Introduction
Shigella is a common cause of bacterial
dysentery, and can be classified into four serogroups: S.
dysenteriae, S. flexneri, S. boydii and S. sonnei
(1). Among these groups, S.
flexneri is the most dominant pathogen found in developing
countries (2,3). The low-infectivity inoculum facilitates
person-to-person spread by fecal-oral contact, which is the
predominant mode of transmission (1). The clinical symptoms of shigellosis
mainly include fever, diarrhea, abdomen convulsions and purulent
bloody stools (4). The lesions are
mainly confined to the colon, and result in mucosal congestion,
edema and inflammatory cell infiltration between the ulcers
(5) Bacteremia with Shigella
is rare, especially in adults (6).
In addition to the common route of oral infection, Shigella
can also cause damage to multiple organs via the route of
extra-intestinal infection, such as kidney and spleen abscesses
induced by bacteremia (7,8), perihepatitis-salpingitis caused by
unhygienic sexual practices, and vulvovaginitis induced by fecal
contamination (9,10). In particular, when Shigella
infection is combined with intestinal perforation, appendicitis and
peritonitis, clinical manifestations of abdominal infection, such
as high fever and septicemia, may occur with a high mortality rate
(11). It was reported that 13 out
of 57 children with surgical complications of Shigellosis died,
despite having received antimicrobial therapy (12). In addition to the clinical reports,
animal experiments have shown that diarrhea symptoms disappear
within 24 h after oral infection with Shigella, while
persistent diarrhea symptoms and mortality occur after
intraperitoneal injection of Shigella (13). Therefore, the clinical manifestations
of parenteral Shigella infection are more serious compared
with those of intestinal infection, and the pathogenic mechanisms
underlying these differences require further investigation.
Due to its long co-evolution with the host species,
the gut microbiota influences numerous aspects of host physiology,
including digestion, metabolism, immunity and behavior (14). In addition, the gut microbiota plays
crucial roles in resisting the invasion and clearance of pathogenic
microorganisms (15). Previous
studies have shown that infectious diarrhea, such as that caused by
Salmonella, Escherichia coli, parasites and viruses, is
usually accompanied by changes in the intestinal flora (16–19).
However, it remains unclear whether the gut microbiota in mice can
be influenced by S. flexneri infection. In order to
investigate the changes in intestinal flora after Shigella
infection, and to identify the different clinical manifestations
after oral or peritoneal infection based on gut microbiota changes,
mice were challenged through two infective routes in the present
study. The diversity and abundance of intestinal flora between
these two groups were compared at different infection stages using
16S ribosomal RNA (rRNA) high-throughput sequencing.
The results from the present study provided an
insight into Shigella infection-induced gut microbiome
changes, and identified potential biomarkers for Shigella
infection, thereby facilitating the development of therapeutics for
the prevention and treatment of shigellosis.
Materials and methods
Animals
Specific pathogen-free BALB/c mice (female; n=30;
weight, 18–22 g; age 6–8 weeks) were purchased from the Shanghai
Laboratory Animal Center CAS, and housed at 22±2°C with 12 h
light/dark cycle under specific pathogen-free conditions in the
experimental facility for ≥1 week prior to use in the experiments.
The mice received a standard batch of food and water ad
libitum. The present study was approved by The Institutional
Animal Care and The Ethic Committee of Fujian Medical
University.
S. flexneri treatment
The standard strain of S. flexneri (cat. no.
12022; American Type Culture Collection) was inoculated in Gram
Negative Enrichment Broth (HuanKai Microbial Sci. and Tech. Co.,
Ltd.) overnight at 37°C with shaking at 180 rpm on an air table.
After the culture reached the logarithmic growth stage (0.4–0.5
optical density at 600 nm), the suspension was centrifuged at 825 ×
g for 1 min at 4°C to collect the bacterial precipitate. Then, this
was suspended in normal saline buffer. The mice were randomly
divided into three groups (n=10 in each group): i) Control group
with normal saline gavage; ii) intraperitoneal (IP) injection
group, 5×107 colony-forming units (CFU) of
Shigella given by IP injection; and iii) oral gavage group,
5×108 CFU of Shigella given orally. Each mouse
was administered the treatment at a volume of 0.1 ml. The
administered bacterial doses were confirmed by plating serial
dilutions onto Salmonella-Shigella agar plates (HuanKai
Microbial Sci. and Tech. Co., Ltd.). After treatment with S.
flexneri, the mice received sterilized food and water ad
libitum for 7 days, and were then sacrificed. During this
period, body weight, general physical activity, fur ruffling,
abdominal swelling and body temperature of the mice in each group
were recorded every day. To calculate the survival rates of mice
infected with Shigella in the groups, Olfert's guidelines on
humane endpoints (20) were adopted
as follows: When a mouse lost >20% of its body weight, showed
signs of coma and lethargy, and had a body temperature <34°C,
the mouse was considered to have reached a humane endpoint and was
euthanized. The remaining mice were euthanized 7 days after
Shigella infection.
Fecal sample collection
To evaluate S. flexneri shedding in the
feces, one piece of fecal pellet was collected and weighed (0.1–0.3
g) from each mouse at the time points of 0, 6, 12, 24 and 48 h
after infection. Then, these were suspended in sterile PBS by
extensive vortexing to prepare a uniform fecal suspension, which
was cultivated on Salmonella-Shigella agar (HuanKai
Microbial Sci. and Tech. Co., Ltd.) by incubation overnight at 37°C
and the number of colonies was counted. For 16S high-throughput
sequencing, five fresh feces samples per day were randomly
collected from five mice in each group using aseptic technique on
days 2, 3 and 4 after the administration of Shigella. Three
pieces of feces from each mouse were combined to be a single sample
to meet the requirement of sample size. In total, 45 samples of
feces were collected which were immediately frozen in liquid
nitrogen and stored at −80°C for further analysis.
DNA extraction, PCR amplification and
16S rRNA sequencing
Bacterial genomic DNA was extracted from the stool
samples using a QIAamp Fast DNA Stool Mini kit (cat. no. 51604;
Qiagen GmbH), according to manufacturer's protocol. The
hypervariable (V)3-V4 region of the bacterial 16S rRNA was
amplified by PCR (ABI GeneAmp® 9700, USA) with
Phusion® High-Fidelity DNA Polymerase (New England
BioLabs, Inc.) under the following conditions: Initial denaturation
at 95°C for 5 min; followed by 30 cycles of 95°C for 30 sec, 55°C
for 30 sec and 72°C for 30 sec; and a final extension at 72°C for 4
min. The resulting PCR product was stored at 4°C. The universal
primers used in the PCR reaction were 341F
(5′-CCTAYGGGRBGCASCAG-3′) and 806R (5′-GGACTACNNGGGTATCTAAT-3′).
The 5′-end of 314F carried a special sequence tag to encode each
sample. The obtained PCR products were separated by electrophoresis
using 2% agarose gel, and purified using the GeneJET Gel Extraction
kit (Thermo Fisher Scientific, Inc.). Then, DNA libraries were
constructed using an Ion Plus Fragment Library kit (Thermo Fisher
Scientific, Inc.) and sequenced using an Ion S5 XL system (Thermo
Fisher Scientific, Inc.), according to manufacturer's protocol. Raw
FASTQ files were pretreated and quality-filtered using the
Quantitative Insights Into Microbial Ecology (QIIME) bioinformatics
pipeline (version 1.17; http://qiime.org/). Next, the sample data were
separated from the reads obtained according to the barcodes, and
the barcodes and primer sequences were removed to obtain raw reads.
After data filtering, the remaining high-quality clean data were
assembled based on tags and sequence overlaps.
Data analysis
For the analysis of microbial diversity, operational
taxonomic units (OTUs) were generated for all sequences based on
different similarity levels (21).
In the present study, the OTUs were clustered according to a
similarity cutoff value of 97% using UPARSE software (version 7.1;
http://drive5.com/uparse/), while USEARCH
(version 10.0.240, http://www.drive5.com/usearch/) was used to identify
and remove chimeric sequences. SSUrRNA database of SILVA132
(http://www.arb-silva.de) was selected for species
annotation analysis, where the threshold was set as 0.8–1). The
taxonomy of 16S rRNA gene sequences was analyzed using the
Ribosomal Database Project (RDP) classifier (version 2.11;
http://sourceforge.net/projects/rdp-classifier/), and
the sequences were classified to different levels, including
phylum, class, order, family, genus and species. Subsequently, the
composition and relative abundance of each sample at each
classification level were calculated. The OTU profiling table and
α-diversity indices, including Chao1, Shannon and Simpson (22), were calculated using QIIME. The
β-diversity among different groups was analyzed by principal
coordinate analysis (PCoA) based on weighted UniFrac distances
(23). Bray-Curtis dissimilarities
were used to construct a hierarchical cluster, while non-metric
multidimensional scaling (NMDS) was plotted based on the weighted
UniFrac dissimilarity (24). Linear
discriminant analysis Effect Size (LEfSe) was used to find
the biomarkers with statistical differences between groups, which
was presented by Histogram of Linear Discriminant Analysis
distribution and cladogram (25).
Data are presented as the mean ± SEM. The statistical analyses were
performed with a one-way ANOVA followed by Tukey's post-hoc test,
while Kruskal-Wallis analysis followed by Dunn's multiple
comparisons test was used for data with variance inhomogeneity
using SPSS 19.0 (IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference. GraphPad Prism (version 7.0;
GraphPad Software, Inc.) was used to compare mouse survival rates
between the groups using a Mantel-Cox test, and to generate a
graphical representation of the results.
Results
General condition of mice after
infection
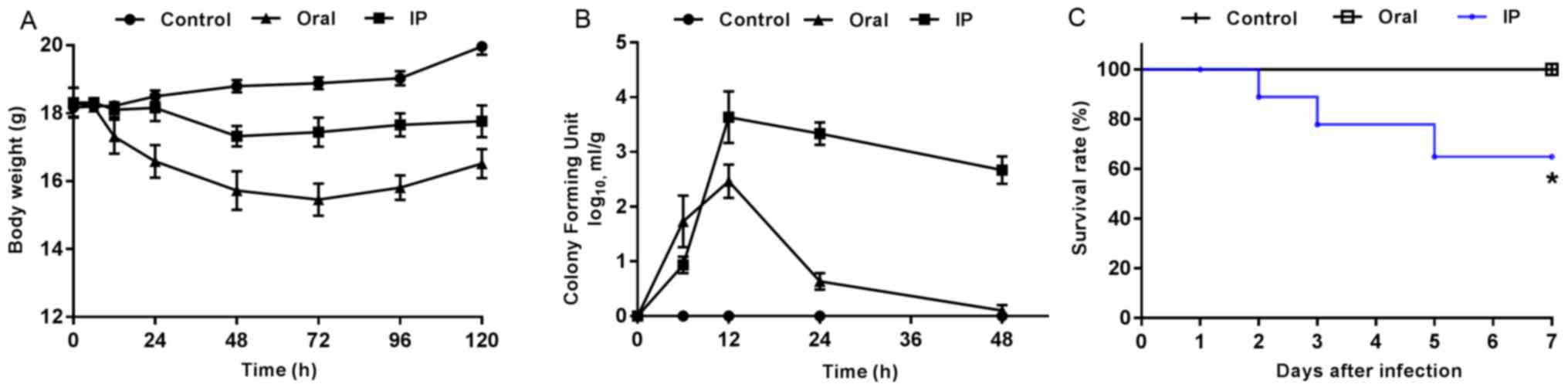
The present results suggested that the animals
challenged with S. flexneri via IP and oral routes exhibited
marked changes in physical activity, food intake, fur ruffling and
abdominal swelling. At 2 h after the oral administration of S.
flexneri, mice began to show diarrhea symptoms, which were
evident at 6 h and gradually disappeared after 24 h. At 12 h after
IP injection, mice began to present with diarrhea, some of which
lasted until day 7. Furthermore, pronounced abdominal edema was
observed, and mice presented with high irritability upon touching
of the abdomen. Compared with the control group, all mice in the
infection groups presented with varying degrees of weight loss
within 3 days after S. flexneri infection (Fig. 1A). The mice infected orally with
S. flexneri had greater weight loss compared with both the
controls and the IP group. In addition, the CFU value of S.
flexneri detected in the feces of the oral group peaked at 12
h, and then exhibited a rapid decline (Fig. 1B). In the IP group, the maximum CFU
value also appeared at 12 h, but was followed by a slower rate of
decline compared with that in the oral group. During the
experiment, three mice in the IP group reached the humane endpoint
which were euthanized and excluded from the further experiments,
while none of the mice in the control or oral groups died. The
present results suggested that the differences between the survival
rates in the three groups was statistically significant (Fig. 1C; P=0.019).
α-diversity
In the present study, 3,517,768 high-quality reads
were generated from the 45 samples by filtering the chimera. In
total, ~78,172 reads were obtained from each sample for further
analysis, and the average read length was 415 bp. Then, OTU
clustering was performed with 97% identity and 1,468 OTUs were
obtained by removing those with low abundance (threshold, 0.005%).
These were then annotated with the species from the Silva132
database. The present results suggested that a total of 738
(50.27%) OTU annotations were made at the genus level. Furthermore,
there were significant differences in α-diversity between the IP
and control groups on day 4 (P<0.05 and P<0.01; Table I). The Shannon index and Chao1 were
lower in the IP group on day 4 compared with the control group,
while there were no significant differences between the oral group
and control group.
 | Table I.α-diversity indices of the gut
microbiota from the three groups at different time points. |
Table I.
α-diversity indices of the gut
microbiota from the three groups at different time points.
| Time | Group | Average length of
sequence, bp | Effective
reads | Shannon index | Simpson index | Chao1 |
|---|
| Day 2 | C | 415 | 80140 | 6.74±0.58 | 0.97±0.02 | 575.21±36.30 |
|
| IP | 413 | 80133 | 6.51±0.35 | 0.98±0.01 | 552.17±20.87 |
|
| Oral | 415 | 80146 | 6.71±0.20 | 0.98±0.01 | 545.29±35.02 |
| Day 3 | C | 417 | 82278 | 6.64±0.21 | 0.97±0.01 | 574.90±116.48 |
|
| IP | 418 | 80872 | 5.96±0.41 | 0.98±0.01 | 517.84±127.05 |
|
| Oral | 418 | 72138 | 6.65±0.51 | 0.97±0.01 | 532.22±155.22 |
| Day 4 | C | 415 | 80225 | 6.73±0.31 | 0.97±0.01 | 600.88±96.70 |
|
| IP | 415 | 80170 |
6.02±0.21a | 0.97±0.01 |
424.79±29.74b |
|
| Oral | 413 | 80151 | 6.49±0.33 | 0.97±0.01 | 634.16±82.14 |
Taxonomic differences between groups
at different time points
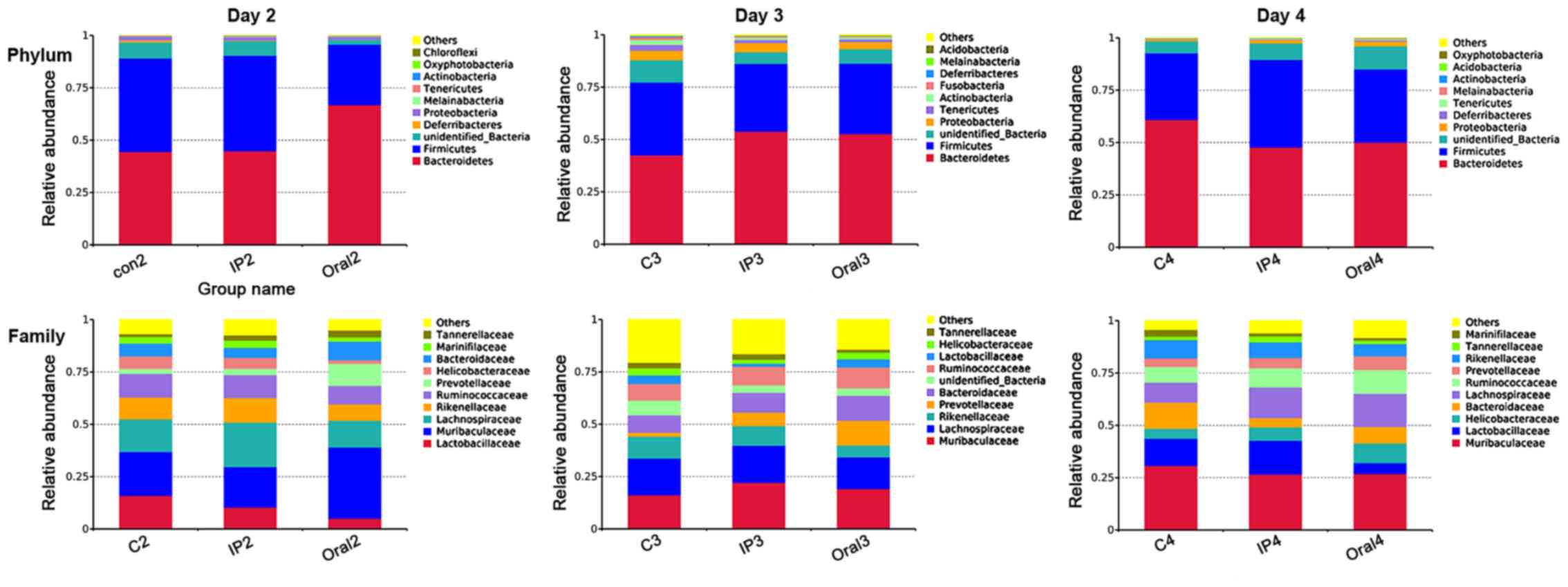
The relative abundances of OTUs from different
groups at different time points were compared, and the relative
abundances of the top ten bacterial species at the level of phylum
and family are presented in Fig. 2.
The present results suggested that almost all OTUs belonged to the
following five phyla: Bacteroidetes, Firmicutes, Proteobacteria,
Deferribacteres and unidentified_Bacteria, accounting for >99%
of the total OTUs. The main taxa at the phylum level, family level
and genus level at different time points (days 2, 3 and 4) in the
three groups were compared (Fig. 3).
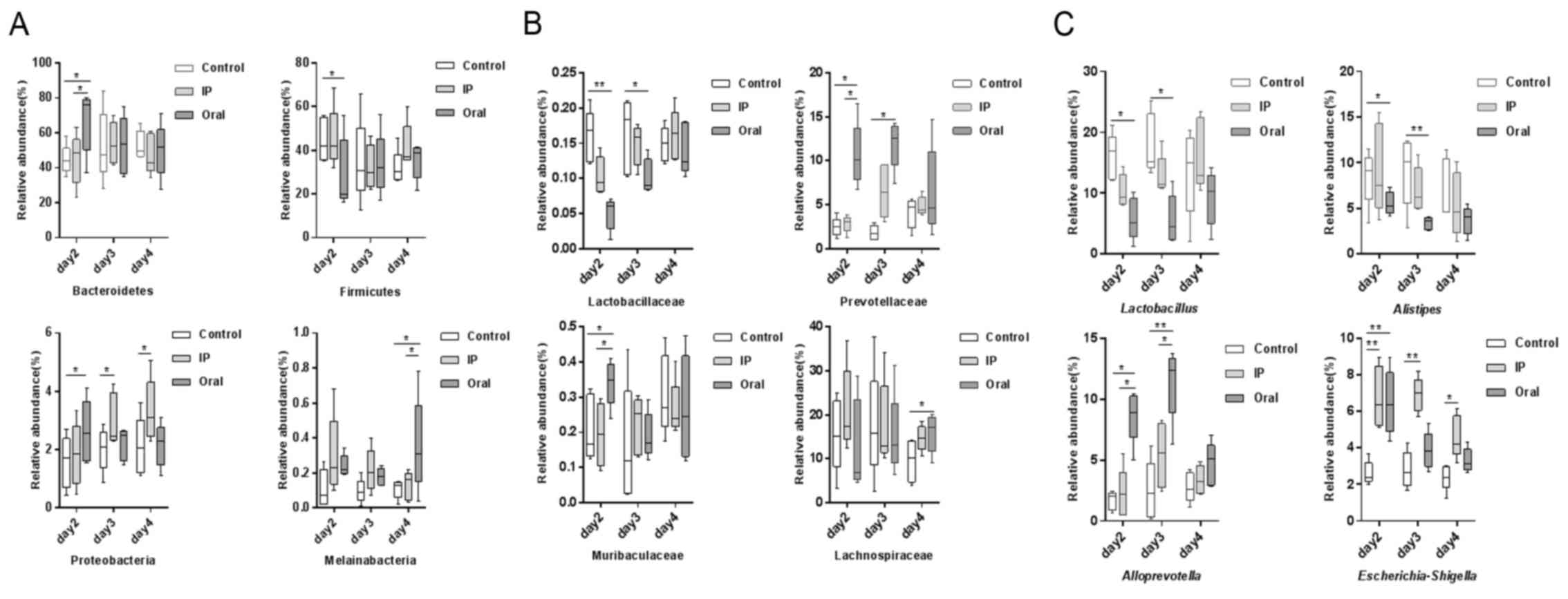
At the phylum level, the oral group showed a higher relative
abundance of Bacteroidetes and Proteobacteria on day 2 compared
with the control group and IP group (P<0.05), but had lower
abundance of Firmicutes compared with the control group
(P<0.05). The present results suggested that on day 4, the
relative abundance of Melainabacteria in the oral group was
significantly higher compared with the control group and IP group
(P<0.05). At the family level, on days 2 and 3 the relative
abundance of Lactobacillaceae in the oral group was significantly
lower compared with the control group (P<0.01 and P<0.05,
respectively); while it was lower in the IP group compared with the
control group, the difference was not statistically significant. On
days 2 and 3, the relative abundance of Prevotellaceae bacteria in
the oral group was higher compared with the control group
(P<0.05). The relative abundance of Muribaculaceae, belonging to
the phylum Bacteroidetes, on day 2 in the oral group was
significantly higher compared with the control group and IP group
(P<0.05). In addition, the present results suggested that the
relative abundance of Lachnospiraceae on day 4 in the oral group
was significantly higher compared with the control group. At the
genus level, the relative abundances of Lactobacillus and
Alistipes in the oral group on days 2 and 3 were
significantly lower compared with the control group (P<0.05 and
P<0.01). By contrast, the relative abundance of
Alloprevotella was significantly higher in the oral group
compared with the control group and IP group (P<0.05 and
P<0.01). The proportion of Escherichia-Shigella was
significantly higher in the IP group and oral group on days 2, 3
and 4 compared with the control group (P<0.05 and
P<0.01).
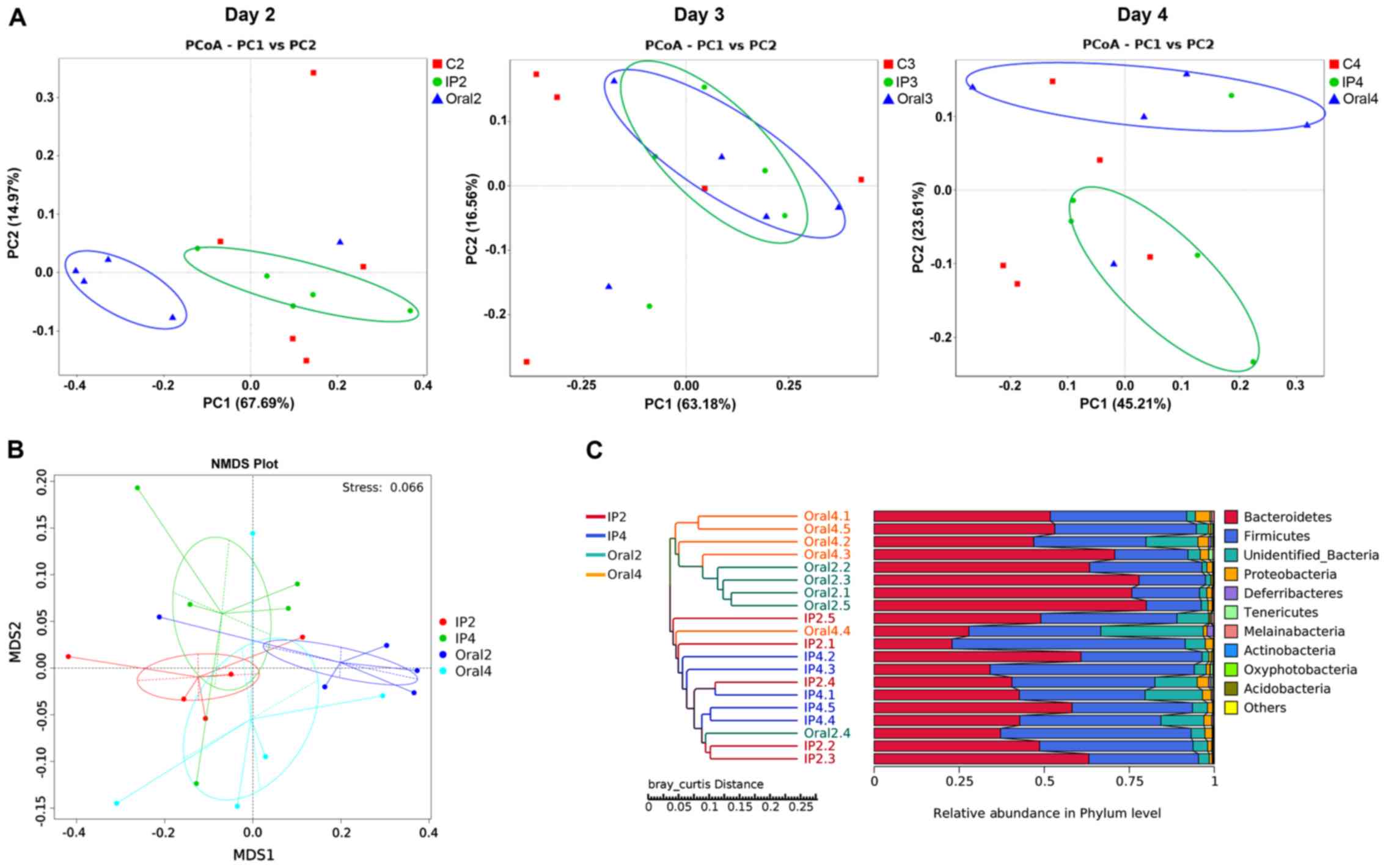
β-diversity
β-diversity describes the microbial community
composition and can be used to evaluate the differences among
microbial communities (22). In the
present study, PCoA analysis was performed based on the weighted
UniFrac distance, and then the main coordinate combination with the
highest contribution rate was selected for plotting. The present
results suggested that there were different distribution styles
between the IP and oral groups at each sampling point (days 2, 3
and 4). On day 2, PC1 represented 67.69% of the variation between
all the groups, and the main coordinate analysis could distinguish
the oral group from the other two groups (Fig. 4A). Furthermore, the coordinates of
the samples in the IP group and control group were similar. On day
3, the coordinates for the IP group overlapped with those of the
oral group (Fig. 4A). On day 4, the
positions of the IP group and the oral group were separated, and
their coordinates were different compared with the control group,
indicating a PC1 value of 45.17% between the three groups (Fig. 4A). NMD analysis showed the degree of
isolation of intestinal flora between the groups at different time
points. The present results identified varying degrees of location
differences between the IP group and the oral group on days 2 and 4
(Fig. 4B). The hierarchical cluster
analysis of OTUs indicated that the IP and oral groups were
well-separated. The present results suggested that the samples in
the oral group (Oral2 and Oral4) were clustered according to the
time points, while samples in the IP group (IP2 and IP4)
intersected the cluster tree to a certain extent (Fig. 4C).
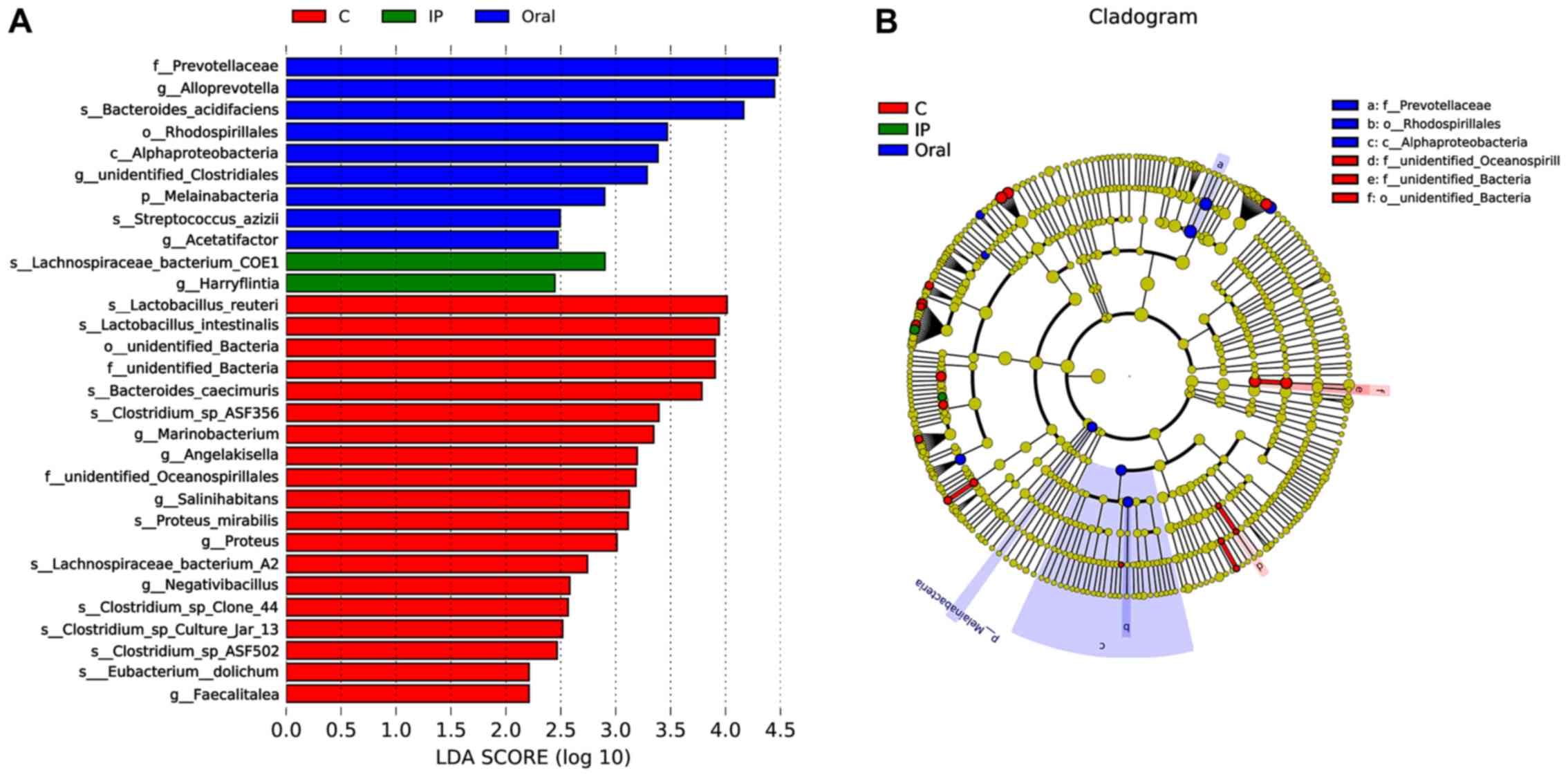
LEfSe analysis
LEfSe, the computational approach to biomarker class
comparisons contributes to the understanding of microbial
communities (25). In order to
assess the gut microbial responses associated with S.
flexneri infection at the taxonomic level, microbial clade
differences were determined using LEfSe analysis. The present
results suggested that a total of 19 bacterial taxa, such as
Lactobacillus reuteri, Lactobacillus intestinalis, Bacteroides
caecimuris and Faecalitalea, had a higher abundance in
the control group compared with the IP group and oral group
(Fig. 5A). However, the relative
abundance of nine taxa of bacteria, such as Prevotellaceae,
Alloprevotella and Bacteroides acidifaciens, in the
oral group was higher compared with the control group and IP group
(Fig. 5A). In addition, only two
taxa of bacteria in the IP group were in higher abundance compared
with the other groups. The present results suggested that three
dominant bacteria were found in the control group, while
Prevotellaceae, Rhodospirillales and Alphaproteobacteria were
dominant in the oral group (Fig.
5B).
Discussion
Infections with Shigella spp. are usually
self-limiting and confined to the mucosa of the distal ileum and
the colon (26). However, in rare
cases, parenteral infection can lead to bacteremia, multiple organ
infections and abscesses (7). In
order to investigate the differences in the effects of
Shigella intestinal infection and parenteral infection on
gut microbiota, and the possible role of gut microbiota in the
different infection routes, a Shigella infection mouse model
was established by IP injection and gavage in the present study.
The present results suggested that adult mice infected by oral
infection and intraperitoneal injection exhibited different disease
outcomes. Oral infection gradually self-cured within 3 days, while
the IP infection developed into persistent diarrhea and other
symptoms of dysentery. In the IP group, S. flexneri was
constantly detected in the feces and a certain proportion of mice
died, which is consistent with previous results from Yang et
al (13) and Sharma et al
(27) Furthermore, 4 day old mice
(28) and young mice treated with
antibiotics (29) are frequently
used to construct animal models of shigellosis via oral
administration. However, both these models neglect the role of the
underdeveloped gut microbiome in the pathogenesis of shigellosis.
By contrast, the adult mice included in the present study had a
developed gut microbiome, which may better reflect the in
vivo effects of Shigella on the gut microbiome.
α-diversity indexes, including Chao1, Shannon,
Simpson, Good's coverage and the Abundance-based Coverage
Estimator, can be used to analyze the abundance and diversity of
microbial communities (30). In the
present study, no significant change in α-diversity was identified
after oral Shigella infection in mice. A possible
explanation may be that Shigella was cleared in a relatively
short time under the action of the intestinal immune system of mice
(31); hence, the effect of
Shigella on the intestinal flora was short-lived and
limited. Only Bacteroidetes, Firmicutes and Proteobacteria with
high relative abundance were significantly affected, while other
bacteria with low abundance were less affected. Therefore, the
present results suggested that there was no significant change in
the α-diversity of the gut microbiota after oral infection. In
addition, the α-diversity in the IP group was unchanged until day
4, which was 3 days after Shigella infection induction. In
the present study the dose challenge in the IP group was set as
5×107 CFU in order to reduce the incidence of mortality;
this dosage was lower than that used by both Yang et al
(13) and Sharma et al
(27), at 5×108 CFU and
108 CFU, respectively. In these previous studies, all
mice in the IP group died within 7 days (13,27).
Although in the present study, the symptoms of dysentery were
pronounced and persistent, a dose-dependent relationship may exist
between S. flexneri and the diversity of the gut
microbiota.
To investigate the effect of Shigella
infection on the relative abundance of specific microbiome taxa,
relative abundances of the species in each group on days 2, 3 and 4
were analyzed in the present study. The present results suggested
that the bacteria taxa of the oral group significantly varied
compared with the control and IP groups. On days 2 and 3, the
relative abundances of Prevotellaceae and Alloprevotella
were higher compared with the IP group and control group, while
Firmicutes, Lactobacillaceae and Lactobacillus had lower
abundances compared with the IP group and control group. On day 4,
there was no significant difference between the oral group and the
other two groups. Therefore, the present results suggested that the
effect of oral infection on microbiome taxa in mice was relatively
rapid, since this route of infection directly exposes the gut
microbiome to S. flexneri. Probiotics such as
Lactobacillus may inhibit the proliferation of
Shigella, and are constantly consumed when fighting against
Shigella, which results in significant decreases in their
abundance (32). Several mechanisms
have been suggested for the inhibitory activity of lactic acid
bacteria against pathogenic bacteria, especially Gram-negative
pathogens (33). These mechanisms
include the production of organic acids, hydrogen peroxide and
bacteriocin, as well as the competition for colonization sites with
pathogenic bacteria (32,33). The protective effects of
Lactobacilli, probiotic bacteria, are greater against
invasive bacteria such as S. sonnei, when compared to
non-invasive bacteria such as Vibrio cholerae (34). However, the mechanism of the
inhibitory effect of lactic acid bacteria on Shigella has
been mostly investigated in vitro (35,36), and
it is not fully understood how these inhibitory effects are exerted
on pathogenic bacteria in the complex intestinal microecological
environment. These inhibitory effects may be associated with
decreases in host gene expression, microRNA regulation and a
substantial reshaping of the Listeria-monocytogenes
transcriptome (37). In the present
study, on days 2 and 3, the abundance of Prevotellaceae
significantly increased in the oral group compared with the control
group. A similar trend was also observed in the IP group,
indicating that the abundance of Prevotellaceae was affected by
Shigella infection. Certain Prevotella strains have
been reported to serve as clinically pivotal pathobionts that
participate in human diseases by promoting chronic inflammation,
such as in intestinal disorders with HIV infection (38), irritable bowel syndrome (39), rheumatic arthritis (40) and periodontitis (41). Therefore, the increased abundance of
Prevotella after Shigella oral infection observed in
the present study likely co-induces inflammatory responses via
pathogenic bacteria and recruitment of inflammatory cells. However,
the specific mechanism needs to be further investigated.
The present PCoA analysis results suggested that at
the initial stage of infection (day 2), the coordinates of the
samples in the IP group and the oral group were divided into two
distinct groups. At day 3, the distribution differences between the
two groups reduced, but the gap between the two groups broadened at
day 4, indicating that Shigella infection could affect the
structure and composition of the intestinal flora at different time
points. In addition, this effect correlated with the different
symptoms and disease outcomes. For example, on the second day
following oral infection of Shigella appeared to be the
initial stage of shigellosis, but the symptoms had completely
disappeared at day 4, while days 2–4 after IP infection indicated
the progression of symptoms. The present results suggested that
there were differences in shigellosis outcomes between the two
infective pathways, based on the examination of intestinal flora.
In addition, it has been shown that the invasion of Shigella
into the large intestine via the abdominal cavity is by migration
in the serosa and muscular layer, rather than via the blood
circulation, suggesting that the early invasion of the pathogen
from the abdominal cavity into the intestinal tract can escape the
endogenous defense system of the host (13). The present results suggested that the
influence of Shigella infection on intestinal flora is weak
and slow at the early stage, but gradually becomes more pronounced
with the development of bacteremia.
The present LEfSe analysis results suggested that
Lactobacillus reuteri and Faecalitalea may be
biomarkers of the control group. Previous studies have demonstrated
that these bacterial groups are probiotics with certain protective
effects, such as eliminating infections, attenuating both GI
diseases and diseases in remote tissues, producing lactate and
butyrate (42,43). The direct supplementation and
prebiotic modulation of Lactobacillus reuteri may be an
attractive preventive and therapeutic strategy against inflammatory
diseases (43). The strain
Lactobacillus reuteri WHH1689 has no lactose utilization
capability and exhibits a high survival rate during storage at room
temperature in drinkable yogurts (44). In addition, thisstrain has shown
great resistance to conditions that simulate the gastrointestinal
tract, including strong adherence to HT-29 cells and inhibitory
activities against Escherichia coli, S. flexneri, Salmonella
paratyphi β and Staphylococcus aureus (44). Faecalibacterium prausnitzii
can inhibit the release of IL-8 and exert anti-inflammatory effects
(45). Faecalibacterium
prausnitzii can also inhibit the invasion of pathogenic
bacteria via colonization resistance (46). Therefore, further research and
clinical intervention evaluations are required to investigate the
use of probiotics for the prevention and treatment of
Shigella infection. One of the biomarkers in the IP group
was Lachnospiraceae bacterium COE1. The members of the
Lachnospiraceae family are known to influence the development of
obesity and diabetes in mice with a genetic susceptibility to
obesity (47). In addition,
long-term high-fat feeding causes obesity-related inflammation of
the ileum and colon, and increases the expression of catenin, a
colon cancer risk factor, in the colon accompanied by an increase
of Lachnospiraceae and Streptococcaceae abundance in the hindgut of
C57BL/6 mice (48). Therefore, the
increased abundance of Lachnospiraceae may affect intestinal
metabolites, which reflect the intestinal metabolic response to
Shigella infection.
In conclusion, the present results suggested that
S. flexneri infection in mice can influence the profile of
the gut microbiota, and the change of some specific taxa may
reflect the results of Shigella-microbiota interaction, such
as the decrease abundance of probiotic Lactobacillus and the
increased abundance of Prevotellaceae. The oral and IP challenges
of S. flexneri exerted different effects on the intestinal
flora, including on the diversity, relative abundance and
composition of the microbiota. The present results suggested that
intestinal flora may serve as a barrier to Shigella
transoral infection, while parenteral infection results in serious
clinical manifestations due to the absence of gut microbiome
inhibition. In future studies, metabonomics, metagenome and
transcriptomics are required to characterize the precise mechanism
of interaction between Shigella, the host and the gut
microbiota.
Acknowledgements
Not applicable.
Funding
This study was financially supported by a grant from
The Open Project of Key Laboratory of Environmental Factors and
Cancer of Fujian Medical University (grant no. GWSZD-201801).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JY and WC participated in raising the animals, DNA
extraction and drafting of the manuscript. PX performed the
statistical analysis and bioinformatics analysis. WZ participated
in the study design and revised the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The current study was approved by The Institutional
Animal Care and Use Committee of Fujian Medical University. All
animal care and experimental procedures were conducted according to
the institutional ethical guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kotloff KL, Riddle MS, Platts-Mills JA,
Pavlinac P and Zaidi AKM: Shigellosis. Lancet. 391:801–812. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chang Z, Zhang J, Ran L, Sun J, Liu F, Luo
L, Zeng L, Wang L, Li Z, Yu H and Liao Q: The changing epidemiology
of bacillary dysentery and characteristics of antimicrobial
resistance of Shigella isolated in China from 2004–2014. BMC Infect
Dis. 16:6852016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Taneja N and Mewara A: Shigellosis:
Epidemiology in India. Indian J Med Res. 143:565–576. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mattock E and Blocker AJ: How do the
virulence factors of shigella work together to cause disease? Front
Cell Infect Microbiol. 7:642017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takeuchi A: Early colonic lesions in
experimental Shigella infection in rhesus monkeys: Revisited. Vet
Pathol Suppl. 7:1–8. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kotloff KL, Winickoff JP, Ivanoff B,
Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK and Levine MM:
Global burden of Shigella infections: Implications for vaccine
development and implementation of control strategies. Bull World
Health Organ. 77:651–666. 1999.PubMed/NCBI
|
|
7
|
Al-Soub H, Al-Maslamani M, Al-Khuwaiter J,
El-Deeb Y and El-Shafie SS: Shigella flexneri perinephric abscess
and bacteremia. Ann Saudi Med. 25:419–421. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Drow DL, Mercer L and Peacock JB: Splenic
abscess caused by Shigella flexneri and Bacteroides fragilis. J
Clin Microbiol. 19:79–80. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Amstey MS and Gandell DL:
Salpingitis-perihepatitis in a patient with cervical Shigella
sonnei. Obstet Gynecol. 55 (3 Suppl):70S–71S. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jasper JM and Ward MA: Shigella
vulvovaginitis in a prepubertal child. Pediatr Emerg Care.
22:585–586. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Upadhyay AK and Neely JA: Toxic megacolon
and perforation caused by Shigella. Br J Surg. 76:12171989.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miron D, Sochotnick I, Yardeni D, Kawar B
and Siplovich L: Surgical complications of shigellosis in children.
Pediatr Infect Dis J. 19:898–900. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang JY, Lee SN, Chang SY, Ko HJ, Ryu S
and Kweon MN: A mouse model of shigellosis by intraperitoneal
infection. J Infect Dis. 209:203–215. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sommer F and Backhed F: The gut
microbiota-masters of host development and physiology. Nat Rev
Microbiol. 11:227–238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vogt SL and Finlay BB: Gut
microbiota-mediated protection against diarrheal infections. J
Travel Med. 24 (Suppl 1):S39–S43. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bhattacharjee S, Kalbfuss N and Prazeres
da Costa C: Parasites, microbiota and metabolic disease. Parasite
Immunol. 392017.doi: 10.1111/pim.12390.
|
|
17
|
Munoz-Vargas L, Opiyo SO, Digianantonio R,
Williams ML, Wijeratne A and Habing G: Fecal microbiome of
periparturient dairy cattle and associations with the onset of
Salmonella shedding. PLoS One. 13:e01961712018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pearson JA, Tai N, Ekanayake-Alper DK,
Peng J, Hu Y, Hager K, Compton S, Wong FS, Smith PC and Wen L:
Norovirus changes susceptibility to type 1 diabetes by altering
intestinal microbiota and immune cell functions. Front Immunol.
10:26542019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun X, Gao Y, Wang X, Hu G, Wang Y, Feng
B, Hu Y, Mu X, Zhang Y and Dong H: Escherichia coli O101-induced
diarrhea develops gut microbial dysbiosis in rats. Exp Ther Med.
17:824–834. 2019.PubMed/NCBI
|
|
20
|
Olfert ED and Godson DL: Humane endpoints
for infectious disease animal models. ILAR J. 41:99–104. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Edgar RC: UPARSE: Highly accurate OTU
sequences from microbial amplicon reads. Nat Methods. 10:996–998.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hugerth LW and Andersson AF: Analysing
microbial community composition through amplicon sequencing: From
sampling to hypothesis testing. Front Microbiol. 8:15612017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lozupone C, Lladser ME, Knights D,
Stombaugh J and Knight R: UniFrac: An effective distance metric for
microbial community comparison. ISME J. 5:169–172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ramette A and Tiedje JM: Multiscale
responses of microbial life to spatial distance and environmental
heterogeneity in a patchy ecosystem. Proc Natl Acad Sci USA.
104:2761–2766. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Segata N, Izard J, Waldron L, Gevers D,
Miropolsky L, Garrett WS and Huttenhower C: Metagenomic biomarker
discovery and explanation. Genome Biol. 12:R602011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ashkenazi S: Shigella infections in
children: New insights. Semin Pediatr Infect Dis. 15:246–252. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sharma D, Yagnik B, Baksi R, Desai N, Padh
H and Desai P: Shigellosis murine model established by
intraperitoneal and intranasal route of administration: A
comparative comprehension overview. Microbes Infect. 19:47–54.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fernandez MI, Thuizat A, Pedron T, Neutra
M, Phalipon A and Sansonetti PJ: A newborn mouse model for the
study of intestinal pathogenesis of shigellosis. Cell Microbiol.
5:481–491. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Q S Medeiros PH, Ledwaba SE, Bolick DT,
Giallourou N, Yum LK, Costa DVS, Oriá RB, Barry EM, Swann JR, Lima
AÂM, et al: A murine model of diarrhea, growth impairment and
metabolic disturbances with Shigella flexneri infection and the
role of zinc deficiency. Gut Microbes. 10:615–630. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim BR, Shin J, Guevarra R, Lee JH, Kim
DW, Seol KH, Lee JH, Kim HB and Isaacson R: Deciphering diversity
indices for a better understanding of microbial communities. J
Microbiol Biotechnol. 27:2089–2093. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shim DH, Ryu S and Kweon MN: Defensins
play a crucial role in protecting mice against oral Shigella
flexneri infection. Biochem Biophys Res Commun. 401:554–560. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Davoodabadi A, Soltan Dallal MM, Lashani E
and Tajabadi Ebrahimi M: Antimicrobial Activity of Lactobacillus
spp. Isolated from fecal flora of healthy breast-fed infants
against diarrheagenic Escherichia coli. Jundishapur J Microbiol.
8:e278522015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang YC, Zhang LW, Ma W, Yi HX, Yang X,
Du M, Shan YJ, Han X and Zhang LL: Screening of probiotic
lactobacilli for inhibition of Shigella sonnei and the
macromolecules involved in inhibition. Anaerobe. 18:498–503. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Alamdary SZ, Bakhshi B and Soudi S: The
anti-apoptotic and anti-inflammatory effect of Lactobacillus
acidophilus on Shigella sonnei and Vibrio cholerae interaction with
intestinal epithelial cells: A comparison between invasive and
non-invasive bacteria. PLoS One. 13:e01969412018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mirnejad R, Vahdati AR, Rashidiani J,
Erfani M and Piranfar V: The antimicrobial effect of lactobacillus
casei culture supernatant against multiple drug resistant clinical
isolates of Shigella sonnei and Shigella flexneri in vitro. Iran
Red Crescent Med J. 15:122–126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Y, Shi X, Hao S, Lu Q, Zhang L, Han
X and Lu W: Inhibition of Shigella sonnei-induced epithelial
barrier disruption by surface-layer associated proteins of
lactobacilli from Chinese fermented food. J Dairy Sci.
101:1834–1842. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Archambaud C, Nahori MA, Soubigou G,
Bécavin C, Laval L, Lechat P, Smokvina T, Langella P, Lecuit M and
Cossart P: Impact of lactobacilli on orally acquired listeriosis.
Proc Natl Acad Sci USA. 109:16684–16689. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Larsen JM: The immune response to
Prevotella bacteria in chronic inflammatory disease. Immunology.
151:363–374. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Su T, Liu R, Lee A, Long Y, Du L, Lai S,
Chen X, Wang L, Si J, Owyang C and Chen S: Altered intestinal
microbiota with increased abundance of Prevotella is associated
with high risk of diarrhea-predominant irritable bowel syndrome.
Gastroenterol Res Pract. 2018:69617832018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Maeda Y and Takeda K: Role of gut
microbiota in rheumatoid arthritis. J Clin Med. 6(pii): E602017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Szafranski SP, Deng ZL, Tomasch J, Jarek
M, Bhuju S, Meisinger C, Kühnisch J, Sztajer H and Wagner-Döbler I:
Functional biomarkers for chronic periodontitis and insights into
the roles of Prevotella nigrescens and Fusobacterium nucleatum; a
metatranscriptome analysis. NPJ Biofilms Microbiomes. 1:150172015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kageyama A, Benno Y and Nakase T:
Phylogenetic and phenotypic evidence for the transfer of
Eubacterium aerofaciens to the genus Collinsella as Collinsella
aerofaciens gen. nov., comb. nov. Int J Syst Bacteriol. 49:557–565.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mu Q, Tavella VJ and Luo XM: Role of
Lactobacillus reuteri in human health and diseases. Front
Microbiol. 9:7572018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen S, Chen L, Chen L, Ren X, Ge H, Li B,
Ma G, Ke X, Zhu J, Li L, et al: Potential probiotic
characterization of Lactobacillus reuteri from traditional Chinese
highland barley wine and application for room-temperature-storage
drinkable yogurt. J Dairy Sci. 101:5780–5788. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sokol H, Pigneur B, Watterlot L, Lakhdari
O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C,
Furet JP, Corthier G, et al: Faecalibacterium prausnitzii is an
anti-inflammatory commensal bacterium identified by gut microbiota
analysis of Crohn disease patients. Proc Natl Acad Sci USA.
105:16731–16736. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Benus RF, Harmsen HJ, Welling GW,
Spanjersberg R, Zijlstra JG, Degener JE and van der Werf TS: Impact
of digestive and oropharyngeal decontamination on the intestinal
microbiota in ICU patients. Intensive Care Med. 36:1394–1402. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kameyama K and Itoh K: Intestinal
colonization by a Lachnospiraceae bacterium contributes to the
development of diabetes in obese mice. Microbes Environ.
29:427–430. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zeng H, Ishaq SL, Zhao FQ and Wright AG:
Colonic inflammation accompanies an increase of β-catenin signaling
and Lachnospiraceae/Streptococcaceae bacteria in the hind gut of
high-fat diet-fed mice. J Nutr Biochem. 35:30–36. 2016. View Article : Google Scholar : PubMed/NCBI
|