Introduction
Non-small cell lung cancer (NSCLC) accounts for ~85%
of all primary lung cancer cases worldwide in 2018 (1). Chemoresistance is responsible for the
high prevalence and mortality of NSCLC, as this phenomenon enhances
NSCLC progression and makes NSCLC difficult to treat (2). The underlying molecular mechanisms of
chemoresistance are not yet understood (3). The aim of the present study was to
elucidate these mechanisms in order to identify a method to
decrease chemotherapy resistance in NSCLC.
MicroRNAs (miRNAs or miRs) are an important
component of epigenetic mechanisms that decrease gene expression at
the post-transcriptional level by binding to 3′-untranslated
regions (UTRs) of target mRNAs (4).
miRNA is an important regulator in a number of biological
processes, such as cell proliferation, invasion, metastasis and
apoptosis (5,6). Abnormal expression of miRNAs has been
demonstrated to be associated with tumor chemotherapy resistance,
including resistance to cisplatin (7). However, how miRNAs regulate cisplatin
resistance is not yet clear. Previous studies have demonstrated the
suppressive role of miR-103a-3p in chemotherapy-resistance in
numerous different types of cancer such as bladder carcinoma,
malignant mesothelioma and glioma (8–11).
However, whether miR-103a-3p regulates cisplatin resistance in
NSCLC remains unknown.
Neurofibromin 1 (NF1) plays a role as a key negative
regulator of the Ras signaling pathway, negatively regulating
mitogen-activated protein kinase-extracellular signal-regulated
kinase (ERK) signaling (12). Recent
studies revealed that primary and acquired chemotherapy-resistance
of lung adenocarcinomas in patients was significantly correlated
with lower NF1expression (12,13).
However, the underlying molecular mechanism of how NF1
downregulates cisplatin resistance in NSCLC remains unknown.
In the present study, it was demonstrated that
expression levels of miR-103a-3p were significantly increased in
patients with cisplatin-resistant NSCLC and can induce cisplatin
resistance in NSCLC cells by directly targeting NF1 to activate ERK
signaling. Furthermore, the results revealed that overexpression of
miR-103a-3p can overcome cisplatin resistance of NSCLC cells.
Materials and methods
Human samples and cell lines
A total of 20 patients (age, 38–69 years; male 12,
female 8) with primary NSCLC who underwent surgical resections from
January 2016 to December 2017 at The Affiliated Tumor Hospital of
Xinjiang Medical University were included in the present study, and
their adjacent normal lung tissues (5 cm away from the tumor
tissue) were obtained. The aforementioned 20 patients' serum was
also included in the present study. All samples were properly
preserved for future use. The tissues were stored in liquid
nitrogen. Venous blood samples of all participants were collected
in EDTAK2 anticoagulation tubes from an antecubital vein, which
were stored in −80°C fridge. The present study was approved by the
Research Ethics Board of the Xinjiang Medical University. All
patients who provided tissues and serum provided written informed
consent and all of them agreed to the use of their samples in
scientific research.
Human lung adenocarcinoma A549 and PC-9 cells, which
were purchased from the American Type Culture Collection, were
cultured in RPMI 1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) with 10 U/ml penicillin and 100 µg/ml
streptomycin at 37°C with 5% CO2. The
cisplatin-resistant A549/PC-9 cell lines were established in the
center laboratory of The Affiliated Tumor Hospital of Xinjiang
Medical University and were preserved in 1 µmol/l cisplatin (cat.
no. BP809; Sigma-Aldrich; Merck KGaA).
Cell transfection
miR-103a-3p inhibitors (100 nM), miR-103a-3p mimics
(50 nM) and negative control (NC), ERK siRNAs, NF1 siRNAs and their
negative controls were synthesized by RiboBio. The final
concentration of siRNAs was 50 nM and the sequences are presented
in Table I. Transfection was
performed using Lipofectamine™ 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. A total
of 10 µl lentivirus (1×108 TU/ml; Shanghai Genechem Co.,
Ltd.) carries a puromycin resistance gene, which can be used to
reject untransfected cells following transfection with lentivirus.
The pre-experimental results revealed that when 2 mg/ml puromycin
was added to the culture medium then cultured at 37°C for 48-h,
A549 cell viability was 0% without any transfection. Total RNA and
protein were prepared 72-h after transfection and were used for
reverse transcription-quantitative PCR (RT-qPCR) or western blot
analysis.
 | Table I.Transfection reagent sequences. |
Table I.
Transfection reagent sequences.
| Name | Sequence |
|---|
| miR-103a-3p mimics,
F |
5′-AGCAGCAUUGUACAGGGCUAUGA-3′ |
| miR-103a-3p mimics,
R |
5′-AUAGCCCUGUACAAUGCUGCUUU-3′ |
| miR-103a
inhibitor |
5′-TCATAGCCCTGTACAATGCTGCT-3′ |
| Inhibitor
control |
5′-CAGTACTTTTGTGTAGTACAA-3′ |
| si NF1, F |
5′-AGATGAAACGATGCTGGTCAAA-3′ |
| si NF1, R |
5′-CCTGTAACCTGGTAGAAATGCGA-3′ |
| si ERK, F |
5′-GGACCAGGUCAACCACAUU-3′ |
| si ERK, R |
5′-AAUGUGGUUGAGCUGGUCC-3′ |
Cell viability assay
A549 and PC9 cells were seeded into 96-well plates
(5×103 cells/well) either directly or 24 h after
transfection and allowed to attach overnight. Freshly prepared
cisplatin was then added at 5, 10, 20, 50 and 100 µM at 37°C. After
24-h, cell viability was assessed using a Cell Counting Kit-8 (cat.
no. HY-K0301; MedChemExpress) according to the manufacturer's
protocol.
RNA preparation and RT-qPCR
Total RNA was isolated from NSCLC tissues or cell
lines using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). RNA was reverse transcribed into cDNA
(All-in-One™ miRNA First-Strand cDNA Synthesis Kit for miRNA; cat.
no. AMRT-0020 and All-in-One™ First-Strand cDNA Synthesis Kit for
mRNA; cat. no. AORT-0020; GeneCopoeia, Inc.) under the following
protocol: 37°C for 60 min then 85°C for 5 min. miRs from serum were
isolated using the miRNeasy Serum/Plasma kit (Qiagen) according to
the manufacturer's protocol.
Mature miR-103a-3p and the RNU6 (GeneCopoeia, Inc.)
endogenous control were analyzed according to the manufacturer's
protocol. The expression levels of miR-103a-3p were quantified in
relation to the expression of RNU6 using the 2−ΔΔCq
method (14). Thermocycling
condition were as follows; 95°C for 10 sec; 58°C for 20 sec; 72°C
for 10 sec and 40 cycles. For the analysis of NF1 expression, RT
and qPCR were performed using a High-Capacity cDNA Reverse
Transcription kit and QuantiTect SYBR Green PCR kit (Thermo Fisher
Scientific, Inc.), respectively. The thermocycling conditions were
as follows: 95°C for 10 sec; 60°C for 20 sec; 72°C for 10 sec and
40 cycles. The expression levels of NF1 were quantified in relation
to the expression levels of GAPDH using the 2−ΔΔCq
method (14). The primer sequences
were described in Table II. All
primers were obtained from Guangzhou RiboBio Co., Ltd.
| Table II.Primer sequences. |
Table II.
Primer sequences.
| Name | Sequence |
|---|
| U6, F |
5′-GCTTCGGCAGCACATATACTAAAAT-3′ |
| U6, R |
5′-CGCTTCACGAATTTGCGTGTCAT-3′ |
| GAPDH, F |
5′-TGAAGGTCGGAGTCAACGGATTTGGT-3′ |
| GAPDH, R |
5′-CATGTGGGCCATGAGGTCCACCAC-3′ |
| NF1, F |
5′-CGAATGGCACCGAGTCTTAC-3′ |
| NF1, R |
5′-GACCAGTTGGACGAGCCC-3′ |
Protein extraction and western
blotting
Total protein was collected using
radioimmunoprecipitation assay buffer (cat. no. R0278; Sigma
Aldrich; Merck KGaA) containing protease inhibitors (Merck KGaA).
The supernatant protein concentration was measured using a
bicinchoninic acid kit (Beijing Dingguo Changsheng Biotechnology
Co., Ltd.). A total of 30 µg of protein per lane was separated via
10% SDS-PAGE electrophoresis and transferred to PVDF membranes
(Bio-Rad Laboratories, Inc.). After the membrane was transferred,
PVDF was blocked with 5% skim milk powder at room temperature for 1
h. Membranes were then incubated with primary antibodies against
ERK (1:1,000; cat. no. 9102), p-ERK
(Thr202/Tyr204) (1:2,000; cat. no. 4370), NF1
(1:100; cat. no. 14623), (all from Cell Signaling Technology, Inc.)
and GAPDH (1:10,000; cat. no. 60004-1-Ig; ProteinTech Group, Inc.)
overnight at 4°C, followed by incubation with secondary antibodies
conjugated to horseradish peroxidase for 2 h at room temperature
(goat anti rabbit; Thermo Fisher Scientific, Inc.; 1:10,000 and
goat anti mouse; Thermo Fisher Scientific, Inc; 1:10,000). Protein
bands were developed using Enhanced chemiluminescence kit (Bio-Rad
Laboratories, Inc.), with images taken by imager (ChemiDoc™ Touch
Imaging System; Bio-Rad Laboratories, Inc.). Density analysis was
performed using Quantity One (Bio-Rad Laboratories, Inc.; Software
version 4.6.2).
Immunohistochemistry (IHC) in NSCLC
xenograft specimens
Specimens were formalin (10%) fixed for 24 h in 4°C
and paraffin-embedded tumor tissues (3 µM) were examined to ensure
a tumor content of >75% by a pathologist. IHC was performed
using Antigen Retrieval Dako Target Retrieval solution (pH 6.0) and
Histostain-Plus 3rd Gen IHC Detection kit (Invitrogen; Thermo
Fisher Scientific, Inc.) on FFPE slides according to the
manufacturer's protocols. Xylene was used for dewaxing and then
samples were blocked with 100% goat serum (cat. no. E510009-0100)
Sangon Biotech Co., Ltd.) for 30 min at 37°C. The sections were
stained with human rabbit Ki-67 antibody (1:10; cat. no. TA801577
OriGene Technologies, Inc.) overnight at 4°C. The secondary
antibody working solution kit containing DAB (Maxim Biotech, Inc.)
was added to the tissue and incubated for 30 min in 37°C and the
slides were reviewed using a light microscope (magnification,
×100).
Target gene prediction
National Center for Biotechnology Information
(https://www.ncbi.nlm.nih.gov) and
TargetScan databases (http://www.targetscan.org/vert_72; http://starbase.sysu.edu.cn/) were downloaded and
analyzed to comprehensively screen miR-103a-3p target genes.
Luciferase reporter assay
QuickMutation kit (cat. no. D0206; Beyotime
Institute of Biotechnology) was used to process the wild type
plasmid (Promega Corporatino) to obtain a mutant plasmid. The NF1
promoter region was cloned using the following primer sequence:
5′-TTAGGTTTAAAATTGGTTAAATTAATGGTG-3′ was inserted into a luciferase
reporter plasmid (pRL-TK; Promega Corporation). A wild-type 3′-UTR
and a mutant 3′-UTR of NF1 that contained the predicted miR-103a-3p
target sequence were amplified using PCR as described previously.
Either the wild-type 3′-UTR or the mutant 3′-UTR of NF1 was
incorporated into a luciferase miRNA expression reporter vector
(pMIR-REPORT) at MluI and HindIII sites. miR-103a-3p and the miRNA
expression reporter vector with wild-type or mutant NF1 3′-UTR and
the pRL-TK were transiently co-transfected into the cells using
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The luciferase activity
was measured following incubation for 24 h at 37°C with Dual
luciferase reporter gene system according to the manufacturer's
protocol (Promega Cor). The luciferase activity was normalized to
the activity of Renilla luciferase. The experiment was
performed in triplicate.
Xenografts
Animal experiments were performed on female BALB/C
nude mice, (6 weeks of age; average weight 18 g). The mice were
kept in specific pathogen-free conditions, with a 12-h light/dark
cycle and had free access to food and water, The room temperature
was 26–28°C, and the relative temperature was maintained at
40–60%.
A549/cisplatin cells were transfected with control
lentivirus or miR-103a-3p inhibitors expression lentivirus as
previously described. After drug (puromycin, 2 mg/ml) screening for
transfection, 1×107 cells in 100 µl of
phosphate-buffered saline were subcutaneously injected into left
side of each mouse. When the tumors reached ~100 mm3,
mice were treated with or without cisplatin (3 mg/kg body weight; 6
mice per group) by intraperitoneal injection every 3 days. After 4
weeks of treatment, the mice, average weight 20 g, were sacrificed
by cervical dislocation (maximum tumor volume was 1,300
mm3), and the tumor weight was measured. The methods of
the animal models used in the present study were approved by the
Research Ethics Board of The Affiliated Tumor Hospital of Xinjiang
Medical University.
Statistical analysis
All data are presented as the mean ± standard
deviation. One-way analysis of variance followed by Tukey's post
hoc test was used to evaluate the comparisons of multiple groups
the SAS statistical software package (version 6.12; SAS Institute,
Inc.). All experiments were performed in triplicate at minimum.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Cisplatin resistance is closely
associated with miR-103a-3p overexpression in NSCLC cells
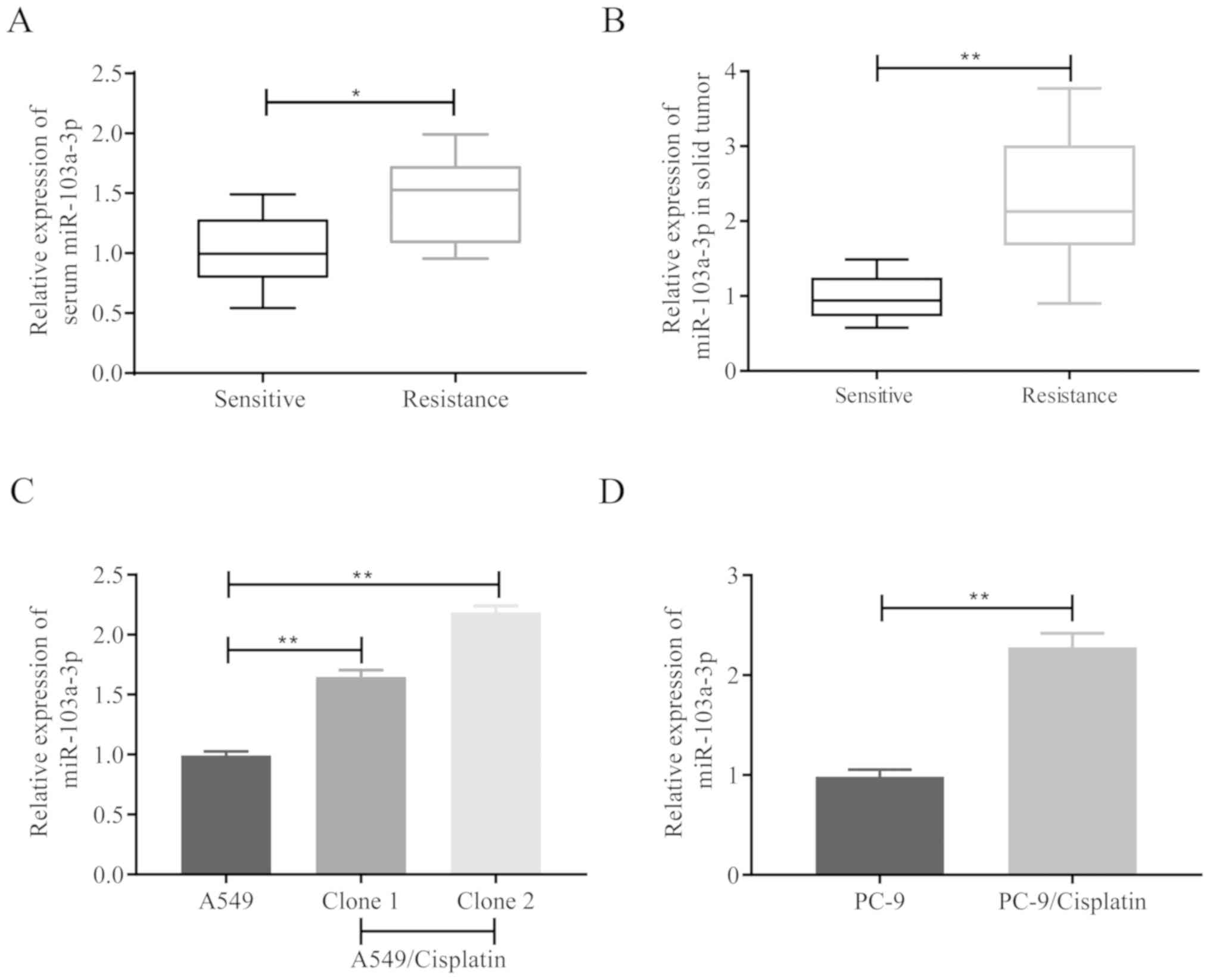
The miR-103a-3p expression levels in 20 human NSCLC
samples (10 cisplatin-resistant samples and 10 cisplatin-sensitive
samples) from different patients were analyzed in the present
study, in order to investigate the association between miR-103a-3p
levels and cisplatin resistance. It was revealed that miR-103a-3p
was significantly increased in the samples from patients with
cisplatin-resistant NSCLC in both serum (Fig. 1A) and solid tumor (Fig. 1B). A549/cisplatin had increased
remarkably compared to parental cell A549 (Fig. 1C) in vitro, and PC-9/cisplatin
demonstrated also changed (Fig. 1D).
These results demonstrated that miR-103a-3p exhibits high
expression levels in NSCLC cells and could affect the development
of cisplatin resistance.
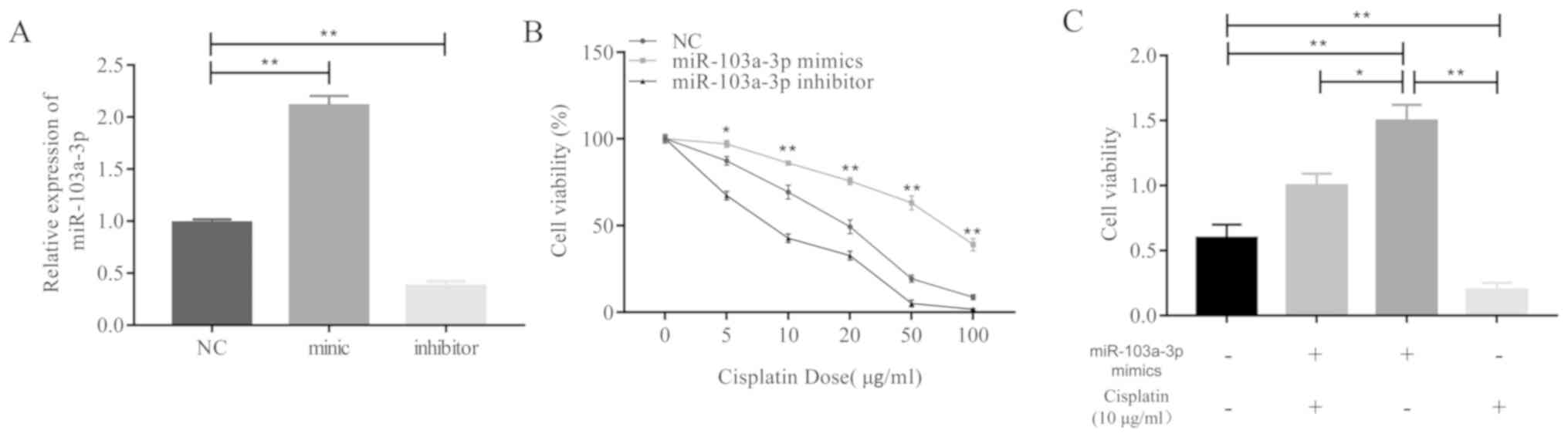
In order to investigate this hypothesis, miR-103a-3p
overexpressed A549 cells were treated with cisplatin and cell
viability assays were performed. The results revealed that the
miR-103a-3p expression levels increased following treatment with
miR-103a-3p mimics or decreased following treatment with inhibitors
(Fig. 2A). High expression levels of
miR-103a-3p caused A549 cells to exhibit significantly greater
levels of resistance to cisplatin treatment compared to the control
and miR-103a-3p inhibitors group (Fig.
2B). miR-103a-3p mimics reversed the inhibitory effect of
cisplatin on A549 cells (Fig. 2C).
Overall, these results demonstrate that high expression levels of
miR-103a-3p significantly contribute to the development of
cisplatin resistance in NSCLC cells.
miR-103a-3p activates the ERK
signaling pathway, leading to cisplatin resistance in NSCLC
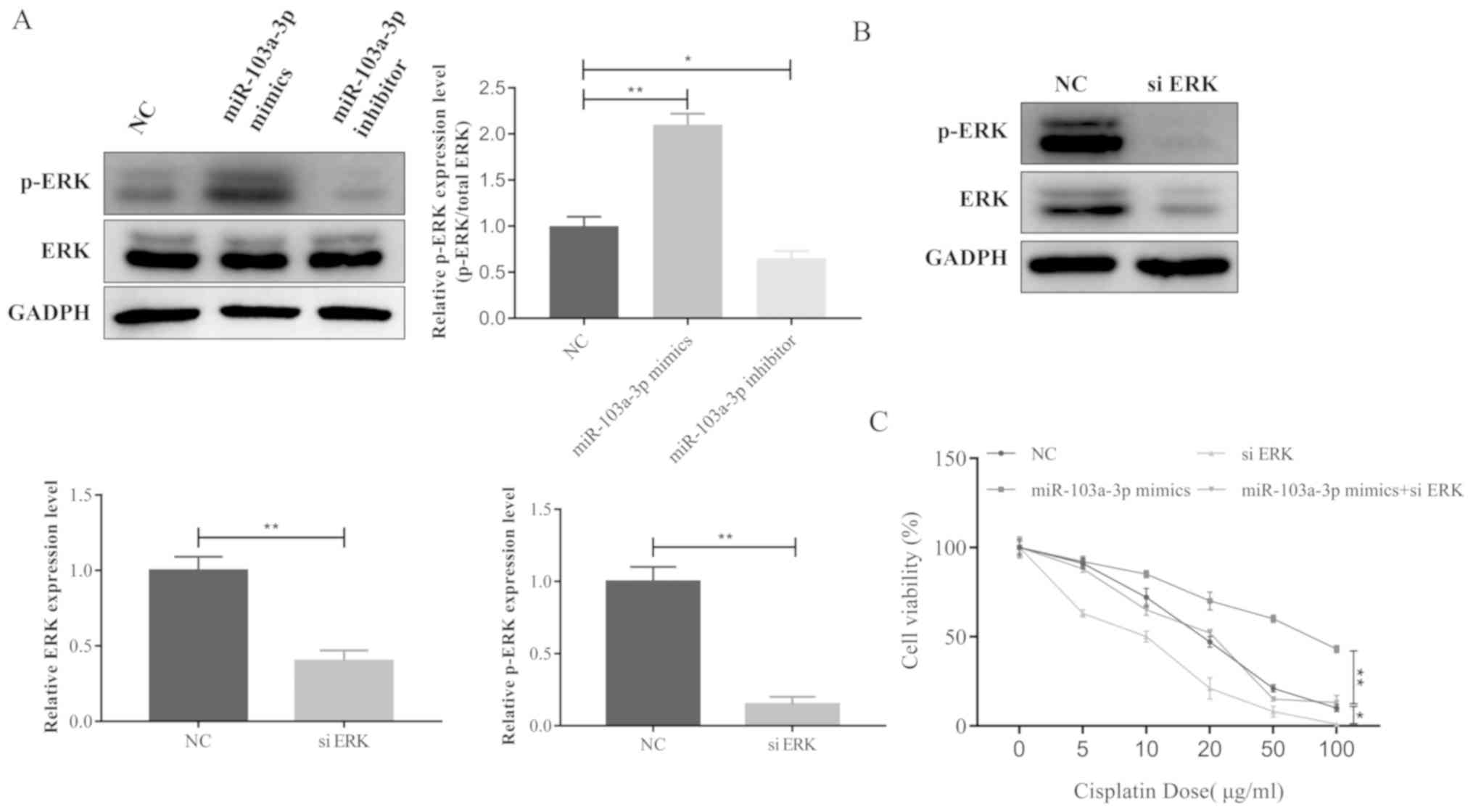
A number of studies have demonstrated that the ERK
signaling pathway contributes to the development of cisplatin
resistance in cancer (11–13). The present study therefore
investigated whether miR-103a-3p was able to affect ERK signaling
in NSCLC. p-ERK was significantly increased when miR-103a-3p was
overexpressed compare to the NC and inhibitors group, whereas
phosphorylation of ERK was decreased by the miR-103a-3p inhibitor
compared to the NC and mimics group (Fig. 3A). These results indicate that
miR-103a-3p serves an important role in the ERK signaling pathway
leading to cisplatin resistance of NSCLC cells. ERK was
knocked-down with siRNA to observe ERK expression (Fig. 3B). In addition, the cell viability
assay data revealed that miR-103A-3p did not induce cisplatin
resistance following ERK silencing in NSCLC cells (Fig. 3C). Taken together, these data
demonstrate that miR-103a-3p induces cisplatin resistance in NSCLC
cells by activating the ERK signaling pathway.
miR-103a-3p induces ERK signaling in
NSCLC cells by targeting NF1 expression
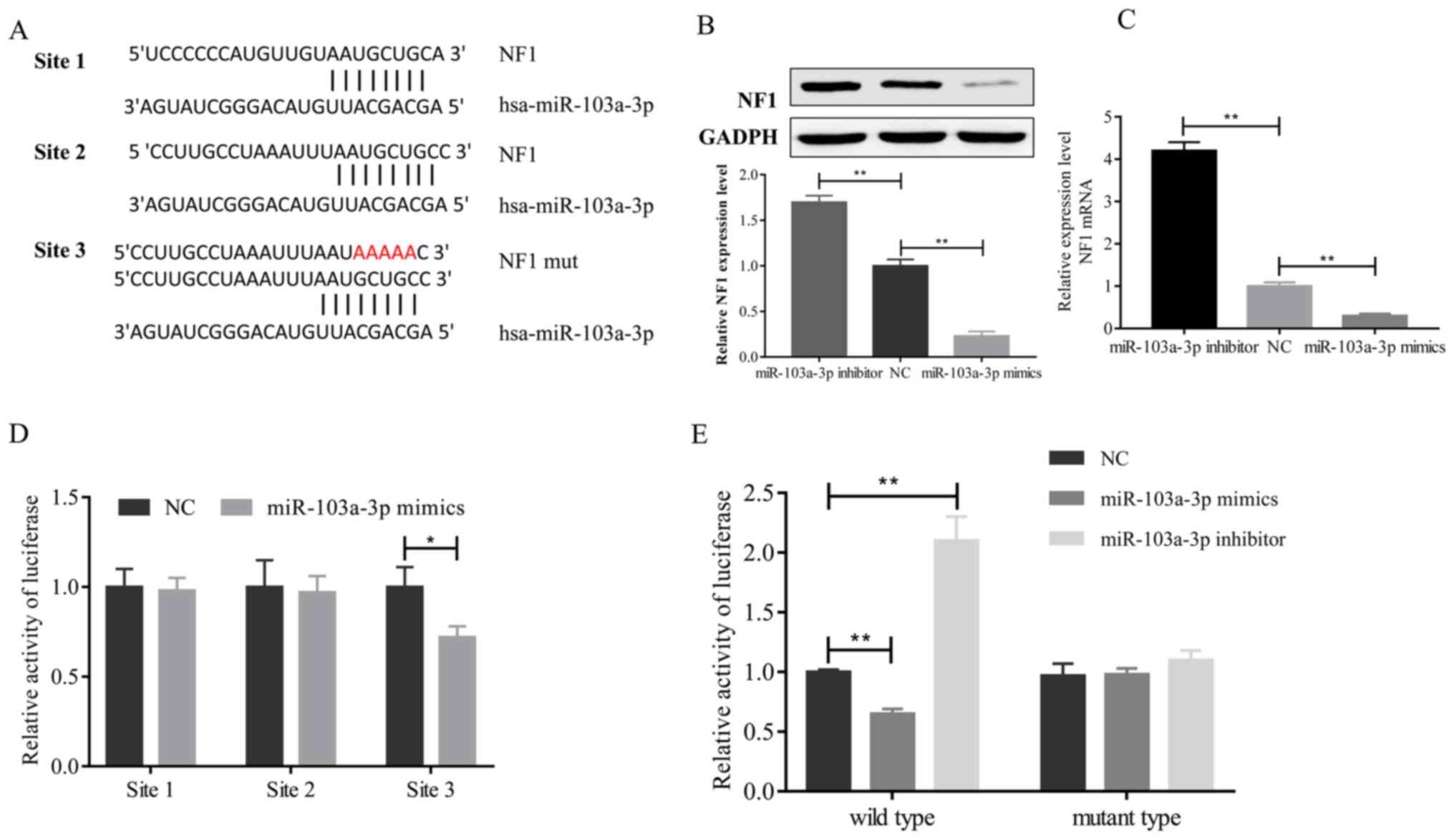
In order to investigate the role of miR-103a-3p in
the regulation of ERK signaling, the National Center for
Biotechnology Information (https://www.ncbi.nlm.nih.gov) and TargetScan databases
(http://www.targetscan.org/vert_72;
http://starbase.sysu.edu.cn/) to
comprehensively to screen miR-103a-3p target genes and identified
NF1 as a tentative target of miR-103a-3 A different 3′-UTR of the
NF1 gene was constructed in the present study, which contained
three sites that interacted with miR-103a-3p (Fig. 4A). In order to confirm whether
miR-103a-3p decreases NF1 expression in A549 cells, NF1 expression
was measured following overexpression or inhibition of miR-103a-3p
in A549 cells. At the protein (Fig.
4B) or mRNA (Fig. 4C) levels,
miR-103a-3p negatively regulated NF1 expression in NSCLC cells. In
addition, the present study assessed the binding of miR-103a-3p to
NF1 3′-UTR by synthesizing each site of the NF1 3′-UTR that could
interact with miR-103a-3p into the firefly luciferase reporter
plasmid. These sites were then transfected into A549 cells with
miR-103a-3p mimics or control oligonucleotides. The luciferase
assay results revealed that the signal was significantly decreased
following transfection with the third site of the NF1 3′-UTR
(Fig. 4D). These results indicate
that miR-103a-3p directly interacts with the third site of the NF1
3′-UTR. A three-nucleotide mutation site of miR-103a-3p was
inserted into the 3′-UTR to confirm whether the expression of
NF1-luciferase is dependent on the 3′-UTR sequence (third binding
site) complementary to the miR-103a-3p seed sequence, as presented
in Fig. 4A. The data from the
present study indicate that the 3′-UTR mutation had no effect on
miR-103a-3p overexpression or luciferase activity, but wild-type
3′-UTR significantly repressed the luciferase activity following
miR-103a-3p overexpression in A549 cells. Inhibition of miR-103a-3p
significantly enhanced the luciferase activity associated with the
wild-type 3′-UTR (Fig. 4E).
Furthermore, it was demonstrated that miR-103a-3p regulates ERK
signaling by targeting NF1.
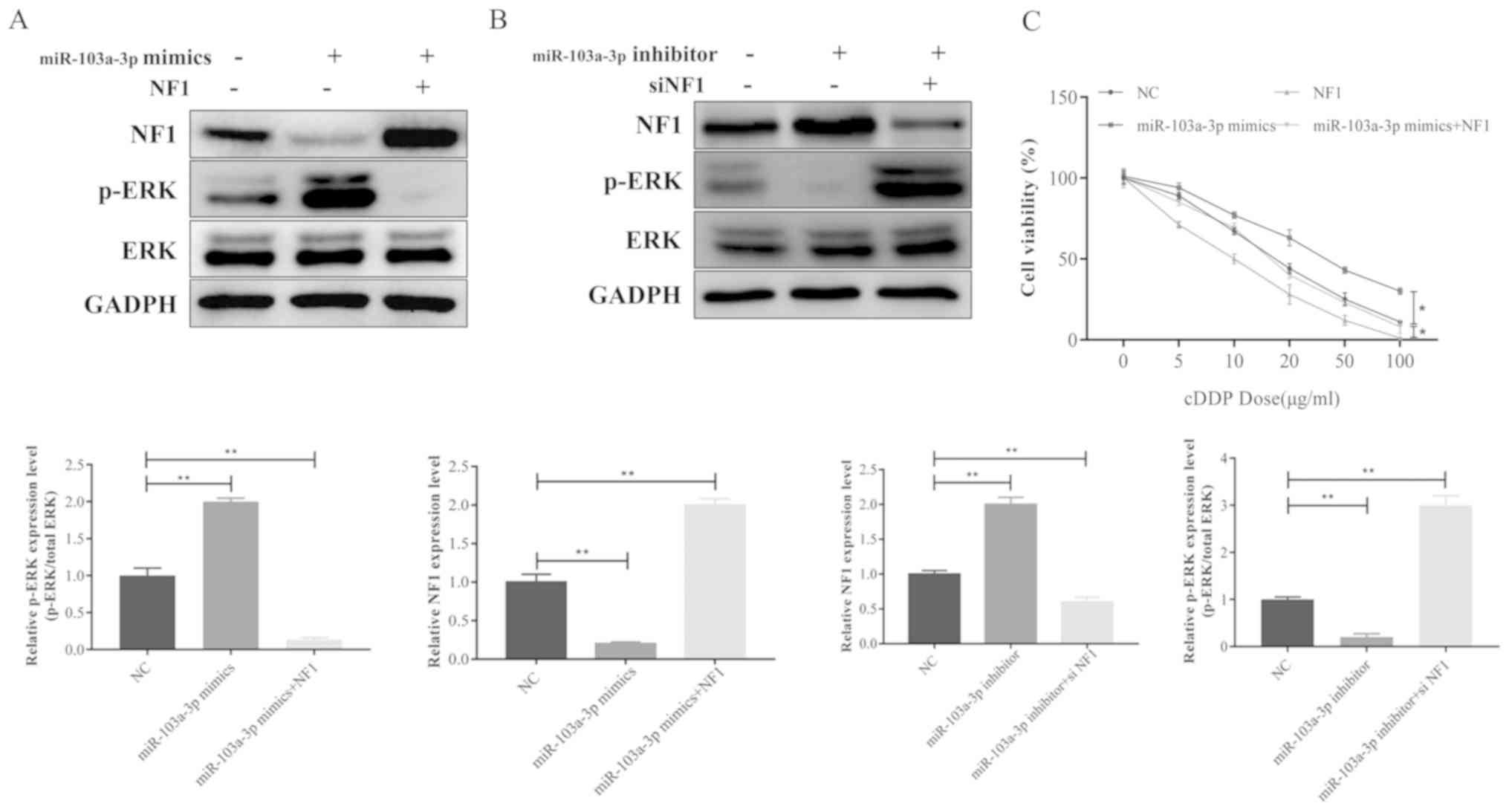
Overexpression of NF1 reversed miR-103a-3p-induced
upregulation of phosphor-ERK in A549 cells (Fig. 5A). In contrast, silencing NF1
reverses the inhibition of miR-103a-3p on ERK phosphorylation
(Fig. 5B). Consistent with these
results, the cell viability assay indicated that miR-103a-3p
overexpression and cisplatin-induced resistance were overcome by
NF1 overexpression in A549 cells (Fig.
5C). In summary, these data indicate that miR-103a-3p regulates
cisplatin resistance of NSCLC cells via NF1, which activates ERK
signaling.
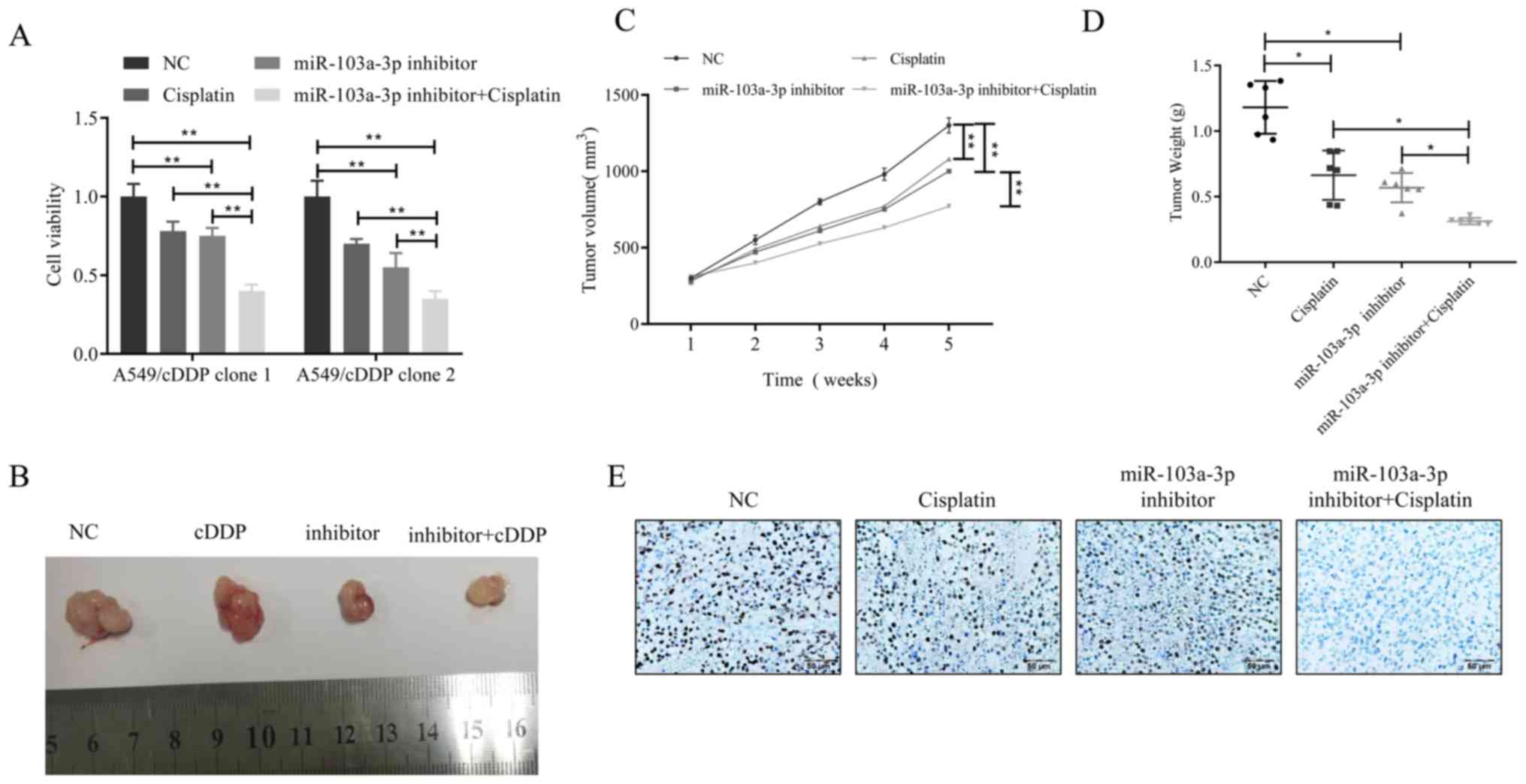
Inhibition of miR-103a-3p can reverse
cisplatin resistance of NSCLC cells in vivo
The results of the present study indicate that
overexpression of miR-103a-3p induces cisplatin resistance, but
whether miR-103a-3p interference can overcome cisplatin resistance
in NSCLC cells remains unclear. To demonstrate this hypothesis,
cell viability changes in two different cisplatin-resistant A549
cell clones treated with miR-103a-3p inhibitor and cisplatin were
then examined. The results revealed that the combination of
miR-103a-3p inhibitor and cisplatin was more effective than
cisplatin only treatment (Fig. 6A).
Furthermore, a xenograft model generated by the lentiviral
transfected cisplatin-resistant cell line A549/cisplatin was used
to observe whether this theory is correct in vivo. When the
average volume of the xenograft tumor reached 100 mm3,
the mice received 4 weeks of cisplatin treatment. The results
revealed that in the A549/cisplatin xenograft model, the
combination of miR-103a-3p inhibitor and cisplatin was superior to
cisplatin or miR-103a-3p inhibitor alone in inhibition of
A549/cisplatin xenograft growth (Fig.
6B). The combination of the two can significantly slowed tumor
growth in volume and in weight (Fig. 6C
and D). The expression levels of Ki-67 were markedly lower in
the miR-103a-3p inhibitor combined with cisplatin treatment group
compared to the control (Fig.
6E).
Discussion
Over the past 10 years, NSCLC has become a prevalent
malignancy worldwide, but cisplatin-based chemotherapy resistance
is one obstacle standing in the way of treatment (15). A number of studies have demonstrated
that dysregulation of specific miRs results in the development of
chemoresistance in a number of different types of cancer (16,17). In
the present study, NF1 was identified as a target of miR-103a-3. It
was revealed that miR-103a-3p overexpression could decrease NF1,
improve cell viability and desensitize NSCLC to cisplatin both
in vivo and in vitro. Chen et al (12) reported that miR-641 can contribute to
erlotinib resistance in NSCLC cells by targeting NF1. miR and NF1
play an important role in NSCLC treatment resistance. Furthermore,
the present study demonstrates the association between miR-103a-3p
and the development of cisplatin chemoresistance in NSCLC.
There are numerous reasons underlying drug
resistance, which include factors such as increases in drug efflux,
alterations in drug targets, DNA repair, cell cycle regulation and
evasion of apoptosis (12,18). It has previously been demonstrated
that selective regulation of miR activity can improve
responsiveness to chemotherapy (18)
miR-103a-3p expression has been demonstrated in several different
cancer cell lines such as bladder carcinoma cell and glioma cell
line (8–10), and miR-103a-3p has been indicated to
be important in proliferation and metastasis (8,10). In
the present study, it was revealed that miR-103a-3p was
significantly increased in patients with NSCLC who acquired
resistance to cisplatin treatment, as well as increased cisplatin
resistance in NSCLC cell lines. It was also demonstrated that
overexpression of miR-103a-3p can decrease NF1 levels, desensitize
A549/cisplatin cells to cisplatin, and promote tumor growth in a
nude mice model. In addition, it was revealed that miR-103a-3p is
partially complementary to the 3′-UTR of the NF1 mRNAs using
bioinformatics (TargetScan) and that miR-103a-3p can affect
luciferase activity due to canonical binding to the NF1 3′-UTR.
Thus, the present study clearly established an inverse association
between miR-103a-3p and NF1. Furthermore, overexpression of NF1 can
reverse high ERK phosphorylation levels, which had been induced by
overexpression of miR-103a-3. On the other hand, low
phosphorylation levels, which had been caused by inhibition of
miR-103a-3p, were increased via inhibition of NF1.
miR-103a-3p levels are highly expressed in breast
cancer, pancreatic cancer and ovarian cancer, and are closely
associated with tumor invasion and metastasis (19–22).
There are some inconsistent results, however; Fasihi et al
(23) assessed whether
hsa-miR-103a-3p plays an important role in colorectal carcinoma via
regulation of the Wnt signaling pathway. They hypothesized that
miR-103a-3p overexpression resulted in cell cycle progression and
decreased apoptotic rate in SW480 cells. A single miR may disrupt
multiple pathways involved in regulating cancer cell survival or
drug response. Thus, the effect of miR-103a-3p was determined by
the function of target genes in the present study. However, the
molecular mechanism underlying the differing functions of
miR-103a-3p in different cancers remains unclear; in addition,
whether miR-103a-3p affect the therapeutic effect of tyrosine
kinase inhibitors treatment in NSCLC requires further
examination.
In summary, the present study combined clinical and
experimental studies to establish a role for miR-103a-3p in
regulating cisplatin chemoresistance in NSCLC. Upregulated
expression of miR-103a-3p dramatically enhances the sensitivity of
NSCLC cells to cisplatin chemotherapy. These results are also
helpful for developing potential therapeutics for the treatment of
NSCLC chemoresistance.
Acknowledgements
We thank all patients who participated in this
study. We thank Professor Yunquan Guo (Department of Pathology,
Affiliated Tumor Hospital, Xinjiang Medical University) for his
support in pathology.
Funding
The study was supported by the foundation of
Xinjiang Uygur Autonomous Region Natural Science (grant. no.
2018D01C277).
Availability of data and materials
The datasets used and/or analyzed during the
currenty study are available from the corresponding author on
reasonable request.
Authors' contributions
SY conceived and designed this study. HZ was
responsible for doing the main experimental. HZ and JY were jointly
involving in extracting data and writing the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Research
Ethics Board of The Affiliated Tumor Hospital of Xinjiang Medical
University (Urumqi, China). All patients who provided tissues and
serum provided written informed consent and all of them agreed to
the use of their samples in scientific research. The methods of the
animal models used in the present study were approved by the
Research Ethics Board of The Xinjiang Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hildebrandt MA, Gu J and Wu X:
Pharmacogenomics of platinum-based chemotherapy in NSCLC. Expert
Opin Drug Metab Toxicol. 5:745–755. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen Z, Fillmore CM, Hammerman PS, Kim CF
and Wong KK: Non-small-cell lung cancers: A heterogeneous set of
diseases. Nat Rev Cancer. 14:535–546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel D: MicroRNAs: Genomics, biogenesis,
mechanism, and function. Cell. 116:281–297. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xie Z, Cai L, Li R, Zheng J, Wu H, Yang X,
Li H and Wang Z: Down-regulation of miR-489 contributes into NSCLC
cell invasion through targeting SUZ12. Tumour Biol. 36:6497–6505.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xia Y, Wu Y, Liu B, Wang P and Chen Y:
Downregulation of miR-638 promotes invasion and proliferation by
regulating SOX2 and induces EMT in NSCLC. FEBS Lett. 588:2238–2245.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ye Z, Yin S, Su Z, Bai M, Zhang H, Hei Z
and Cai S: Downregulation of miR-101 contributes to
epithelial-mesenchymal transition in cisplatin resistance of NSCLC
cells by targeting ROCK2. Oncotarget. 7:37524–37535. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu M, Xue Y, Zheng J, Liu X, Yu H, Liu L,
Li Z and Liu Y: Linc00152 promotes malignant progression of glioma
stem cells by regulating miR-103a-3p/FEZF1/CDC25A pathway. Mol
Cancer. 16:1102017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhong Z, Lv M and Chen J: Screening
differential circular RNA expression profiles reveals the
regulatory role of circTCF25-miR-103a-3p/miR-107-CDK6 pathway in
bladder carcinoma. Sci Rep. 6:309192016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou H and Rigoutsos I: MiR-103a-3p
targets the 5′UTR of GPRC5A in pancreatic cells. RNA. 20:1431–1439.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weber DG, Casjens S, Johnen G, Bryk O,
Raiko I, Pesch B, Kollmeier J, Bauer TT and Brüning T: Combination
of MiR-103a-3p and mesothelin improves the biomarker performance of
malignant mesothelioma diagnosis. PLoS One. 9:e1144832014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen J, Cui JD, Guo XT, Cao X and Li Q:
Increased expression of miR-641 contributes to erlotinib resistance
in non-small-cell lung cancer cells by targeting NF1. Cancer Med.
7:1394–1403. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liao J, Lin J, Lin D, Zou C, Kurata J, Lin
R, He Z and Su Y: Down-regulation of miR-214 reverses erlotinib
resistance in non-small-cell lung cancer through up-regulating LHX6
expression. Sci Rep. 7:7812017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ettinger DS, Akerley W, Borghaei H, Chang
AC, Cheney RT, Chirieac LR, D'Amico TA, Demmy TL, Govindan R,
Grannis FW Jr, et al: Non-small cell lung cancer, version 2.2013. J
Natl Compr Canc Netw. 11:645–653. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bian HB, Pan X, Yang JS, Wang ZX and De W:
Upregulation of microRNA-451 increases cisplatin sensitivity of
non-small cell lung cancer cell line (A549). J Exp Clin Cancer Res.
30:202011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liang W, Wei X, Li Q, Dai N, Li CY, Deng
Y, Jiang X, Tan XR, Dai XY, Li MX, et al: MicroRNA-765 enhances the
anti-angiogenic effect of CDDP via APE1 in osteosarcoma. J Cancer.
8:1542–1551. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Garofalo M and Croce CM: MicroRNAs as
therapeutic targets in chemoresistance. Drug Resist Updat.
16:47–59. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boren T, Xiong Y, Hakam A, Wenham R, Apte
S, Wei Z, Kamath S, Chen DT, Dressman H and Lancaster JM: MicroRNAs
and their target messenger RNAs associated with endometrial
carcinogenesis. Gynecol Oncol. 110:206–215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo Y, Chen Z, Zhang L, Zhou F, Shi S,
Feng X, Li B, Meng X, Ma X, Luo M, et al: Distinctive microRNA
profiles relating to patient survival in esophageal squamous cell
carcinoma. Cancer Res. 68:26–33. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen HY, Lin YM, Chung HC, Lang YD, Lin
CJ, Huang J, Wang WC, Lin FM, Chen Z, Huang HD, et al: miR-103/107
promote metastasis of colorectal cancer by targeting the metastasis
suppressors DAPK and KLF4. Cancer Res. 72:3631–3641. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brewster BL, Rossiello F, French JD,
Edwards SL, Wong M, Wronski A, Whiley P, Waddell N, Chen X, Bove B,
et al: Identification of fifteen novel germline variants in the
BRCA1 3′UTR reveals a variant in a breast cancer case that
introduces a functional miR-103 target site. Hum Mutat.
33:1665–1675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fasihi A, M Soltani B, Atashi A and Nasiri
S: Introduction of hsa-miR-103a and hsa-miR-1827 and hsa-miR-137 as
new regulators of Wnt signaling pathway and their relation to
colorectal carcinoma. J Cell Biochem. 119:5104–5117. 2018.
View Article : Google Scholar : PubMed/NCBI
|