Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic and
progressive fibrotic lung disease, with a median patient survival
time of 2.5–3.5 years and an increasing worldwide incidence
(1). IPF is characterized by tissue
remodeling, fibroblast proliferation and extracellular matrix
accumulation in the lung parenchyma. The fibrotic response in IPF
appears to be driven by sequential alveolar epithelium injury and
the subsequent abnormal wound-healing response, which involves
multiple cells and factors (1–3).
Chemokine expression is stimulated by infection and
inflammation, which serves an important role in the attraction,
recruitment and activation of leukocytes and immune cells (4,5). C-X-C
Motif Chemokine Ligand 12 (CXCL12) represents the natural ligand
for C-X-C motif chemokine receptor 4 (CXCR4) and has been indicated
to play a role in the metastasis of CXCR4-expressing cells
(6–8). Fibrocytes originate from the bone
marrow, and express hematopoietic (CD34+) and mesenchymal markers
[collagen I+ (ColI+) and vimentin+] (9). Increasing experimental evidence has
demonstrated that CD45+ ColI+ CXCR4+ fibrocytes can migrate to the
lungs, following bleomycin injury, in a CXCL12-dependent manner
(10–12). Furthermore, the pathophysiological
axis of CXCR4/CXCL12 is an important mediator of the activation of
fibrocytes to produce excess extracellular matrix components and to
further differentiate into contractile myofibroblasts (10–13).
Experimental evidence has revealed that the CXCR4/CXCL12 axis is an
important mechanism by which fibrocytes participate in the
pathophysiological process of lung fibrosis (10–13).
However, an additional study demonstrated that CD45+ ColI+ CXCR4+
fibrocytes make a negligible contribution to lung fibrosis
(14). Therefore the direct role of
this biological axis in mediating pulmonary fibrosis remains to be
accurately determined.
Within hypoxic areas of malignant tumors, it has
been demonstrated that CXCL12 is secreted by a variety of cells,
including tumor cells, stromal cells and tumor-associated
fibroblasts (4,5). It has been indicated that CXCL12
activates specific tumor cells via autocrine and paracrine
signaling, and enhances their proliferation by upregulating CXCR4
expression (15–17). Furthermore, CXCR4 can facilitate
tumor cell adhesion and resistance to apoptosis (17,18). In
conclusion, it can be suggested that CXCR4 is associated with the
proliferation, cell trafficking and metastasis of cancer cells.
It was hypothesized that the CXCR4/CXCL12 axis is
highly active in IPF and contributes to pathogenesis by directly
targeting human lung fibroblasts through autocrine mechanisms.
Furthermore, inhibition of CXCR4 may attenuate lung injury and
fibrosis pathologically and physiologically. The current study
revealed, through both in vitro and in vivo
experiments, that autocrine CXCR4/CXCL12 contributes to lung
fibrosis by directly modulating the activities of lung
fibroblasts.
Patients and methods
Patient samples
Tissues from 4 IPF patients were collected by
surgical lung biopsy between October 2016 and March 2018. Patients
were diagnosed through histological evidence of usual interstitial
pneumonia. IPF was diagnosed in accordance with the current
guidelines of the American Thoracic Society and the European
Respiratory Society (1). Control
samples from 4 patients who had been diagnosed with primary
spontaneous pneumothorax and received thoracoscopy for stapling air
leakage, were collected between November 2016 and May 2018. Patient
details are shown in Table I.
 | Table I.General data of included
subjects. |
Table I.
General data of included
subjects.
| Patient | Age (yr) | Sex (M/F) | Smoker (Y/N) | SCD
(year/month) | Diagnosis |
|---|
| 1 | 68 | M | N | 2016/Oct | IPF |
| 2 | 64 | M | N | 2017/Nov | IPF |
| 3 | 64 | M | N | 2017/Nov | IPF |
| 4 | 58 | M | N | 2018/Mar | IPF |
| 5 | 52 | M | N | 2016/Nov | Pneumothorax |
| 6 | 21 | F | N | 2018/Feb | Pneumothorax |
| 7 | 16 | M | N | 2017/Dec | Pneumothorax |
| 8 | 30 | M | N | 2018/May | Pneumothorax |
Primary human lung fibroblast culture
and proliferation assay
Human lung fibroblasts (HLFs) were derived from the
lung tissues of 3 patients with IPF (patient nos. 2-4, described in
Table I). Control lung fibroblasts
were derived from histologically normal lung tissue samples of 3
patients (patient nos. 6-8, described in Table I). HLFs were cultured at 37°C in a 5%
CO2 incubator using the tissue explant adherent method
and morphologically observed. Immunofluorescence staining was used
to ensure the identification and purity of the primary-cultured
HLFs. The culture procedure and cell characterization were
conducted as previously described (19).
The proliferation of normal HLFs was evaluated using
a MTT assay (Sigma-Aldrich; Merck KGaA). A total of
5×103 lung fibroblasts were seeded into each well of a
96-well plate with DMEM (HyClone; GE Healthcare Life Sciences)
containing 2% FBS (HyClone; GE Healthcare Life Sciences), and
cultured for 108 h at 37°C. The medium was then replaced with 200
µl of PBS containing 250 µg/ml MTT every 12 h. Subsequently, the
plates were cultured for an additional 4 h at 37°C. The PBS in the
plate was then carefully removed and the trapped MTT crystals were
solubilized with 200 µl of DMSO (Sigma-Aldrich; Merck KGaA) at
37°C. Absorbance was measured 10 min later using a microtiter plate
reader (model Infinite M200; Tecan Group Ltd.) at 490 nm. Each
experiment was performed in triplicate.
To investigate the effects of CXCL12 on cell
proliferation and expression of CXCR4 and colI, normal HLFs were
cultured in 96-well plates (5×103 cells/well) or
six-well plates (1×105 cells/well) in DMEM with 10% FBS
at 37°C. After 24 h of culture, the cells were starved in FBS-free
DMEM overnight and then incubated for 72 h with a variety of
concentrations of CXCL12 (0, 0.2, 1, 5, 25 and 125 ng/ml; R&D
Systems, Inc.) in DMEM with 2% FBS at 37°C.
The current study was approved by the ethics
committee of the China-Japan Friendship Hospital. All included
patients signed informed consent for participation in the present
study.
Western blot analysis
The fourth passages of fibrotic HLFs (collected from
patient nos. 2-4, described in Table
I) or normal HLFs (collected from patient nos. 6-8, described
in Table I) and the whole lung
tissues from 4 IPF patients and 4 control patients (patient nos.
1-8, described in Table I) were used
to determine the expression of CXCR4 and ColI, using a RIPA assay
buffer (150 mM NaCl; 10 mM NaF; 1.5 mM MgCl2; 10%
glycerol; 1% Triton X-100; 4 mM EDTA; 0.1% SDS; 50 mM HEPES; 1%
deoxycholate; pH 7.4) containing complete proteinase and
phosphatase inhibitor cocktails (Roche Diagnostics). The
cytoplasmic extracts were obtained from lysates after
centrifugation at 10,000 × g for 10 min at 4°C. Protein
concentrations were assessed using a BCA kit (Pierce; Thermo Fisher
Scientific, Inc.). An equal amount of protein (30 µg for HLFs and
100 µg for lung lysates) were subjected to 10% SDS-PAGE gel and
transferred onto a PVDF membrane (EMD Millipore) at 4°C, 200 mA for
2 h. Membranes were subsequently blocked with 5% non-fat dry milk
or BSA (Sigma-Aldrich; Merck KGaA) in TBS (10 mM Tris-HCl; pH 7.6;
150 mM NaCl; 0.1% Tween-20) at room temperature for 1 h and then
incubated at 4°C overnight with the indicated primary antibodies
supplied by Abcam: Rabbit anti-CXCR4 antibody (1:500; cat. no.
ab181020), rabbit anti-collagen I antibody (1:500; cat. no.
ab138492) and mouse anti-β-actin monoclonal antibody (1:1,000; cat.
no. ab8226). Immunoreactive bands were detected via incubation for
1 h with the appropriate secondary, HRP-labeled antibodies supplied
by ProteinTech Group, Inc.: HRP-conjugated Affinipure Goat
Anti-Rabbit IgG (1:2,000; cat. no. SA00001-1), HRP-conjugated
Affinipure Goat Anti-Mouse IgG (1:2,000; cat. no. SA00001-2), and
visualized using an ECL buffer (KPL; Kirkegaard & Perry
Laboratories, Inc.) with ChemiDoc XRS (Bio-Rad Laboratories, Inc.).
Relative protein levels were calculated by densitometry using
Quantity One version 4.6.6 (Bio-Rad Laboratories, Inc.) or Image J
version 1.42q (National Institutes of Health). The study of human
lung tissues was approved by the ethics committee of the
China-Japan Friendship Hospital. All investigated subjects signed
an informed consent form consenting to institutional
guidelines.
Migration assay
In the transwell migration assay, 24-well culture
inserts with porous polycarbonate membrane (8.0 µm pore size; EMD
Millipore) were used. A total of 10×103 Normal HLFs were
seeded into 60 mm dishes and grew to 80% confluence at 37°C in a 5%
CO2 incubator. Cells were then serum-starved in DMEM for
24 h and incubated with CXCL12 (final concentration, 5 ng/ml) in
the absence or presence of the CXCR4 antagonist AMD3100 (100 ug/ml;
Sigma-Aldrich; Merck KGaA) for 1 h at 37 °C prior to stimulation.
Subsequently, the cells were resuspended and loaded into the upper
compartment of the migratory well at a density of 10×103
cells in 100 µl of serum free DMEM. In the lower compartment, 500
µl of DMEM was supplemented with 10% FBS in the absence or presence
of CXCL12 (200 ng/ml). After incubation for 24 h at 37°C, the cells
on the lower side of the chambers were fixed with 4% formaldehyde
solution for 30 min, stained using crystal violet solution
(Sigma-Aldrich: Merck KGaA) for 10 min at room temperature and
counted using a light microscope in 6 randomly selected fields
(magnification, ×100).
Pulmonary fibrosis model building and
AMD3100 treatment
Female C57BL/6 mice (age, 6–8 weeks; weight, 18–22
g; n=10 per experimental group) were purchased from the Animal
Center of Peking University Health Science Center and maintained
under specific pathogen-free conditions at 25±2°C in a 12 h
night/dark cycle. The protocols were approved by the Committee on
the Ethics of Animal Experiment of Capital Medical University.
All animals were randomly divided into three groups.
A total of 5 mg/kg bleomycin (BLM; Haizheng Pharmaceutical Co.,
Ltd.) in 50 µl PBS was intratracheally injected, or the equivalent
volume of PBS was used as a control, following anesthetization with
intraperitoneal injections of pentobarbital sodium (80 mg/kg). Mice
in the AMD3100 treatment group received an intraperitoneal
injection 200 µg of AMD3100 in 250 µl sterile PBS 1 day prior to
bleomycin injection. Mice in the bleomycin group received 250 µl
PBS instead of AMD3100. Mice were euthanized using pentobarbital on
day 3, 7, 14 and 21 following exposure to bleomycin.
Spirometry
Lung function was measured using a method outlined
in a previous study (20). Each
animal in the three groups was weighed and anesthetized with an
intraperitoneal injection of pentobarbital sodium at a dose of 80
mg/kg on day 21. The tracheal intubation was connected to a
computerized small animal ventilator (SCIREQ® flexiVent;
SCIREQ Scientific Respiratory Equipment, Inc.; emka Technologies).
All mice were mechanically ventilated with 21% O2 at 150
breaths/min and a tidal volume of 10 ml/kg. After 3 min of
mechanical ventilation, rocuronium bromide (0.6 mg/kg) was injected
intraperitoneally to paralyze all animals. The volume signal
generated using a computer was applied to the airway opening. After
a deep inspiration under 30 cm H2O airway pressure, the
quasi-static pressure-volume (P-V) curve was performed in
situ, and the impedance of the respiratory system was measured
under positive end-expiratory pressures of 2 cm H2O. The
parameter K of the P-V curve and relevant data of airway resistance
(R) and compliance (C) were obtained using the flexiVent
system.
Bronchoalveolar lavage fluid
After testing the lung function, mice were
euthanized using pentobarbital overdose and the whole lung was
removed via thoracotomy. Bronchoalveolar lavage fluid (BALF) from
the whole lung was collected using an 800 µl aliquot of PBS, and
this was repeated twice. The collected fluids were centrifuged at
4°C for 5 min at 160 × g. The supernatants were collected and
stored at −80°C until cytokine analyses. According to the
manufacturers protocol, the concentrations of CXCL12 in HLFs
supernatant, and in the mouse BALF and lung homogenates, were
determined using human and mouse CXCL12 Quantikine ELISA kits (cat.
nos. DSA00 and MCX120; R&D Systems, Inc.).
Collagen assay in lung tissue
The total amount of collagen in mouse lung tissues
was determined using the SIRCOL Collagen Assay kit (Biocolor Ltd.)
according to the manufacturers protocol. Extracts derived from left
lung lobe homogenates were incubated with Sirius red dye at room
temperature for 30 min. Then absorbance at 540 nm was determined
using a spectrophotometer (Model, Infinite M200; Tecan Group Ltd.).
The amount of collagen is presented in µg per mg of wet tissue.
Histological examination
The excised lungs were perfused with saline and
inflated with 1 ml of 4% paraformaldehyde. Lungs were then ligated
at the trachea and immersed in 4% paraformaldehyde at 4°C for 24 h.
The fixed lung was embedded in paraffin, cut into 5 µm sections,
and stained with hematoxylin and eosin for 5 min or Massons
trichrome for 30 min at room temperature following a standard
protocol. For grading lung fibrosis, a numerical fibrotic scale was
used (Ashcroft score) (21). All
histological specimens were randomly numbered and examined by three
pathologists who were blinded to the treatment. The severity of
fibrosis in each lung section was assessed as a mean fibrotic score
from observation under a light microscopic in 6 randomly selected
fields (magnification, ×200).
Statistical analysis
Continuous data were presented as the mean ± SEM.
Differences between two groups were analyzed using a two-tailed
unpaired Student's t-test, while one-way ANOVA with a Bonferroni
post-hoc test was used for comparisons of multiple groups. All
analyses were performed using SPSS version 13.0 (SPSS, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Clinical samples indicate a high
expression level of chemokine receptor CXCR4 in IPF lungs
Increasing experimental evidence has indicated that
CXCR4/CXCL12 is associated with the pathogenesis of lung fibrosis
(10–13). Therefore, it was hypothesized that
the level of CXCR4 would be significantly increased in lung tissue.
To determine this, the current study aimed to examine whether CXCR4
was upregulated in IPF. Proteins extracted from lung tissues of
patients with IPF and non-IPF controls were analyzed for CXCR4
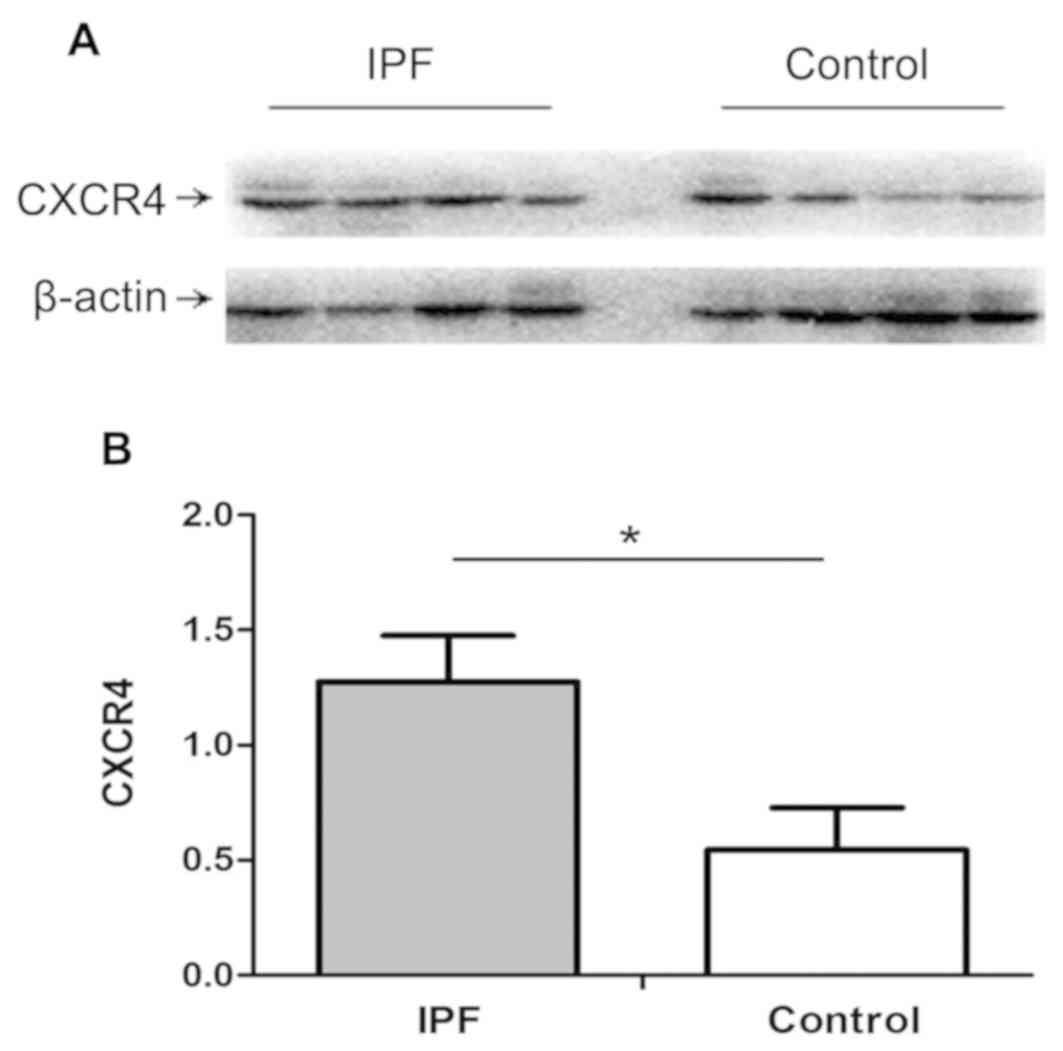
expression using western blot analysis. As presented in Fig. 1, compared with normal controls,
subjects diagnosed with IPF exhibited higher expression of CXCR4 in
lung tissues (P<0.05).
Proliferation of primary human lung
fibroblasts
To determine the role of CXCR4/CXCL12 in pulmonary
fibrosis, primary human lung fibroblasts were cultured from lung
tissues of 3 patients with IPF (fibrotic HLFs) and 3 patients with
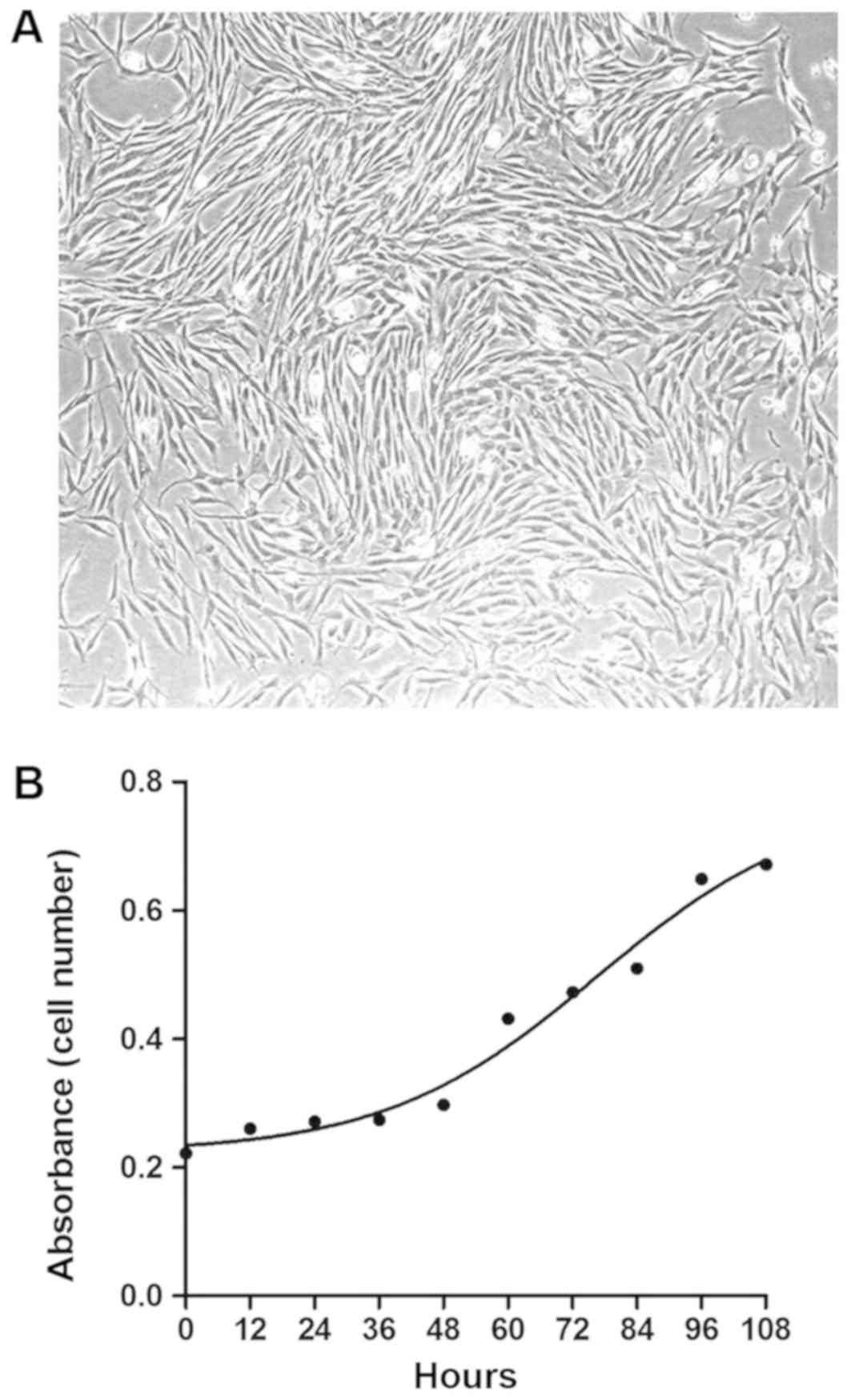
primary spontaneous pneumothorax (normal HLFs). As presented in
Fig. 2A and in previous data
(19), primary HLFs had a typical
spindle-shaped appearance under a phase-contrast light microscope
and were identified by positive staining for vimentin, fibronectin
and collagen III, and negative staining for vWF, ProSP-C and α-SMA
(19). The growth curve of the
fourth normal HLF passages was presented in Fig. 2B, indicating that HLFs grow
relatively rapidly, with an approximate doubling time of 72 h.
Therefore, this time-point was subsequently used to assess the
proliferation effect of CXCL12 on HLFs.
High expression of CXCR4 and collagen
and autocrine secretion of CXCL12 by fibrotic HLFs
The cellular sources and targets of CXCR4-CXCL12
remain to be determined. Therefore, the current study examined the
positive regulation of the CXCR4/CXCL12 axis in passage 4 HLFs.
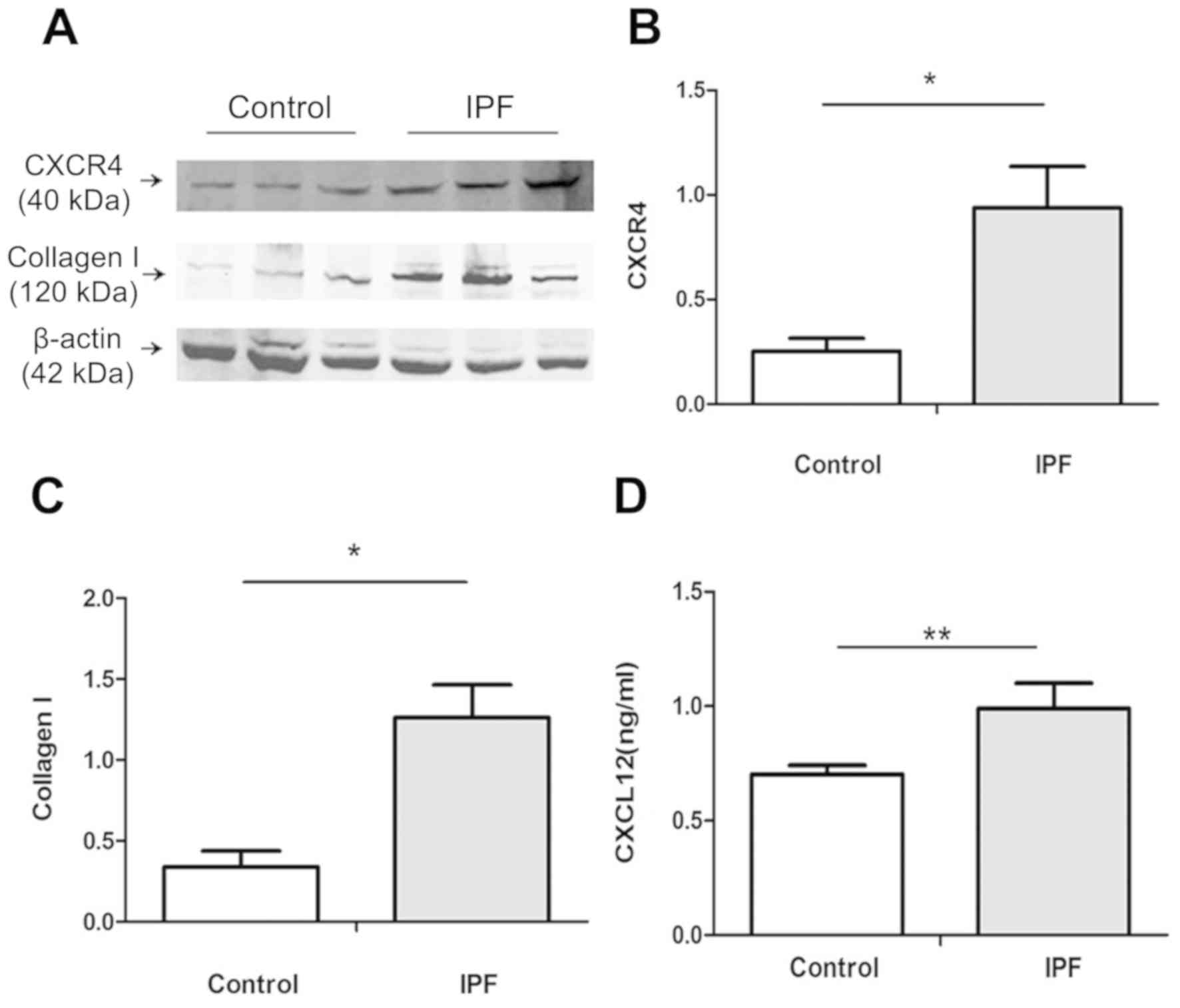
Whole cell lysates were subjected to western blot analysis. As
presented in Fig. 3A-C, CXCR4 and
collagen I expression were significantly upregulated in fibrotic
HLFs compared with normal HLFs (P<0.05). The supernatant of
fibrotic and normal HLFs was collected for ELISA to determine the
extracellular level of CXCL12. As presented in Fig. 3D, fibrotic and normal HLFs secreted
CXCL12. The secretion level of CXCL12 in fibrotic HLFs was
significantly higher compared with normal HLFs (P<0.01). These
results indicated that HLFs (especially fibrotic HLFs) are
potential cellular sources and targets of CXCL12.
Significant inhibition of HLFs
proliferation, and CXCR4 and collagen I protein production
following blocking of CXCR4
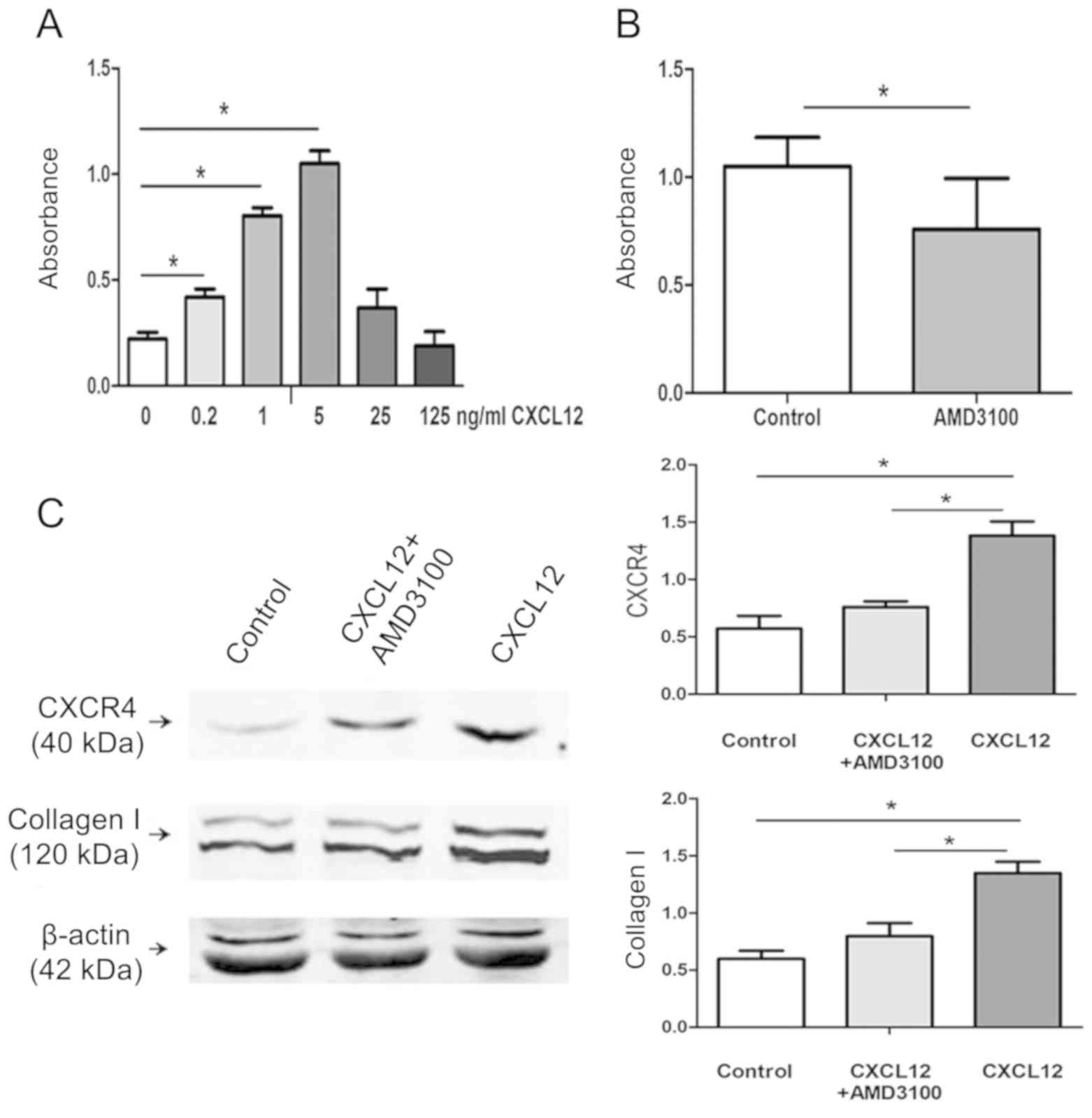
To determine whether CXCL12 directly induced HLFs
proliferation and CXCR4 and collagen I protein production, normal
HLFs were exposed to a variety of CXCL12 concentrations (0, 0.2, 1,
5, 25 and 125 ng/ml) for 72 h, and cell growth and protein
expression were assessed using an MTT assay and western blot
analysis, respectively. As presented in Fig. 4A and B, compared with the control
group (medium without CXCL12), HLFs proliferation was induced by
CXCL12 in a dose-dependent manner at levels below 5 ng/ml. The
maximum growth response was indicated at a dose of 5 ng/ml, and
this proliferation reaction was significantly inhibited following
the use of a CXCR4 receptor antagonist (P<0.05). Subsequently,
HLFs were treated with CXCL12 at a concentration of 5 ng/ml in the
presence or absence of AMD3100, an antagonist of CXCR4, and their
effects on CXCR4 and collagen I expression were determined.
Fig. 4C indicated that compared with
the control group (medium without CXCL12), CXCL12 treatment
significantly induced CXCR4 and collagen I expression, and this
effect was reduced by pre-treatment with AMD3100 (P<0.05). These
results demonstrated that inhibition of CXCR4 signaling can inhibit
HLFs proliferation and CXCR4 and collagen I protein production.
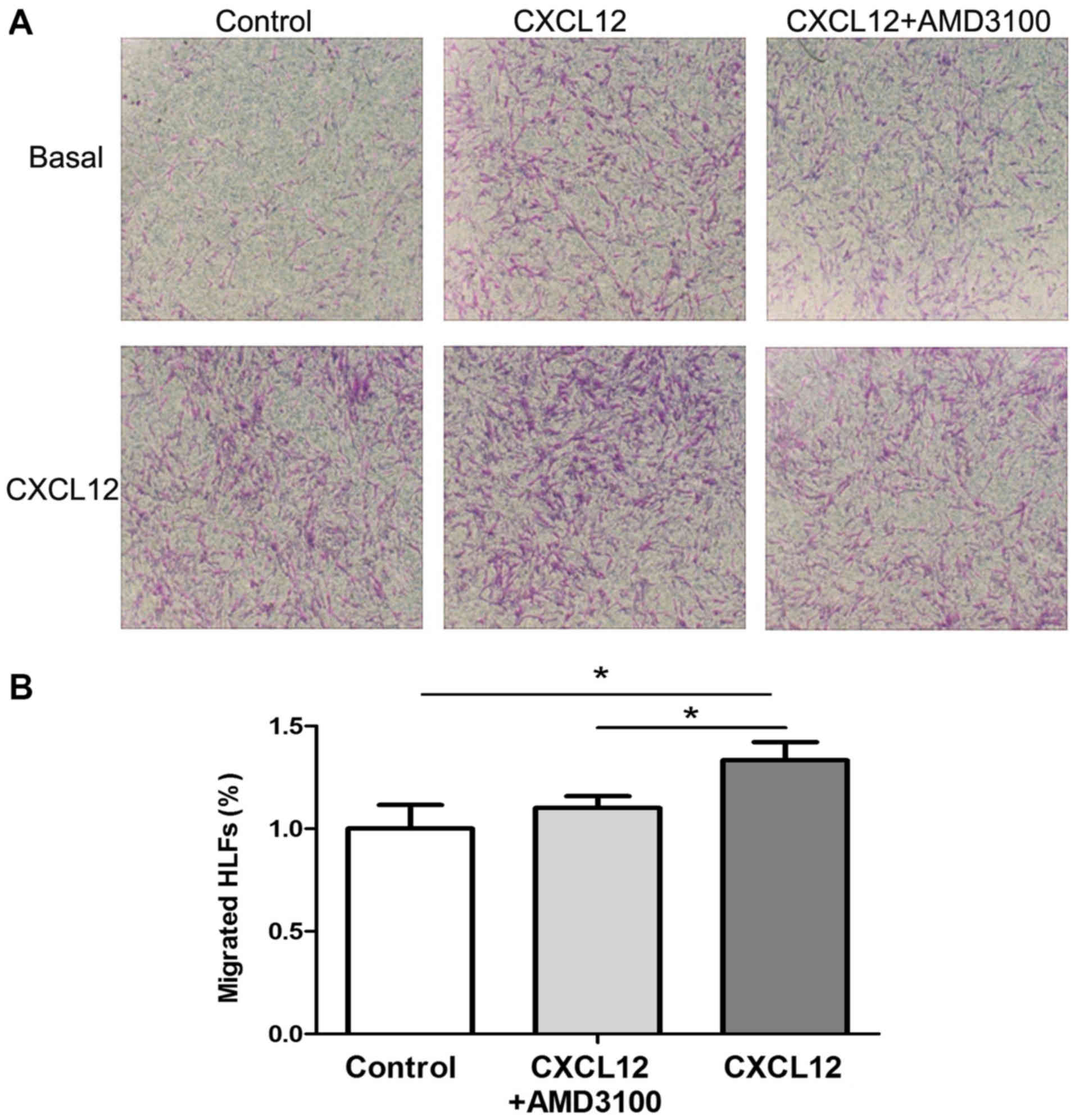
CXCL12 is required for HLFs
migration
The development of lung fibrosis has been suggested
to be associated with fibroblast recruitment into the sites of lung
injury (1–3,9). A
previous study revealed that CXCL12 can be secreted by HLFs
(22). To determine the effect of
CXCL12 on HLFs migration, an in vitro chemotaxis assay was
performed. As indicated in Fig. 5,
CXCL12 incubation significantly increased HLF migration
(P<0.05), which was significantly inhibited by AMD3100
pre-stimulation (P<0.05). The results demonstrated that the
CXCR4/CXCL12 chemokine axis promoted the migration of HLFs.
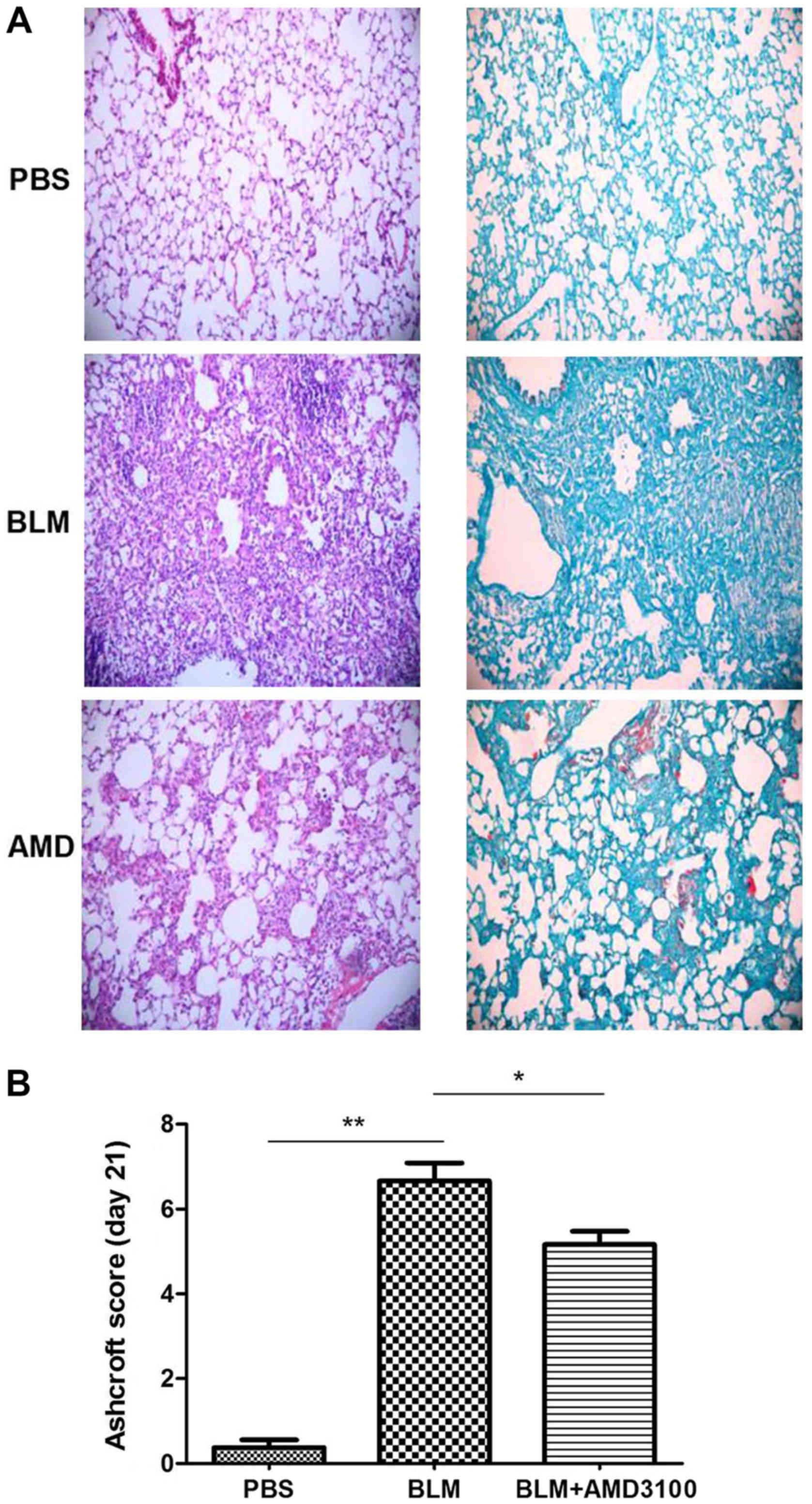
A CXCR4 antagonist attenuates
pulmonary fibrosis and decreases the protein expression of CXCL12
and CXCR4
In accordance with previous studies (10–13), the
current study indicated that AMD3100 treatment significantly
attenuated the BLM-induced pulmonary inflammation and fibrosis, as
determined by histological examination and fibrosis score on day 21
compared with the bleomycin group (Fig.
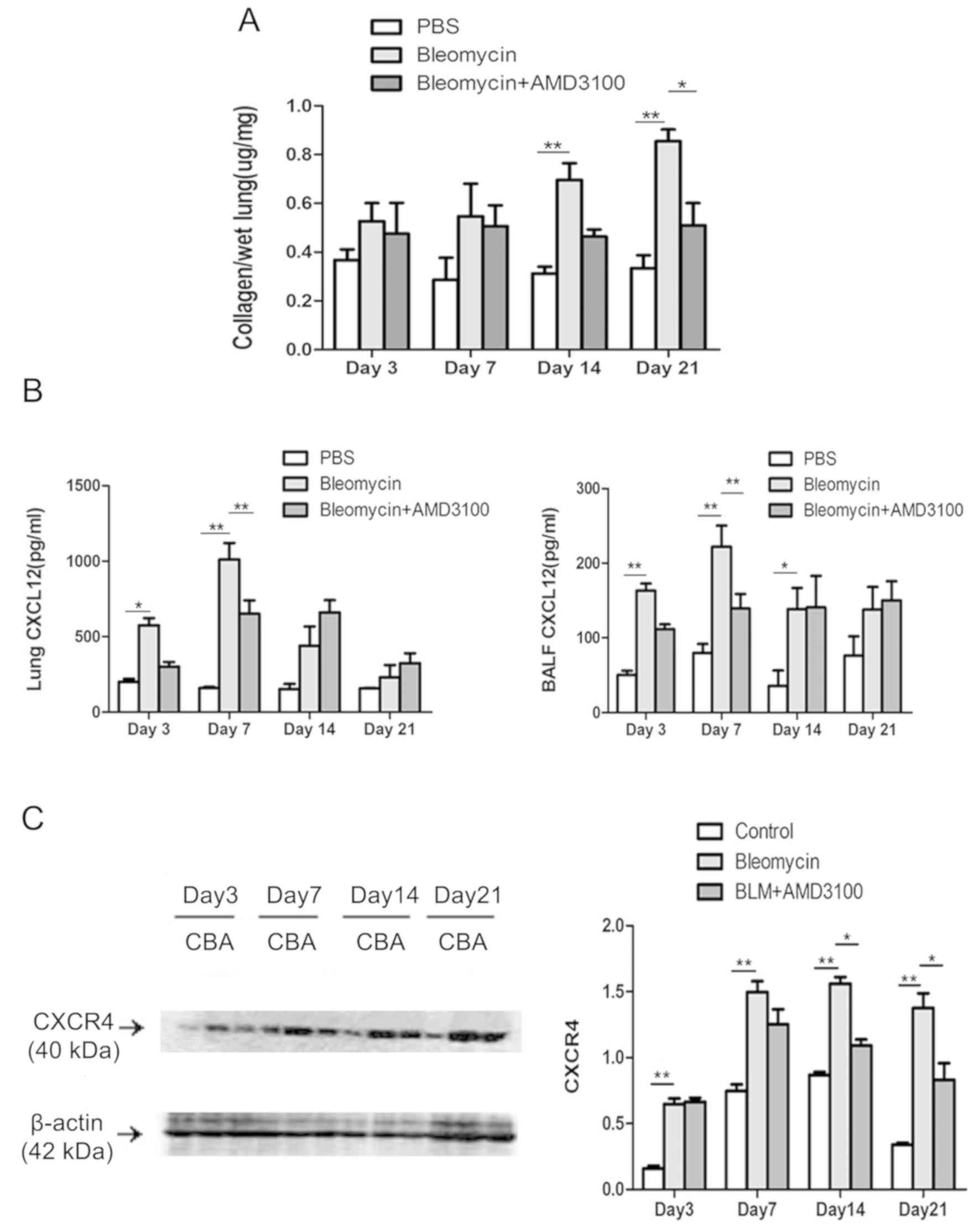
6). To evaluate the therapeutic value of AMD3100 in
vivo, the effect of AMD3100 treatment on lung collagen content
following BLM challenge was also assessed (Fig. 7A). The collagen deposition in
response to BLM was decreased in the BLM + AMD3100 group, which was
more obvious on day 21, compared with BLM group (P<0.05). The
protein levels of CXCL12 and CXCR4 were also determined. As
presented in Fig. 7B, the
concentrations of CXCL12 protein in BALF and lung homogenates from
BLM treated mice increased from day 3, peaked on day 7, and then
decreased gradually to the levels of control group on day 21.
AMD3100 pre-treatment decreased the levels of CXCL12 on day 3 and
day 7 (P<0.01). BLM upregulated the expression of CXCR4 in lung
tissue, which reached a plateau on day 7. AMD3100 treatment
significantly reduced the levels of CXCR4 after day 14 (P<0.01
vs. bleomycin group; Fig. 7C).
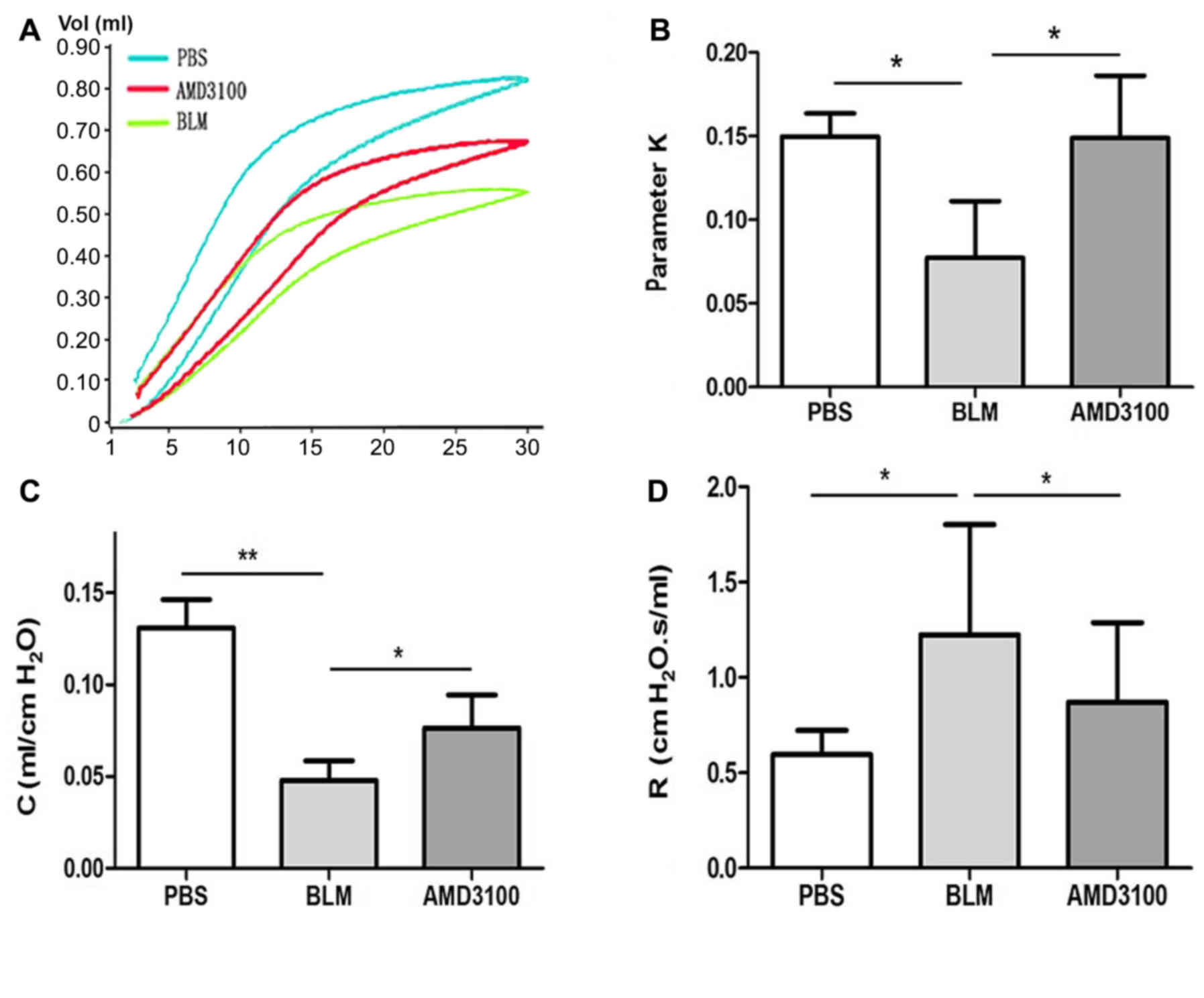
Inhibition of CXCR4 ameliorates lung
function in pulmonary fibrosis
Lung function tests were performed in three groups
of mice on day 21 following exposure to PBS, BLM and AMD3100. The
P-V curve obtained is a typical curve of the first inflation and
deflation under degassing conditions and the second complete curve
to 30 cm H2O. As presented by the P-V curve (Fig. 8A), the maximal lung volume was
decreased after BLM challenge, compared with the control group.
However, AMD3100 treatment appeared to reduce this disparity. The
parameter K, which is an indirect representation of lung
elasticity, indicated the curvature of the upper portion of the
deflation P-V curve. This parameter of BLM group was significantly
lower than that of the PBS group, and treatment with AMD3100
increased the parameter K (Fig. 8B;
P<0.05). A similar variation of parameter K patterns were
observed in terms of compliance. The Quasi-static compliance
indicated the static elastic recoil pressure of the lungs at a
given lung volume. The decrease in compliance was significantly
greater in BLM group compared with the AMD3100 group (Fig. 8C; P<0.05). The resistance of BLM
group was increased, which was significantly inhibited by AMD3100
pretreatment (Fig. 8D; P<0.05).
The aforementioned findings implied that blockage of CXCR4
ameliorated the lung compliance and resistance in pulmonary
fibrosis.
Discussion
IPF is a progressive fibrotic lung disease with a
poor prognosis following the initial diagnosis (1). However, the pathologic profiles for
pulmonary fibrosis are poorly understood (1). Fibroblasts are considered to be the
master switch of IPF by synthesizing ECM in fibroblastic foci (the
histopathological hallmarks of IPF) (1–3). In the
current study, the effect of the CXCR4/CXCL12 axis on the
proliferation and ECM metabolism of primary HLFs was assessed. It
was indicated that CXCL12 aggravated proliferation and migration of
HLFs, and increased collagen release by direct autocrine
stimulation via CXCR4, which could be attenuated by AMD3100
pretreatment.
Chemokines and receptors are recognized universally
to be crucial in the process of leukocyte recruitment, especially
at sites of tissue injury, cell damage and infection (23–25).
CXCR4 is a chemokine receptor for the ligand of CXCL12.
CXCR4/CXCL12 axis is functional in a number of organs including the
lung, heart, kidney and liver. The present study identified high
expression of CXCR4 in lung tissues with pulmonary fibrosis and
indicated that high expression may contribute to lung injury and
fibrosis, and subsequent research confirms this point.
Multiple cell types can express CXCR4/CXCL12 in lung
tissues (especially in fibrotic lungs), including fibroblasts,
epithelial cells, vascular endothelial cells and some inflammatory
cells (26–29). As previously reported, a major
population of CXCR4+ cells was localized close to the fibroblastic
foci in lung tissue sections from patients with IPF (28,29).
CXCL12 was indicated to be upregulated in reactive hyperplastic
alveolar epithelial cells often overlying fibroblastic foci, and
staining for this was also observed in endothelial cells and some
alveolar macrophages, except for the normal areas of the IPF lungs
(28), supporting the notion that
these cells serve a role in the pathogenesis of IPF. The results of
the current study demonstrated that primary human lung fibroblasts
in adult pulmonary fibrosis can express and secrete CXCL12, and can
respond to the extraneous CXCL12 by binding to their own receptor,
CXCR4. This indicated that the autocrine CXCR4/CXCL12 axis resides
in lung fibroblasts and contributed to lung fibrosis by directly
activating lung fibroblasts.
A number of studies have demonstrated that
CXCR4/CXCL12 participates in cancer development (6,8,15–18).
CXCR4 is the only chemokine receptor expressed by the majority of
cancer cells, and its ligand CXCL12 can be secreted by tumor cells
and stromal cells, including tumor-associated fibroblasts (24,25). The
underlying mechanism of this is yet to be determined. Increasing
evidence has demonstrated that binding CXCL12 to CXCR4 stimulates
the proliferation of a variety of tumor cell lines and their
migration and adhesion to ECM components by the activation of
downstream signal transduction pathways. For example, Lin CH et
al (30) demonstrated that
CXCL12, acting through CXCR4 and activating the Rac/ERK and JNK
signaling pathways, could induce the expression of connective
tissue growth factor, which is a profibrotic protein, in human lung
fibroblasts, and potentiate their transdifferentiation into
myofibroblasts. Wang X et al (16) demonstrated that the autocrine
CXCL12/CXCR4 axis can mediate the metastatic property of esophageal
cancer stem cells depending on ERK1/2 signaling pathway. Tian Y
et al (31) indicated that
CXCL12 induced the migration of oligodendrocyte precursor cells via
the CXCR4 dependent MEK/ERK and PI3K/AKT pathways. The increased
expression of FOXM1 has been demonstrated to induce apoptosis
resistance in fibroblasts and contribute to lung fibrosis (32). A previous study has also revealed
that PI3Kα signaling via PDK1/AKT could mediate FGF2-induced FOXM1
upregulation in lung fibroblasts (32). Furthermore, FOXM1 signaling mediated
vascular remodeling and pulmonary hypertension, as previously
reported (27). Collectively, these
studies indicated that PI3K/AKT and MEK/ERK pathways and FOXM1 may
serve as potential post-receptor signaling pathways that mediate
the profibrotic effect of CXCL12 in lung fibroblasts. However, this
still remains to determined in the future.
Previous studies on CXCR4/CXCL12 have focused on
CD45 + Col I + CXCR4 + fibrocytes, which are one of the origins of
the fibroblasts/myofibroblasts (10–13,28,29). A
previous study suggested that fibrocytes are not a necessary source
of collagen during pulmonary fibrosis, and indicated that fibrocyte
may make other contributions to collagen accumulation, including
activating fibroblasts to secrete CXCL12 and aggregating other
fibrotic effector cells and factors to release CXCL12 to act on
fibroblasts (14). Recent studies
using lineage tracing to explore the origins of myofibroblasts in
models of lung fibrosis have concluded that resident lung
fibroblasts are their major source (33,34).
Lung biopsies from patients with IPF indicated an increase in the
number of fibroblasts, which is likely due to increased
proliferation (3). The current study
demonstrated that CXCL12 potentiated proliferation in primary HLFs.
The strategical blocking of the CXCR4/CXCL12 axis may be an
effective approach to inhibit the cellular activities of HLFs.
In line with prior trails (12), the results of the present study
demonstrated that the concentration of CXCL12 was increased late in
BALF and lung homogenates after bleomycin instillation. This
increase in CXCL12 was accompanied by an increase in CXCR4
expression in the lungs with a peak at the third week following
injury. Currently, it is believed that inflammation is associated
with the formation of pulmonary fibrosis, and a variety of
inflammatory cells, including neutrophils and lymphocytes, release
a variety of inflammatory factors, including CXCL12. In this
cascade reaction, the increased concentration of CXCL12 leads to
the expression of its receptor CXCR4, which further promotes the
proliferation of fibroblasts and further aggravates pulmonary
fibrosis (9). Treatment of mice with
AMD3100, the CXCR4 antagonist, decreased the production of
CXCR4/CXCL12 and attenuated bleomycin induced lung fibrosis,
despite incomplete inhibition.
It has been well established that pulmonary fibrosis
is a restrictive ventilatory dysfunction due to reduced lung volume
and decreased compliance (2). With
the alleviation of pulmonary fibrosis, pulmonary function will be
improved. Furthermore, the current study measured the lung function
of fibrotic mice. It was demonstrated that blocking CXCR4 could not
only alleviate pulmonary fibrosis pathologically, but also
physiologically. The whole lung resistance of mice decreased
significantly, and the pulmonary elasticity also improved
significantly. This is the first study exploring the autocrine
mechanism of CXCR4/CXCL12 axis in the pathogenesis of pulmonary
fibrosis; however, the current study has limitations due to the
small number of studied subjects. Future research using a large
cohort and long-term follow-up is required to prove the autocrine
mechanism of CXCR4/CXCL12 axis in the pathogenesis of pulmonary
fibrosis.
In conclusion, the current study demonstrated that
fibroblasts are the cellular sources and targets of CXCL12, due to
the autocrine secretion of excessive amount of CXCL12 and
over-expression of the corresponding receptor CXCR4. The current
study indicated the important role of the CXCR4/CXCL12 chemokine
axis in the proliferation, migration and collagen production of
HLFs in vivo and in vitro. Blocking this axis could
partially attenuate pulmonary fibrosis pathologically and
physiologically. These findings demonstrated that the CXCR4/CXCL12
axis could be a potential therapeutic target that may be used in
the treatment of pulmonary disease.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81430001
and 81470258).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FL designed and performed the experiments, analyzed
the data, drafted and revised the manuscript. XX, JG and XW
participated in the experiments and analysis of the data. HD
contributed to the conception and design of the present study, the
analysis and interpretation of the data, revision of the article
and final approval of the version to be published. All authors have
read and approved this final manuscript.
Ethics approval and consent to
participate
Studies involving human tissues were approved by the
Ethics Committee of The China-Japan Friendship Hospital (approval
no. 2014-KE-71) and written informed consent was obtained from all
investigated subjects. Studies involving animals were approved by
the Committee on the Ethics of Animal Experiment of Capital Medical
University (approval no. 2014-KE-38).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Raghu G, Remy-Jardin M, Myers JL, Richeldi
L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F,
et al: Diagnosis of idiopathic pulmonary fibrosis. An official
ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care
Med. 198:e44–e68. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Richeldi L, Collard HR and Jones MG:
Idiopathic pulmonary fibrosis. Lancet. 389:1941–1952. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wolters PJ, Collard HR and Jones KD:
Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol.
9:157–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Legler DF and Thelen M: Chemokines:
Chemistry, biochemistry and biological function. Chimia (Aarau).
70:856–859. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liekens S, Schols D and Hatse S:
CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell
mobilization. Curr Pharm Des. 16:3903–3920. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li J, Li T, Li S, Xie L, Yang YL, Lin Q,
Kadoch O, Li H, Hou S and Xu Z: Experimental study of the
inhibition effect of CXCL12/CXCR4 in malignant pleural
mesothelioma. J Investig Med. 67:338–345. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miller RJ, Banisadr G and Bhattacharyya
BJ: CXCR4 signaling in the regulation of stem cell migration and
development. J Neuroimmunol. 198:31–38. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He W, Yang T, Gong XH, Qin RZ, Zhang XD
and Liu WD: Targeting CXC motif chemokine receptor 4 inhibits the
proliferation, migration and angiogenesis of lung cancer cells.
Oncol Lett. 16:3976–3982. 2018.PubMed/NCBI
|
|
9
|
Bagnato G and Harari S: Cellular
interactions in the pathogenesis of interstitial lung diseases. Eur
Respir Rev. 24:102–114. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Phillips RJ, Burdick MD, Hong K, Lutz MA,
Murray LA, Xue YY, Belperio JA, Keane MP and Strieter RM:
Circulating fibrocytes traffic to the lungs in response to CXCL12
and mediate fibrosis. J Clin Invest. 114:438–446. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mehrad B, Burdick MD and Strieter RM:
Fibrocyte CXCR4 regulation as a therapeutic target in pulmonary
fibrosis. Int J Biochem Cell Biol. 41:1708–1718. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu J, Mora A, Shim H, Stecenko A, Brigham
KL and Rojas M: Role of the SDF-1/CXCR4 axis in the pathogenesis of
lung injury and fibrosis. Am J Respir Cell Mol Biol. 37:291–299.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Makino H, Aono Y, Azuma M, Kishi M, Yokota
Y, Kinoshita K, Takezaki A, Kishi J, Kawano H, Ogawa H, et al:
Antifibrotic effects of CXCR4 antagonist in bleomycin-induced
pulmonary fibrosis in mice. J Med Invest. 60:127–137. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kleaveland KR, Velikoff M, Yang J, Agarwal
M, Rippe RA, Moore BB and Kim KK: Fibrocytes are not an essential
source of type I collagen during lung fibrosis. J Immunol.
193:5229–5239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barbero S, Bonavia R, Bajetto A, Porcile
C, Pirani P, Ravetti JL, Zona GL, Spaziante R, Florio T and
Schettini G: Stromal cell-derived factor 1alpha stimulates human
glioblastoma cell growth through the activation of both
extracellular signal-regulated kinases 1/2 and Akt. Cancer Res.
63:1969–1974. 2003.PubMed/NCBI
|
|
16
|
Wang X, Cao Y, Zhang S, Chen Z, Fan L,
Shen X, Zhou S and Chen D: Stem cell autocrine CXCL12/CXCR4
stimulates invasion and metastasis of esophageal cancer.
Oncotarget. 8:36149–36160. 2017.PubMed/NCBI
|
|
17
|
Guo S, Xiao D, Liu H, Zheng X, Liu L and
Liu S: Interfering with CXCR4 expression inhibits proliferation,
adhesion and migration of breast cancer MDA-MB-231 cells. Oncol
Lett. 8:1557–1562. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Abraham M, Klein S, Bulvik B, Wald H,
Weiss ID, Olam D, Weiss L, Beider K, Eizenberg O, Wald O, et al:
The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by
downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered
miR-15a/16-1 expression. Leukemia. 31:2336–2346. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu X, Wan X, Geng J, Li F, Wang C and Dai
H: Kinase inhibitors fail to induce mesenchymal-epithelial
transition in fibroblasts from fibrotic lung tissue. Int J Mol Med.
32:430–438. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang J, Li Z, Yao X, Li Y, Reng X, Li J,
Wang W, Gao J, Wang C, Tankersley CG and Huang K: Altered Th1/Th2
commitment contributes to lung senescence in CXCR3-deficient mice.
Exp Gerontol. 48:717–726. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ashcroft T, Simpson JM and Timbrell V:
Simple method of estimating severity of pulmonary fibrosis on a
numerical scale. J Clin Pathol. 41:467–470. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shimizu Y, Dobashi K, Endou K, Ono A,
Yanagitani N, Utsugi M, Sano T, Ishizuka T, Shimizu K, Tanaka S and
Mori M: Decreased interstitial FOXP3(+) lymphocytes in usual
interstitial pneumonia with discrepancy of CXCL12/CXCR4 axis. Int J
Immunopathol Pharmacol. 23:449–461. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Buck AK, Stolzenburg A, Hänscheid H,
Schirbel A, Lückerath K, Schottelius M, Wester HJ and Lapa C:
Chemokine receptor-directed imaging and therapy. Methods.
130:63–71. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Balkwill F: Chemokine biology in cancer.
Semin Immunol. 15:49–55. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Balkwill F: The significance of cancer
cell expression of the chemokine receptor CXCR4. Semin Cancer Biol.
14:171–179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dai Z, Li M, Wharton J, Zhu MM and Zhao
YY: Prolyl-4 Hydroxylase 2 (PHD2) deficiency in endothelial cells
and hematopoietic cells induces obliterative vascular remodeling
and severe pulmonary arterial hypertension in mice and humans
through hypoxia-inducible factor-2α. Circulation. 133:2447–2458.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dai Z, Zhu MM, Peng Y, Jin H, Machireddy
N, Qian Z, Zhang X and Zhao YY: Endothelial and smooth muscle cell
interaction via FoxM1 signaling mediates vascular remodeling and
pulmonary hypertension. Am J Respir Crit Care Med. 198:788–802.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Andersson-Sjöland A, de Alba CG, Nihlberg
K, Becerril C, Ramírez R, Pardo A, Westergren-Thorsson G and Selman
M: Fibrocytes are a potential source of lung fibroblasts in
idiopathic pulmonary fibrosis. Int J Biochem Cell Biol.
40:2129–2140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mehrad B, Burdick MD, Zisman DA, Keane MP,
Belperio JA and Strieter RM: Circulating peripheral blood
fibrocytes in human fibrotic interstitial lung disease. Biochem
Biophys Res Commun. 353:104–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin CH, Shih CH, Tseng CC, Yu CC, Tsai YJ,
Bien MY and Chen BC: CXCL12 induces connective tissue growth factor
expression in human lung fibroblasts through the Rac1/ERK, JNK, and
AP-1 pathways. PLoS One. 9:e1047462014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tian Y, Yin H, Deng X, Tang B, Ren X and
Jiang T: CXCL12 induces migration of oligodendrocyte precursor
cells through the CXCR4-activated MEK/ERK and PI3K/AKT pathways.
Mol Med Rep. 18:4374–4380. 2018.PubMed/NCBI
|
|
32
|
Penke LR, Speth JM, Dommeti VL, White ES,
Bergin IL and Peters-Golden M: FOXM1 is a critical driver of lung
fibroblast activation and fibrogenesis. J Clin Invest.
128:2389–2405. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rock JR, Barkauskas CE, Cronce MJ, Xue Y,
Harris JR, Liang J, Noble PW and Hogan BL: Multiple stromal
populations contribute to pulmonary fibrosiswithout evidence for
epithelial to mesenchymal transition. Proc Natl Acad Sci USA.
108:E1475–E1483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xie T, Liang J, Liu N, Huan C, Zhang Y,
Liu W, Kumar M, Xiao R, D'Armiento J, Metzger D, et al:
Transcription factor TBX4 regulates myofibroblast accumulation and
lung fibrosis. J Clin Invest. 126:3063–3079. 2016. View Article : Google Scholar : PubMed/NCBI
|