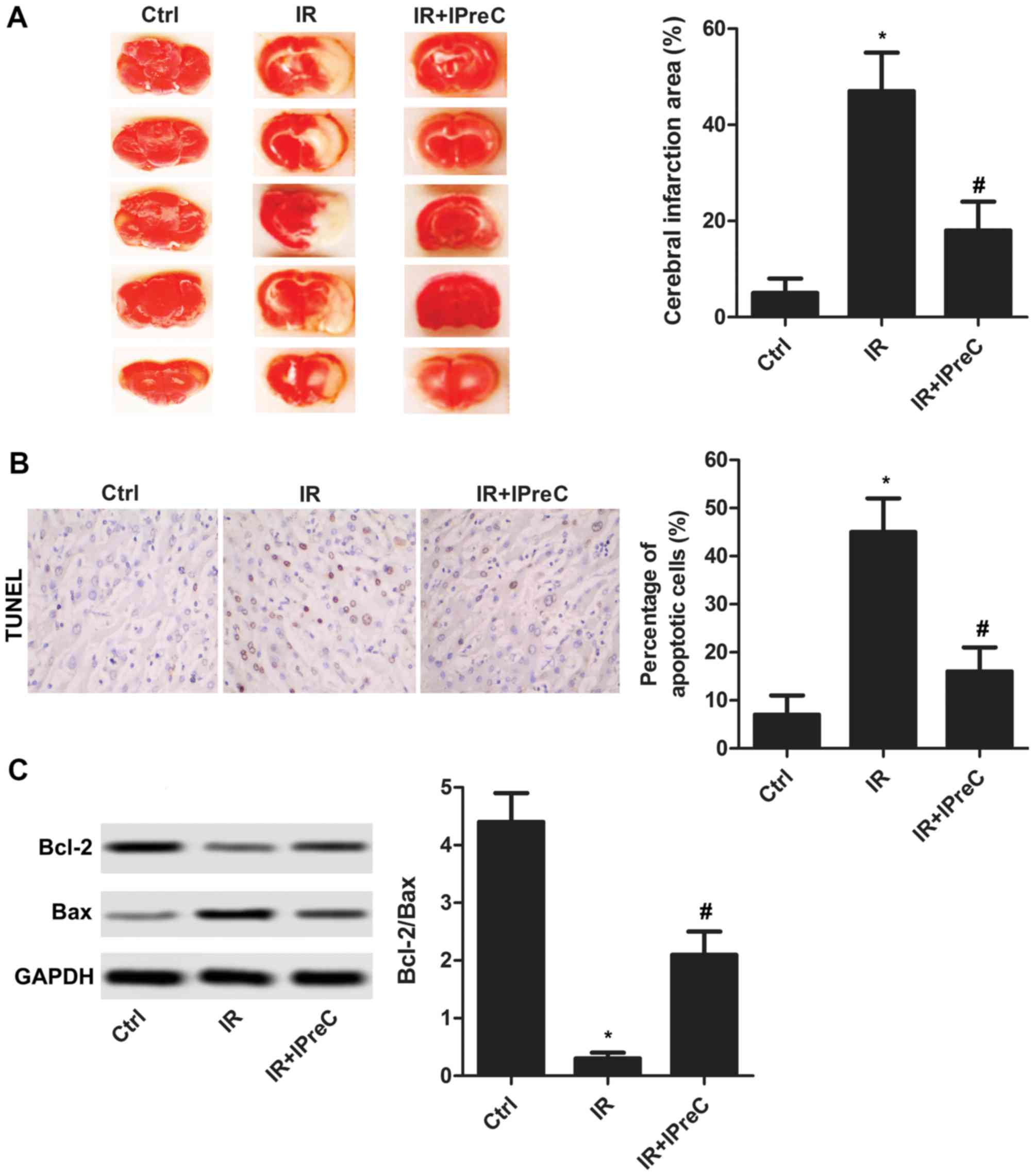 | Figure 1.Remote IPreC alleviates cell
apoptosis. (A) Cerebral infarction area was measured using
2,3,5-triphenyl-tetrazolium chloride staining. (B) Cell apoptosis
was detected using TUNEL staining (magnification, 400×). The bar
graph shows the statistical analysis of the percentage of apoptotic
cells in the brain according to the result of the TUNEL assay. (C)
Expression levels of Bcl-2 and Bax were detected using western blot
analysis. Experiments were repeated at least three times, and error
bars represent the mean ± standard deviation (*P<0.05, vs. Ctrl
group; #P<0.05, vs. IR group). IPreC, ischemic
preconditioning; IR, ischemia/reperfusion; Ctrl, control; Bcl-2,
B-cell lymphoma 2; Bax, Bcl-2-associated X protein. |
IPreC attenuates free radical
injury
To investigate the association between the
protective effect of IPreC on IR injury and its antioxidant status,
relative expression levels of MDA, LDH, NO and SOD in brain tissue
were measured using biochemical methods. As shown in Fig. 2, the IR rats exhibited a marked
increase in MDA, LDH and NO content, whereas the expression of SOD
was markedly suppressed, and these changes were reversed in IR +
IPreC group. These results demonstrated the antioxidative stress
effect of IPreC in mitigating cerebral IR-induced injury.
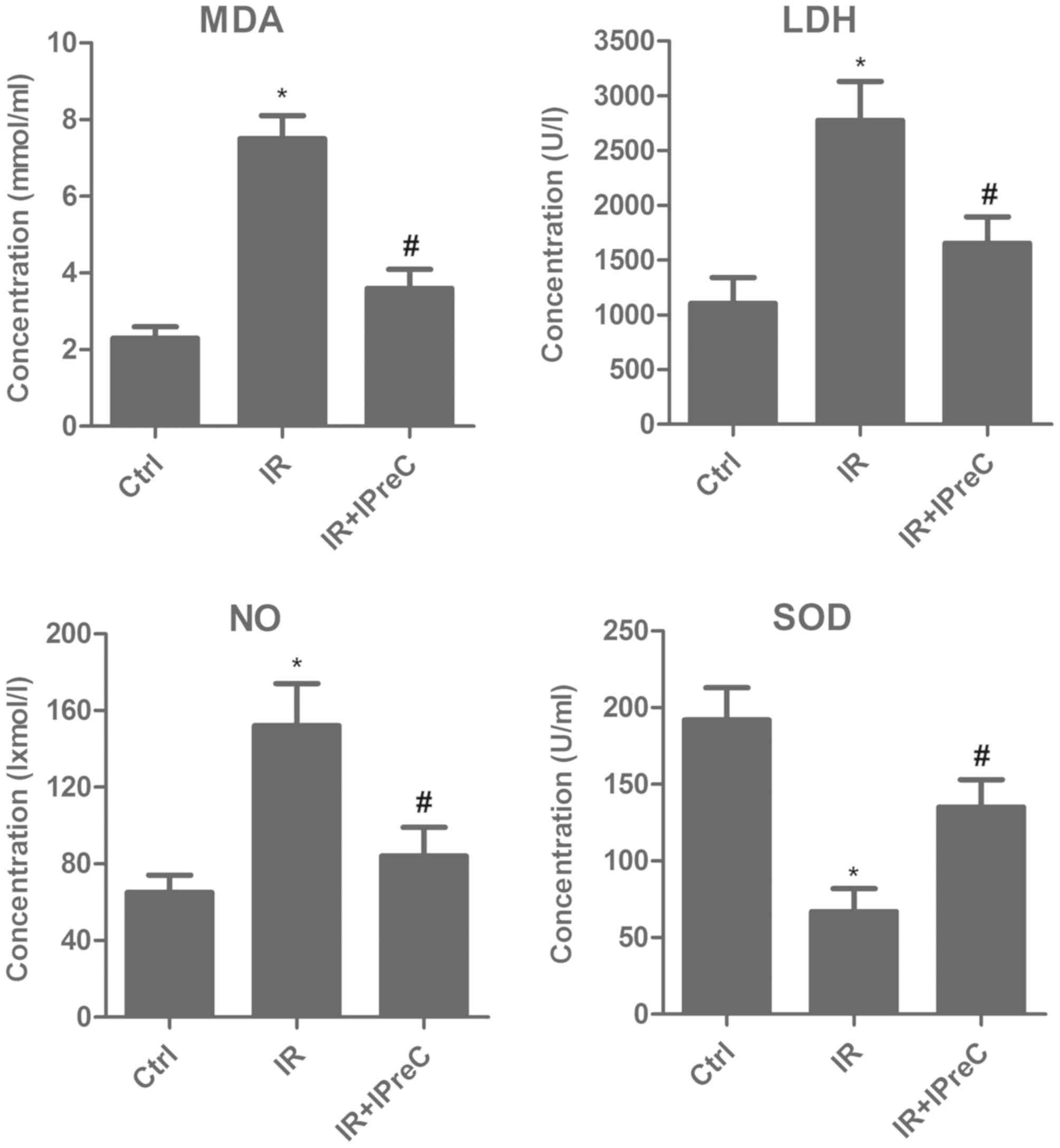 | Figure 2.Remote IPreC attenuates free radical
injury. The levels of MDA, LDH, NO and SOD were detected using
corresponding biochemical methods. Experiments were repeated at
least three times, and error bars represent the mean ± standard
deviation (*P<0.05, vs. Ctrl group; #P<0.05 vs. IR
group). IPreC, ischemic preconditioning; IR, ischemia/reperfusion;
Ctrl, control; MDA, malondialdehyde; LDH, lactate dehydrogenase;
NO, nitric oxide; SOD, superoxide dismutase. |
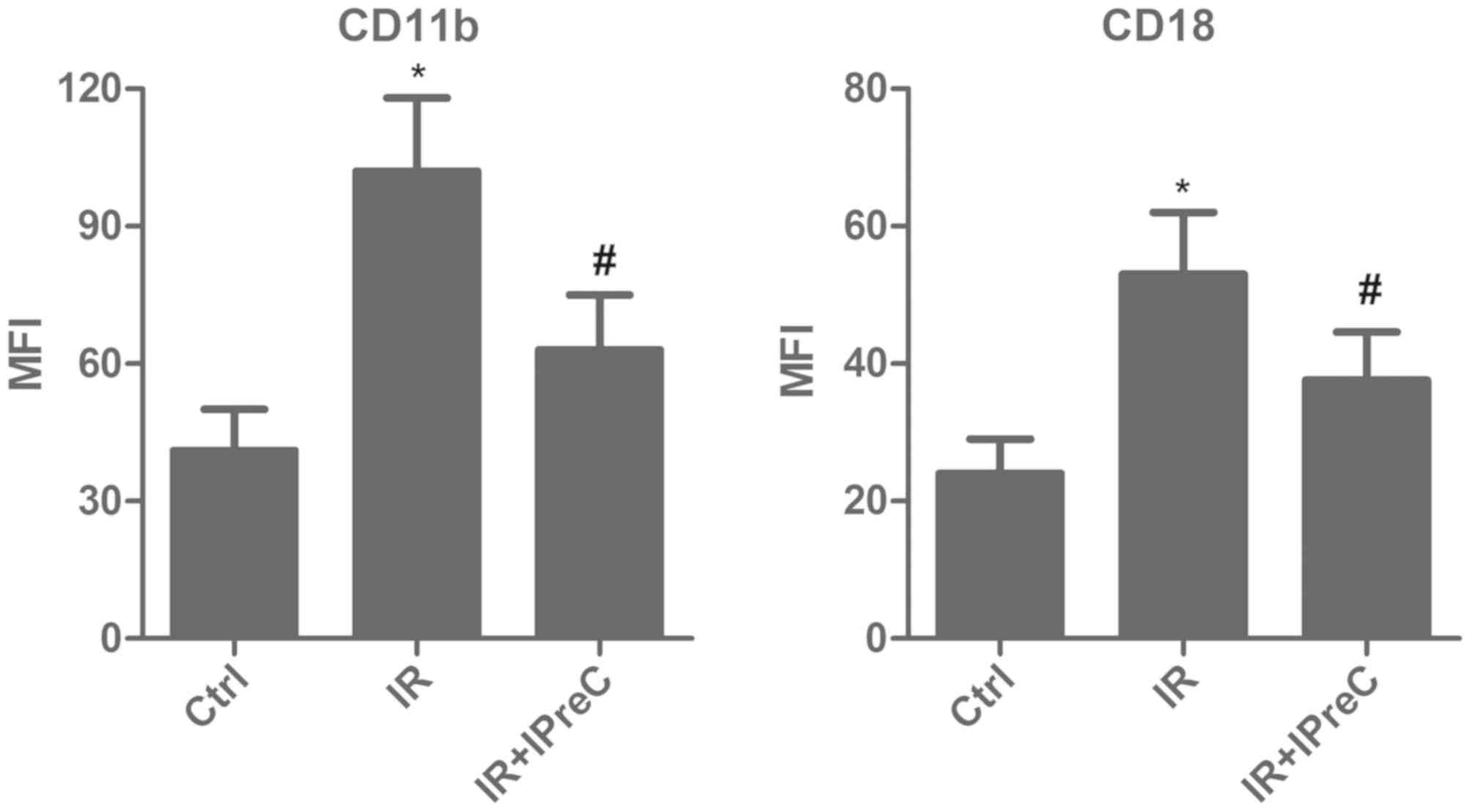
IPreC decreases the levels of CD11b
and CD18
To evaluate whether IPreC can affect the levels of
the CD11b+ and CD18+ sub-population, flow
cytometry and FACS were applied. The data indicated a marked
elevation in the percentage of CD11b+ and
CD18+ cells in the IR group compared with the control
group. However, IPreC reversed these changes (Fig. 3).
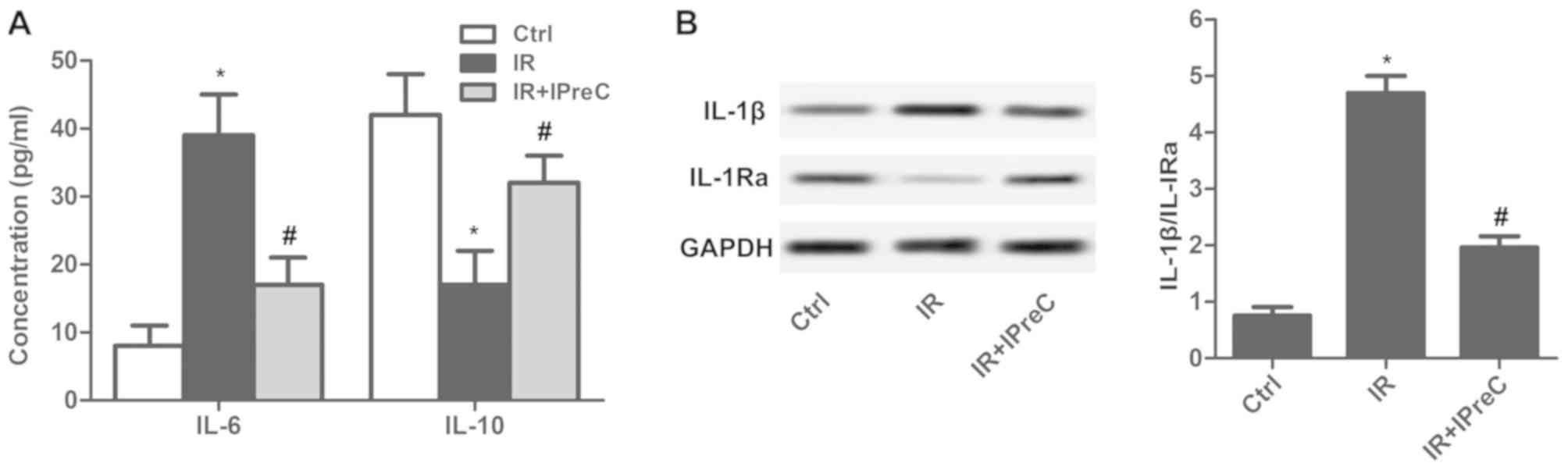
IPreC ameliorates the inflammatory
response
To detect the effect of IPreC on the IR-induced
inflammatory response, inflammatory factors were measured. The
results indicated that the serum level of IL-6 was increased and
that of IL-10 was decreased in the IR group compared with the
control group. A reduced level of IL-6 and elevated level of IL-10
was measured in the IR + IPreC group compared with the IR group
(Fig. 4A). In addition, IPreC
reduced the IR-induced increase in the ratio of IL-1β/ IL-1Ra in
the brain (Fig. 4B). These findings
indicated that IPreC markedly suppressed the IR-induced
inflammatory response.
IPreC inactivates the NF-κB
pathway
To examine the mechanisms of IPreC on the IR-induced
inflammatory reaction, the NF-κB pathway was detected using western
blot analysis. As illustrated in Fig.
5, the increase in the expression levels of TAK1, p-P65/P65 and
TNF-α induced by IR were be significantly suppressed by IPreC.
These results demonstrated that IPreC inactivated the NF-κB
pathway.
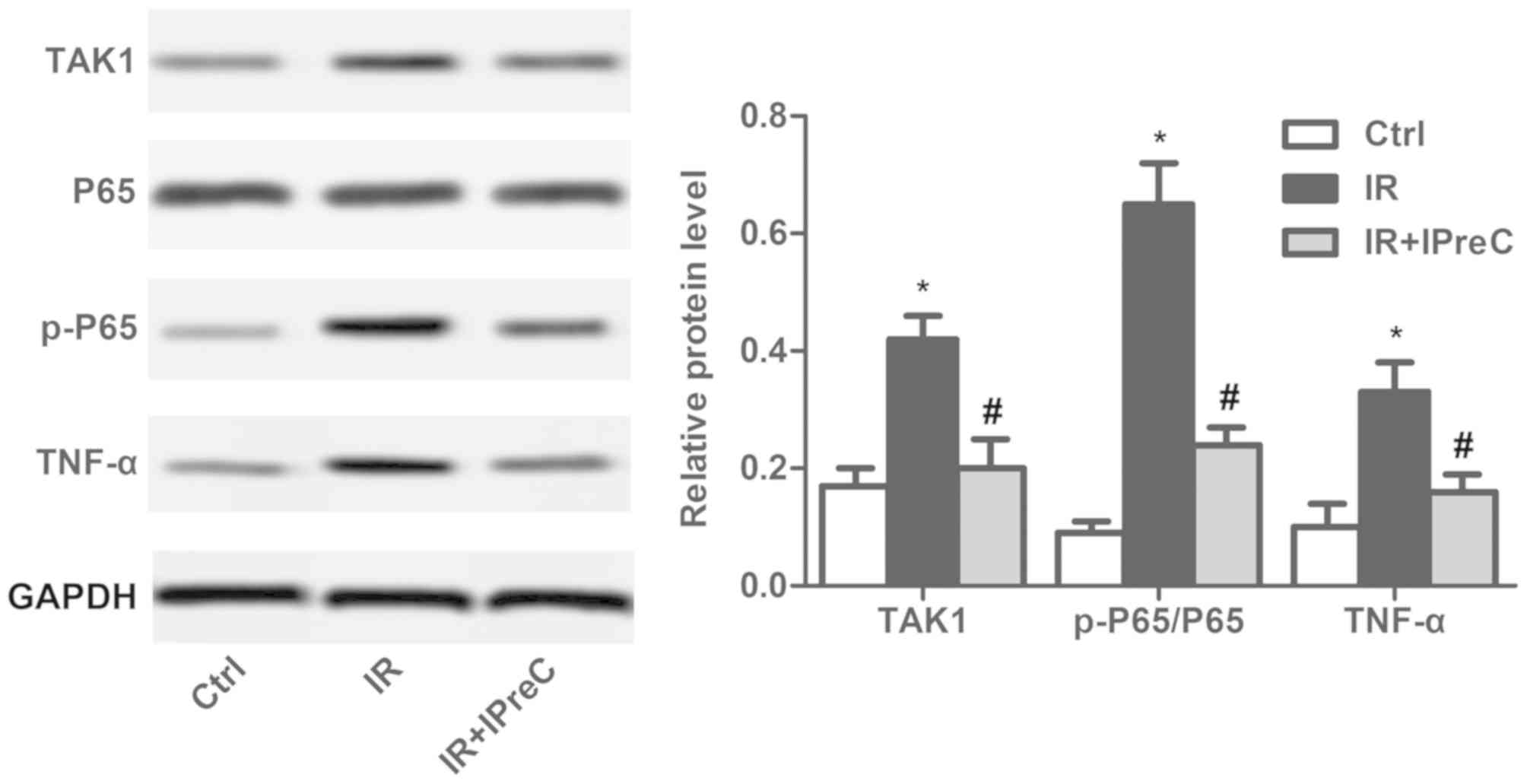 | Figure 5.Remote IPreC inactivates the
NF-κB/TAK1 pathway. The expression levels of TAK1, P65, p-P65, and
TNF-α were measured using western blot analysis. Experiments were
repeated at least three times, and error bars represent the mean ±
standard deviation (*P<0.05, vs. Ctrl group;
#P<0.05, vs. IR group). IPreC, ischemic
preconditioning; IR, ischemia/reperfusion; Ctrl, control; NF-κB,
nuclear factor-κB; TAK1, transforming growth factor-β-activated
kinase 1; p-, phosphorylated; TNF-α, tumor necrosis factor-α. |
Discussion
Cerebral IR is the main cause of mortality and
disability in adults leading to functional and structural injury in
different brain regions (31). It is
becoming increasingly accepted that IPreC is able to defend the
brain against a subsequent longer ischemic insult and does not lead
to neuronal death (8). IPreC
exhibits a tolerance, which is termed ‘ischemic tolerance’
(32). However, the underlying
molecular mechanisms of ischemic tolerance remained to be fully
elucidated.
Apoptosis is involved in trauma, ischemia, and
neurodegenerative diseases, thus being important in brain damage
(33). The results demonstrated that
many neurons in ischemic regions suffered from necrotic and
apoptotic cell death (34). Studies
have also indicated that Bcl-2 family proteins are involved in
apoptotic signaling pathways (35).
Bax is a pro-apoptotic member of the Bcl-2 family and is involved
in inducing cell apoptosis (36).
According to published reports, IPreC can alleviate IR injury via
reducing apoptosis. For example, in IR-induced myocardial injury,
IPreC markedly suppressed the expression of cleaved caspase 3,
cleaved poly (ADP-ribose) polymerase and the Bax/Bcl-2 ratio
(37). Jeong et al (38) demonstrated that IPreC increased the
mRNA level of Bcl-2 in hepatic IR injury. In addition, it was
reported that the decrease in the level of Bcl-2 and the increase
in the level of Bax induced by IR were significantly suppressed by
IPreC in renal IR injury (39).
Similarly, in the present study, IPreC suppressed cell apoptosis
with elevated Bcl-2/Bax level.
As byproducts or intermediates generated from
complicated reactions in living cells, free radicals can lead to
the production of nitrogen species and reactive oxygen (40). Under pathological circumstances, the
excessive accumulation of free radicals and the deficiency of
antioxidants result in tissue injury, namely oxidative damage
(41). When lipids, protein and DNA
are damaged by free radicals, stable oxidized biomolecule products,
including MDA, are produced (42).
Increasing evidence has indicated that IPreC is vital in
alleviating oxidative damage following an IR event. Yang et
al (43) demonstrated that IPreC
reduced the level of MDA and elevated the activity of SOD in serum
and the intestine following an intestinal IR event. In addition,
IPreC decreased the level of NO in the plasma of mice suffering a
kidney IR event (44). The serum
level of LDH was reported to be significantly lower under IPreC
treatment in patients with IR injury following tourniquet release
during total knee arthroplasty (45). Similarly, in the present study, IR
significantly downregulated the level of SOD and increased the
levels of MDA, LDH and NO. However, IPreC reversed these
changes.
A previous study indicated that oxygen free radicals
can also be triggered by infiltrating leukocytes following
reperfusion (46). According to
previous reports, IR triggers microcirculation interplay in various
ways that promote the expression of leukocyte adhesion molecules
CD11b/CD18, promoting the adhesion of leukocytes to venules
(47). The leukocytes adhere to the
venular wall and, in turn, release peroxides and protease that
cause the leakage of serum (48,49).
Therefore, the expression levels of CD11b and CD18 are directly
associated with the extent of post-operative IR injury (50,51).
Kharbanda et al (52)
reported that IPreC inhibited the increased expression of
neutrophil CD11b in humans with forearm ischemia followed by
reperfusion. Chouker et al (51) demonstrated that the expression of
CD18 did not elevate further by IPreC in patients subjected to IR
injury. The present study was in accordance with these results.
IPreC, significantly suppressed the increased CD11b and CD18
expression levels induced by IR.
The IR injury in organs is caused by various
factors, including inflammation. During the inflammatory reaction,
pro-inflammatory factors, including IL-6 and IL-1β, were released,
and the level of anti-inflammatory cytokines (IL-10 and IL-1Ra) was
decreased (53). Previous studies
have indicated that IPreC markedly suppressed the gene expression
level of pro-inflammatory factors (TNF-α, IL-1β, and IL-6) and
chemokines in patients with renal IR injury (54). In addition, IPreC inhibited the
release of IL-1β and enhanced the generation of IL-10 following IR
in normal and steatotic livers (55). IPreC also led to inhibition of the
mRNA and protein levels of IL-1β at 6 h and the increased the
protein level of IL-1Ra at 24 h (56). Similarly, in the present study, IPreC
suppressed the increased IL-6 and IL-1β and decreased IL-10 and
IL-1Ra expression levels induced by IR.
TAK1 is an emerging therapeutic target for
inflammation and fibrosis and the convergence point in cellular
responses to inflammatory stimuli, modulating the expression of
mediators and cell death (57). The
downregulation of TAK1 has been reported to ameliorate IR
injury-induced renal interstitial fibrosis in mice (58). By contrast, the pathogenesis of IR
injury is involved in tissue hypoxia, reactive oxygen species,
complement activation, and the activity of pro- and anti-apoptotic
signaling cascades, all of which are controlled at a certain level
through the activity of the NF-κB pathway (59). NF-κB promotes the release of a host
of inflammatory cytokines and cytotoxins, including TNF-α and IL-1β
(60). The mitogen-activated protein
kinase family member TAK1 is involved in the mechanism of
hypoxia-induced NF-κB (61). It has
been suggested and supported by studies that IPreC can inactivate
the NF-κB pathway in IR injury. For example, combining IPreC with
sevoflurane postconditioning protected rats against myocardial
injury induced by IR partly via inactivating the toll-like receptor
4/myeloid differentiation primary response 88/NF-κB signaling
pathway (37). IPreC markedly
alleviated lipopolysaccharide-induced liver injury via the
inhibition of NF-κB activation in mice (62). Similarly, in the present study, IPreC
significantly suppressed IR-induced increased levels of TAK1,
p-P65/P65 and TNF-α.
Taken together, the results of the present study
suggested that cerebral IR injury may be ameliorated by IPreC via
decreasing free radicals and the inflammatory response. These
results may provide novel insight into the mechanisms of IPreC for
the treatment of cerebral IR injury.
Acknowledgements
The authors would like to thank Dr Xian-Liang Meng
(Binzhou Central Hospital, Binzhou, China) for providing technical
support for the present study.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XLM and DLZ obtained the data regarding the IR
injury model establishment and IPreC. DLZ performed the ELISA assay
and statistical analysis. SHS designed the study and prepared the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental protocols in the present study were
approved by the Animal Ethics Committee of Binzhou Medical College
(Binzhou, China). All experiments were performed in compliance with
relevant laws and guidelines. All experiments were performed
according to the institutional guidelines of Binzhou Central
Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TAK1
|
transforming growth factor-β-activated
kinase 1
|
|
Bcl-2
|
B-cell lymphoma 2
|
|
MDA
|
malondialdehyde
|
|
NO
|
nitric oxide
|
|
SOD
|
superoxide dismutase
|
|
LDH
|
lactate dehydrogenase
|
References
|
1
|
Mestriner RG, Saur L, Bagatini PB,
Baptista PP, Vaz SP, Ferreira K, Machado SA, Xavier LL and Netto
CA: Astrocyte morphology after ischemic and hemorrhagic
experimental stroke has no influence on the different recovery
patterns. Behav Brain Res. 278:257–261. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gomis M and Davalos A: Recanalization and
reperfusion therapies of acute ischemic stroke: What have we
learned, what are the major research questions, and where are we
headed? Front Neurol. 5:2262014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fjetland L and Roy S: Transcarotid
endovascular thrombectomy for acute ischemic stroke. J Vasc Int
Radiol. 29:1006–1010. 2018. View Article : Google Scholar
|
|
4
|
Chouchani ET, Pell VR, Gaude E,
Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord
ENJ, Smith AC, et al: Ischaemic accumulation of succinate controls
reperfusion injury through mitochondrial ROS. Nature. 515:431–435.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hu YQ, Chen W, Yan MH, Lai JJ, Tang N and
Wu L: Ischemic preconditioning protects brain from
ischemia/reperfusion injury by attenuating endoplasmic reticulum
stress-induced apoptosis through PERK pathway. Eur Rev Med
Pharmacol Sci. 21:5736–5744. 2017.PubMed/NCBI
|
|
6
|
Murry CE, Jennings RB and Reimer KA:
Preconditioning with ischemia: A delay of lethal cell injury in
ischemic myocardium. Circulation. 74:1124–1136. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wegener S, Gottschalk B, Jovanovic V, Knab
R, Fiebach JB, Schellinger PD, Kucinski T, Jungehülsing GJ,
Brunecker P, Müller B, et al: Transient ischemic attacks before
ischemic stroke: Preconditioning the human brain? A multicenter
magnetic resonance imaging study. Stroke. 35:616–621. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lehotsky J, Burda J, Danielisova V,
Gottlieb M, Kaplan P and Saniova B: Ischemic tolerance: The
mechanisms of neuroprotective strategy. Anat Rec (Hoboken).
292:2002–2012. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hess DC, Hoda MN and Bhatia K: Remote limb
perconditioning [corrected] and postconditioning: Will it translate
into a promising treatment for acute stroke? Stroke. 44:1191–1197.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thompson JW, Dave KR, Young JI and
Perez-Pinzon MA: Ischemic preconditioning alters the epigenetic
profile of the brain from ischemic intolerance to ischemic
tolerance. Neurotherapeutics. 10:789–797. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yunoki M, Kanda T, Suzuki K, Uneda A,
Hirashita K and Yoshino K: Ischemic tolerance of the brain and
spinal cord: A review. Neurol Med Chir (Tokyo). 57:590–600. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schoen M, Rotter R, Gierer P, Gradl G,
Strauss U, Jonas L, Mittlmeier T and Vollmar B: Ischemic
preconditioning prevents skeletal muscle tissue injury, but not
nerve lesion upon tourniquet-induced ischemia. J Trauma.
63:788–797. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeffries O, Waldron M, Pattison JR and
Patterson SD: Enhanced local skeletal muscle oxidative capacity and
microvascular blood flow following 7-day ischemic preconditioning
in healthy humans. Front Physiol. 9:4632018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dong S, Cao Y, Li H, Tian J, Yi C and Sang
W: Impact of ischemic preconditioning on ischemia-reperfusion
injury of the rat sciatic nerve. Int J Clin Exp Med. 8:16245–16251.
2015.PubMed/NCBI
|
|
15
|
Granger DN and Kvietys PR: Reperfusion
injury and reactive oxygen species: The evolution of a concept.
Redox Biol. 6:524–551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wanchao S, Chen M, Zhiguo S, Futang X and
Mengmeng S: Protective effect and mechanism of Lactobacillus on
cerebral ischemia reperfusion injury in rats. Braz J Med Biol Res.
51:e71722018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Valko M, Leibfritz D, Moncol J, Cronin MT,
Mazur M and Telser J: Free radicals and antioxidants in normal
physiological functions and human disease. Int J Biochem Cell Biol.
39:44–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Matsuda S, Umeda M, Uchida H, Kato H and
Araki T: Alterations of oxidative stress markers and apoptosis
markers in the striatum after transient focal cerebral ischemia in
rats. J Neural Transm (Vienna). 116:395–404. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Angelova PR and Abramov AY: Role of
mitochondrial ROS in the brain: From physiology to
neurodegeneration. FEBS Lett. 592:692–702. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yao Y, Chen L, Xiao J, Wang C, Jiang W,
Zhang R and Hao J: Chrysin protects against focal cerebral
ischemia/reperfusion injury in mice through attenuation of
oxidative stress and inflammation. Int J Mol Sci. 15:20913–20926.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singh V, Krishan P and Shri R:
Antioxidant-mediated neuroprotection by allium schoenoprasum L.
leaf extract against ischemia reperfusion-induced cerebral injury
in mice. J Basic Clin Physiol Pharmacol. 29:403–410. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bohacek I, Cordeau P, Lalancette-Hebert M,
Gorup D, Weng YC, Gajovic S and Kriz J: Toll-like receptor 2
deficiency leads to delayed exacerbation of ischemic injury. J
Neuroinflammation. 9:1912012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chapman SN, Mehndiratta P, Johansen MC,
McMurry TL, Johnston KC and Southerland AM: Current perspectives on
the use of intravenous recombinant tissue plasminogen activator
(tPA) for treatment of acute ischemic stroke. Vasc Health Risk
Manag. 10:75–87. 2014.PubMed/NCBI
|
|
24
|
del Zoppo GJ: Acute anti-inflammatory
approaches to ischemic stroke. Ann N Y Acad Sci. 1207:143–148.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clemens JA, Stephenson DT, Dixon EP,
Smalstig EB, Mincy RE, Rash KS and Little SP: Global cerebral
ischemia activates nuclear factor-kappa B prior to evidence of DNA
fragmentation. Brain Res Mol Brain Res. 48:187–196. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Subedi L, Venkatesan R and Kim SY:
Neuroprotective and anti-inflammatory activities of allyl
isothiocyanate through attenuation of JNK/NF-κB/TNF-α signaling.
Int J Mol Sci. 18:E14232017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang L, Zhang Y, Asakawa T, Li W, Han S,
Li Q, Xiao B, Namba H, Lu C and Dong Q: Neuroprotective effect of
neuroserpin in oxygen-glucose deprivation- and
reoxygenation-treated rat astrocytes in vitro. PLoS One.
10:e01239322015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Giacoppo S, Galuppo M, Iori R, De Nicola
GR, Bramanti P and Mazzon E: (RS)-glucoraphanin purified from
tuscan black kale and bioactivated with myrosinase enzyme protects
against cerebral ischemia/reperfusion injury in rats. Fitoterapia.
99:166–177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Awooda HA, Lutfi MF, Sharara GM and Saeed
AM: Role of N-Nitro-L-Arginine-Methylester as anti-oxidant in
transient cerebral ischemia and reperfusion in rats. Exp Transl
Stroke Med. 5:12013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Altintas O, Antar V, Baran O, Karatas E,
Altintas MO, Kesgin S, Buyukpinarbasili N, Kocyigit A and Asil T:
Neuroprotective effects of hemicraniectomy in malign middle
cerebral artery infarctions: Experimental study. J Neurosurg Sci.
2015.
|
|
31
|
Qi D, Liu H, Niu J, Fan X, Wen X, Du Y,
Mou J, Pei D, Liu Z, Zong Z, et al: Heat shock protein 72 inhibits
c-Jun N-terminal kinase 3 signaling pathway via Akt1 during
cerebral ischemia. J Neurol Sci. 317:123–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Feng Z, Davis DP, Sasik R, Patel HH,
Drummond JC and Patel PM: Pathway and gene ontology based analysis
of gene expression in a rat model of cerebral ischemic tolerance.
Brain Res. 1177:103–123. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Friedlander RM: Apoptosis and caspases in
neurodegenerative diseases. N Engl J Med. 348:1365–1375. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dziodzio T, Biebl M and Pratschke J:
Impact of brain death on ischemia/reperfusion injury in liver
transplantation. Curr Opin Organ Transplant. 19:108–114. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kale J, Osterlund EJ and Andrews DW: BCL-2
family proteins: Changing partners in the dance towards death. Cell
Death Differ. 25:65–80. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang J, Yu P, Chen M, Peng Q, Wang Z and
Dong N: Remote ischaemic preconditioning and sevoflurane
postconditioning synergistically protect rats from myocardial
injury induced by ischemia and reperfusion partly via inhibition
TLR4/MyD88/NF-κB signaling pathway. Cell Physiol Biochem. 41:22–32.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jeong JS, Kim D, Kim KY, Ryu S, Han S,
Shin BS, Kim GS, Gwak MS and Ko JS: Ischemic preconditioning
produces comparable protection against hepatic ischemia/reperfusion
injury under isoflurane and sevoflurane anesthesia in rats.
Transplant Proc. 49:2188–2193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shen S, Zhou J, Meng S, Wu J, Ma J, Zhu C,
Deng G and Liu D: The protective effects of ischemic
preconditioning on rats with renal ischemia-reperfusion injury and
the effects on the expression of Bcl-2 and Bax. Exp Ther Med.
14:4077–4082. 2017.PubMed/NCBI
|
|
40
|
Ray PD, Huang BW and Tsuji Y: Reactive
oxygen species (ROS) homeostasis and redox regulation in cellular
signaling. Cell Signal. 24:981–990. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tothova L and Celec P: Oxidative stress
and antioxidants in the diagnosis and therapy of periodontitis.
Front Physiol. 8:10552017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Flatow J, Buckley P and Miller BJ:
Meta-analysis of oxidative stress in schizophrenia. Biol
Psychiatry. 74:400–409. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang B, Chen Y, Long YH, Fan X, Liu KX,
Wang XB and Zhou J: Intestinal and limb ischemic preconditioning
provides a combined protective effect in the late phase, but not in
the early phase, against intestinal injury induced by intestinal
ischemia-reperfusion in rats. Shock. 49:596–603. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tuorkey MJ: Kidney remote ischemic
preconditioning as a novel strategy to explore the accurate
protective mechanisms underlying remote ischemic preconditioning.
Interv Med Appl Sci. 9:20–26. 2017.PubMed/NCBI
|
|
45
|
Oh CS, Kim SH, Lee J and Rhee KY: Impact
of remote ischaemic preconditioning on cerebral oxygenation during
total knee arthroplasty. Int J Med Sci. 14:115–122. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Stoll G, Jander S and Schroeter M:
Inflammation and glial responses in ischemic brain lesions. Prog
Neurobiol. 56:149–171. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lan W, Harmon D, Wang JH, Ghori K, Shorten
G and Redmond P: The effect of lidocaine on in vitro neutrophil and
endothelial adhesion molecule expression induced by plasma obtained
during tourniquet-induced ischaemia and reperfusion. Eur J
Anaesthesiol. 21:892–897. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Han JY, Miura S, Akiba Y, Higuchi H, Kato
S, Suzuki H, Yokoyama H and Ishii H: Chronic ethanol consumption
exacerbates microcirculatory damage in rat mesentery after
reperfusion. Am J Physiol Gastrointest Liver Physiol.
280:G939–G948. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dehnadi A, Benedict Cosimi A, Neal Smith
R, Li X, Alonso JL, Means TK and Arnaout MA: Prophylactic
orthosteric inhibition of leukocyte integrin CD11b/CD18 prevents
long-term fibrotic kidney failure in cynomolgus monkeys. Nat
Commun. 8:138992017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Healy DG, Wood AE, O'Neill A, McCarthy JF,
Fitzpatrick JM and Watson RW: Can preoperative modelling of
individual neutrophil adhesion responses predict renal morbidity?
Eur J Cardiothorac Surg. 31:1088–1093. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chouker A, Martignoni A, Schauer R, Dugas
M, Rau HG, Jauch KW, Peter K and Thiel M: Beneficial effects of
ischemic preconditioning in patients undergoing hepatectomy: The
role of neutrophils. Arch Surg. 140:129–136. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kharbanda RK, Peters M, Walton B,
Kattenhorn M, Mullen M, Klein N, Vallance P, Deanfield J and
MacAllister R: Ischemic preconditioning prevents endothelial injury
and systemic neutrophil activation during ischemia-reperfusion in
humans in vivo. Circulation. 103:1624–1630. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Halladin NL, Ekelof S, Alamili M, Bendtzen
K, Lykkesfeldt J, Rosenberg J and Gögenur I: Lower limb ischaemia
and reperfusion injury in healthy volunteers measured by oxidative
and inflammatory biomarkers. Perfusion. 30:64–70. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Choi HS, Hwang JK, Kim JG, Hwang HS, Lee
SJ, Chang YK, Kim JI and Moon IS: The optimal duration of ischemic
preconditioning for renal ischemia-reperfusion injury in mice. Ann
Surg Treat Res. 93:209–216. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Serafin A, Rosello-Catafau J, Prats N,
Gelpi E, Rodes J and Peralta C: Ischemic preconditioning affects
interleukin release in fatty livers of rats undergoing
ischemia/reperfusion. Hepatology. 39:688–698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shin JA, Park EM, Choi JS, Seo SM, Kang
JL, Lee KE and Cho S: Ischemic preconditioning-induced
neuroprotection is associated with differential expression of
IL-1beta and IL-1 receptor antagonist in the ischemic cortex. J
Neuroimmunol. 217:14–19. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kilty I and Jones LH: TAK1 selective
inhibition: State of the art and future opportunities. Future Med
Chem. 7:23–33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wu H, Zhou J, Ou W, Li Y, Liu M and Yang
C: TAK1 as the mediator in the protective effect of propofol on
renal interstitial fibrosis induced by ischemia/reperfusion injury.
Eur J Pharmacol. 811:134–140. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Latanich CA and Toledo-Pereyra LH:
Searching for NF-kappaB-based treatments of ischemia reperfusion
injury. J Invest Surg. 22:301–315. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Morishita R, Sugimoto T, Aoki M, Kida I,
Tomita N, Moriguchi A, Maeda K, Sawa Y, Kaneda Y, Higaki J and
Ogihara T: In vivo transfection of cis element ‘decoy’ against
nuclear factor-kappaB binding site prevents myocardial infarction.
Nat Med. 3:894–899. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bandarra D, Biddlestone J, Mudie S, Muller
HA and Rocha S: Hypoxia activates IKK-NF-kappaB and the immune
response in Drosophila melanogaster. Biosci Rep. 34:e001272014.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Shin HJ, Won NH and Lee HW: Remote
ischemic preconditioning prevents lipopolysaccharide-induced liver
injury through inhibition of NF-kappaB activation in mice. J
Anesth. 28:898–905. 2014. View Article : Google Scholar : PubMed/NCBI
|