Introduction
Cardiac fibrosis is a risk factor for the
development of various cardiovascular diseases, including
myocardial infarction, arrhythmia and heart failure (1,2). The
main pathological features of cardiac fibrosis include abnormal
proliferation of cardiac fibroblasts (CFs) and excessive deposition
of extracellular matrix in the interstitium and perivascular region
(3–6). In response to pathological stimuli, CFs
can differentiate into myofibroblasts and increase the secretion of
extracellular matrix proteins, which consequently leads to cardiac
fibrosis (7–9). Despite advancements in the diagnosis
and treatment of cardiac fibrosis, the exact pathogenesis remains
unclear.
MicroRNAs (miRNAs) are a family of endogenous
non-coding RNAs, 22–25 nucleotides in length, which serve as
transcriptional regulators of genes (10). A number of studies have revealed that
miRNAs are associated with cellular processes, including cell
growth, proliferation and differentiation (11–13).
Furthermore, miRNAs have been demonstrated to be key regulators of
cardiovascular disease, including cardiac fibrosis (14–17). For
instance, overexpression of let-7i attenuated angiotensin
II-induced cardiac fibrosis by regulating the expression levels of
interleukin-6 and collagen (18).
Furthermore, it has been reported that increased miR-489 expression
decreased pulmonary fibrosis by targeting MYD88 innate immune
signal transduction adaptor (19).
Transgenic overexpression of miR-489 was also reported to inhibit
cardiac fibrosis following treatment with angiotensin II treatment
(20). However, to the best of our
knowledge, the underlying mechanism of miR-489 in attenuating the
development of cardiac fibrosis has not been previously
reported.
Histone deacetylases (HDACs) include 18 isoforms and
are subdivided into four classes. HDAC2, a member of HDAC class II,
is involved in a number of diseases, including tumorigenesis and
cardiovascular disease (21). It has
been reported that HDAC inhibitors inhibit fibrosis in a number of
organs, such as the lungs and liver (22,23). In
addition, overexpression of HDAC2 promotes cardiac hypertrophy
(24). As miRNAs regulate the
expression of downstream target genes, miR-489 may suppress cardiac
fibrosis by downregulating the expression of HDAC2.
The present study revealed that miR-489 inhibited
isoproterenol (ISO)-induced cardiac fibrosis in Sprague-Dawley (SD)
rats by downregulating the expression of HDAC2. The results may
guide the development of novel therapeutic agents for cardiac
fibrosis.
Materials and methods
Animal experiments
A total of 30 SD rats (8 weeks old) were obtained
from the Laboratory Animal Center of Soochow University. The mice
were maintained under 22°C, 50% relative humidity, with a 12-h
light/dark cycle and received food and water ad libitum. All
animal experiments were approved by the Ethics Committee of the
Third Affiliated Hospital of Soochow University (Changzhou, China).
The rats were randomly divided into two groups, as follows: i)
Cardiac fibrosis group, which consisted of animals subcutaneously
injected with 5 mg/kg/day ISO (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for 10 days; and ii) control group, which
consisted of animals injected with an equivalent volume of saline.
Animal health and behavior, such as body temperature, weight loss,
behavioral changes and pathological changes, were monitored every
day. On day 11, the rats were sacrificed by cervical dislocation
following anesthesia with sodium pentobarbital (50 mg/kg),
following which their hearts were immediately isolated. Part of the
cardiac tissue specimens were cut into 5-µm thick sections and
fixed in 4% formaldehyde solution and stained with hematoxylin and
eosin (H&E) or Masson solution for 5–10 min at room temperature
to detect pathological changes in the cardiac tissues. The
remaining section was used for RNA analysis.
Isolation of rat CFs and ISO
treatment
Heart tissues were collected from SD rats,
homogenized into 1-mm3 sections, placed in D-Hank's
solution and subsequently digested using a mixed enzyme solution
(trypsin: Collagenase ratio, 2:1). The cells were centrifuged at
room temperature and 800 × g for 10 min and cultured in Dulbecco's
modified Eagle medium (Corning, Inc., Corning, NY, USA) containing
10% fetal bovine serum (Corning, Inc.), 100 U/ml penicillin and 100
µg/ml streptomycin at 37°C for 2 h. Adherent cells were obtained
after discarding the non-adherent cells and were identified as CFs
by their characteristic spindle shaped appearance using an inverted
phase contrast microscope and immunohistochemical staining for
vimentin (25,26). CFs at passage 3 were used in
subsequent experiments. The cells were divided into two groups, and
treated with 10 µM ISO or saline for 24 h, respectively.
Transfection with small interfering
RNA (siRNA), mimics and inhibitor
siRNAs targeting HDAC2 (siHDAC2), siRNA-negative
control (siNC), miR-489 mimic, miR-NC, miR-489 inhibitor and NC
inhibitor were synthesized by GenePharma Co., Ltd. (Shanghai,
China). For HDAC2 overexpression, full length HDAC2 cDNA was
amplified by PCR reaction from the cDNA library of HeLa cells (cat.
no. R71407; Invitrogen; Thermo Fisher Scientific, Inc.) and
subcloned into the pcDNA3.1 vector (Invitrogen; Thermo Fisher
Scientific, Inc.), with empty pcDNA3.1 serving as control, and then
transfected into cells using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Transfection with siHDAC2 or siNC (10 nM),
miR-489 or NC mimics (10 nM), miR-489 inhibitor or NC inhibitor (10
nM) and co-transfection with miR-489 mimics and HDAC2 were
performed using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The cells were subsequently split 36 h after transfection and
treated with ISO for 24 h. The transfection sequences were as
follows: siHDAC2, 5′-GUAUCAUCAGAGAGUCUUATT-3′; siNC,
5′-UUUGUACUACACAAAAGUACUG-3′; miR-489 mimic,
5′-GUGACAUCACAUAUACGGCAGC-3′; NC mimics,
5′-UUCUCCGAACGUGUCACGUUU-3′; miR-489 inhibitor,
5′-GCUGCCGUAUAUGUGAUGUCAC-3′; NC inhibitor,
5′-CAGUCCUUUUGUGUAGUACAA-3′.
Bioinformatics prediction and
dual-luciferase reporter assay
The bioinformatics prediction software TargetScan
version 7.2 (www.targetscan.org) was used to predict the potential
target genes of miR-489. Next, a dual-luciferase reporter assay was
conducted to validate the predicted targets. Wild-type (WT) and
mutant (MUT) HDAC2 were cloned into a pGL3 plasmid to construct
pGL3-HDAC2-WT and pGL3-HDAC2-MUT vectors, respectively.
Subsequently, miR-489 mimics and vectors were co-transfected into
293T cells (American Type Culture Collection) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.), and luciferase activity was measured using the
Dual-Luciferase Reporter System (Promega Corporation, Madison, WI,
USA) at 48 h after transfection.
Determination of serum myocardial
injury markers
The rats were anaesthetized and sacrificed by
cervical dislocation following anesthesia with sodium pentobarbital
(50 mg/kg). Then, the blood samples were collected from the carotid
artery of rats and centrifuged at 3,000 × g for 10 min at room
temperature. The activities of creatine kinase (CK) and CK isozyme
(CK-MB) were measured using the MD-100 multifunctional automatic
biochemistry analyzer (Sanhe Medical Equipment Co., Ltd., Sanhe,
China). Additionally, the concentration of cardiac troponin I
(cTnI) was measured using the VITROS Immuno Diagnostic kit
(Ortho-Clinical Diagnostics, Inc., Raritan, NJ, USA). All assays
were performed according to the manufacturer's protocol.
Cell viability assay
Cell viability was detected using an MTT assay
(KeyGen Biotech Co., Ltd.) according to the manufacturer's
protocol. In brief, CFs were seeded into 96-well plates (3,000
cells/well) and transfected with the miR-489 mimic, miR-489
inhibitor or corresponding NC. After 36 h transfection, the cells
were treated with ISO for 24 h and total of 50 µl MTT solution was
added in each sample well, which were subsequently incubated for 4
h at 37°C. Next, 200 µl dimethyl sulfoxide was added into each
well, and the optical density was measured at a wavelength of 570
nm.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from cardiac tissues and CFs
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. RNA
concentrations were measured using a NanoDrop™ 2000
spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). For miRNA expression, RT reactions were performed using a One
Step PrimeScript miRNA cDNA Synthesis kit (Takara Biotechnology
Co., Ltd.) at 37°C for 30 min according to the manufacturer's
protocols, followed by qPCR with SYBR® Premix Ex Taq
(Takara Biotechnology Co., Ltd.). For mRNA expression, cDNA was
synthesized from total RNA using a PrimeScript™ RT Reagent kit
(Takara Biotechnology Co., Ltd.). qPCR amplifications were then
performed using SYBR® Green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). For miR-489 detection,
the following thermocycling conditions were used: An initial
denaturation step at 95°C for 2 min, followed by 40 cycles of
denaturation at 95°C for 10 sec and annealing at 60°C for 1 min.
For the detection of mRNA, the thermocycling conditions were as
follows: Initial denaturation at 95°C for 15 sec, denaturation at
94°C for 30 sec, annealing at 60°C for 20 sec, and extension at
72°C for 40 sec for 40 cycles. Fold changes in expression levels
were calculated using the 2−ΔΔCq method (27). The primer sequences used in qPCR were
as follows: miR-489 forward, 5′-ACACTCCAGCTGGGGTGACATCACATA-3′, and
reverse, 5′-TGGTGTCGTGGAGTCG-3′; HDAC2 forward,
5′-GCTATTCCAGAAGATGCTGTTC-3′, and reverse,
5′-GTTGCTGAGCTGTTCTGATTTG-3′; collagen I (Col1A1) forward,
5′-CAGAGCACGATGTCCTGAGA-3′, and reverse,
5′-GCAAATGTGAGCTTCTGTGC-3′; α-smooth muscle actin (α-SMA) forward,
5′-GGAGTGATGGTTGGAATGG-3′, and reverse, 5′-ATGATGCCGTGTTCTATCG-3′;
GAPDH forward, 5′-CAAGCTCATTTCCTGGTATGAC-3′, and reverse
5′-CAGTGAGGGTCTCTCTCTTCCT-3′; and U6 forward,
5′-CTCGCTTCGGCAGCACATATACTA-3′, and reverse,
5′-ACGAATTTGCGTGTCATCCTTGCG-3′. HDAC2, collagen I and α-SMA mRNA
levels were normalized to the internal reference gene GAPDH, while
miR-489 levels were normalized to U6.
Statistical analysis
Statistical analyses were performed using SPSS
software (version 18.0; SPSS, Inc.). Comparisons of parameters
between two groups were performed using a paired Student's t-test.
Comparisons among multiple groups were performed by one-way
analysis of variance, followed by Tukey's test. Data are presented
as the mean ± standard deviation of at least three independent
experiments. P<0.05 was considered to indicate a statistically
significant difference.
Results
Pathological changes and levels of
fibrosis-associated mRNAs in vivo and in vitro
H&E and Masson staining were performed to
confirm that the model of ISO-induced cardiac fibrosis was
successfully constructed in the rats (Fig. 1A). Furthermore, the serum levels of
CK, CK-MB and cTnI (28,29), which serve as diagnostic markers of
myocardial damage, were measured. The data revealed that the levels
of CK, CK-MB and cTnI were significantly increased in the serum of
ISO-treated rats compared with those in the control rats (Fig. 1B-D). Furthermore, RT-qPCR indicated
that miR-489 expression was significantly decreased in heart
tissues obtained from the ISO-treated rats as compared with the
control group (Fig. 1E). By
contrast, the mRNA expression levels of Col1A1 and α-SMA were
markedly increased in heart tissues obtained from ISO-treated rats
compared with the control group (Fig.
1F). Consistent with these in vivo experimental results,
miR-489 expression was found to be decreased in ISO-treated CFs
compared with the control group, while the expression levels of
Col1A1 and α-SMA were significantly increased in the ISO-treated
CFs (Fig. 1G and H).
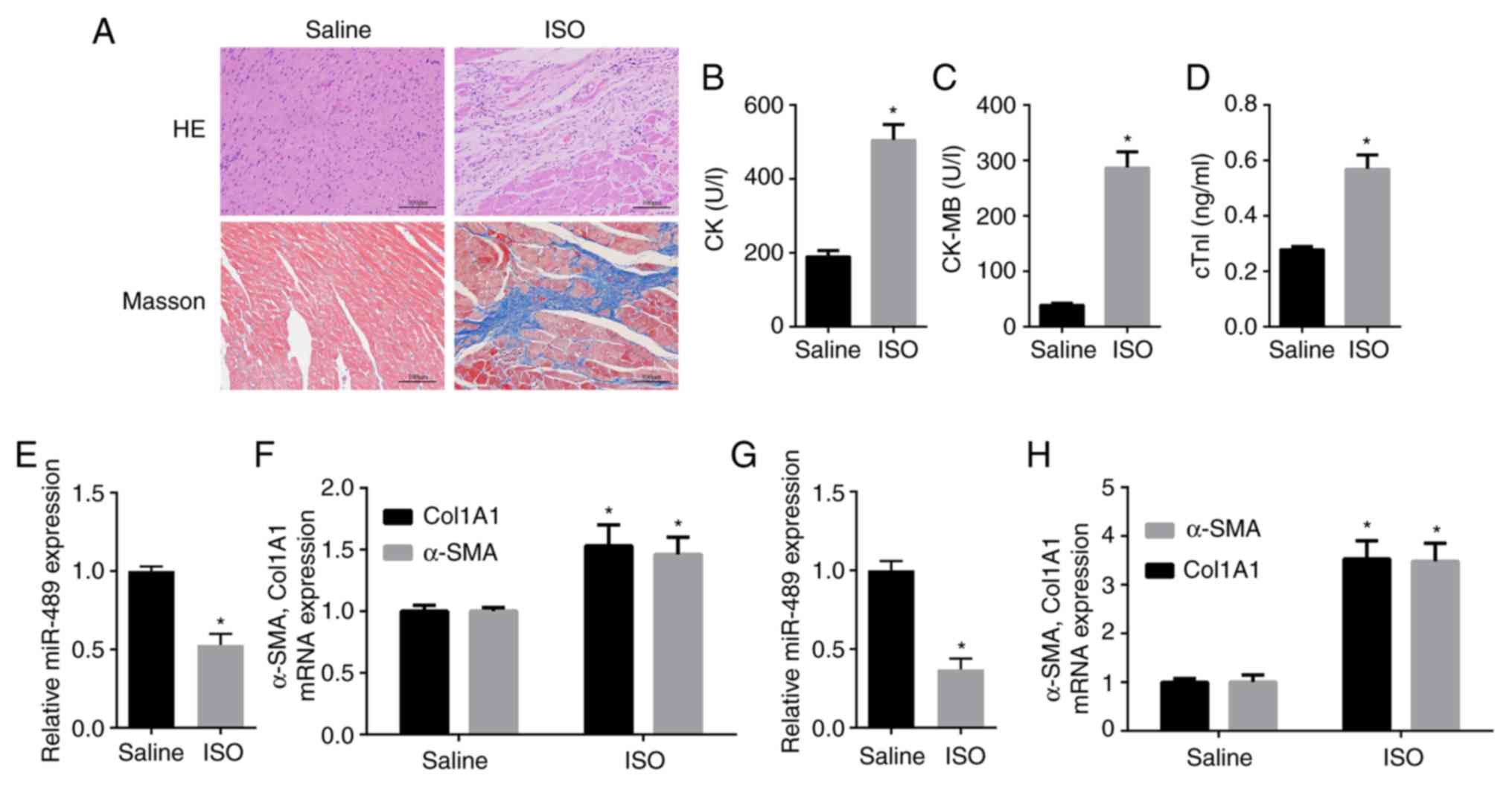 | Figure 1.Pathological changes in myocardial
tissue and expression of fibrosis-associated markers in ISO-treated
rat heart tissues and CFs. (A) H&E and Masson staining revealed
myocardial collagen deposition in myocardial tissues subsequent to
ISO injection (scale bar, 100 µm). (B) CK activity, (C) CK-MB
activity and (D) cTnI concentration were measured in the serum of
ISO-treated and normal control rats. (E) miR-489 expression, and
(F) Col1A1 and α-SMA mRNA levels in heart tissues obtained from the
control and ISO-treated rats, as well as (G) miR-489, and (H)
Col1A1 and α-SMA levels in the control and ISO-treated CFs were
determined by reverse transcription-quantitative polymerase chain
reaction. The data are presented as the mean ± standard deviation.
*P<0.05 vs. saline group. ISO, isoproterenol; CFs, cardiac
fibroblasts; CK, creatine kinase; CK-MB, CK isozyme; cTnI, troponin
I; miR, microRNA; Col1A1, collagen I; α-SMA, α-smooth muscle
actin. |
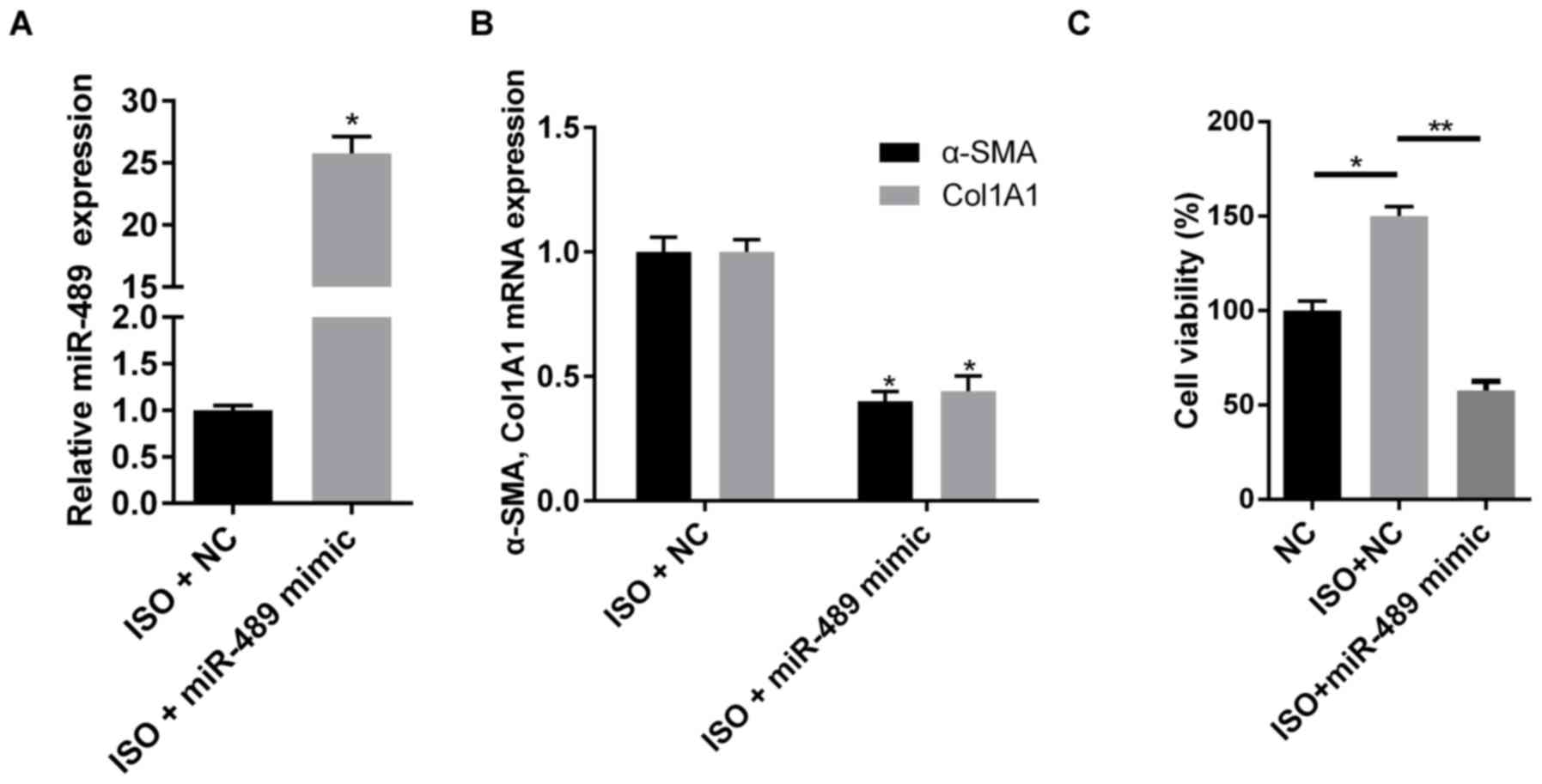
miR-489 inhibits the viability and
differentiation of CFs
To investigate the effect of miR-489 on cardiac
fibrosis, miR-489 mimics were transfected into rat CFs to induce
miRNA overexpression (Fig. 2A). As
shown in Fig. 2B, overexpression of
miR-489 decreased the mRNA expression levels of Col1A1 and α-SMA.
In addition, overexpression of miR-489 significantly inhibited the
cell viability induced by ISO (Fig.
2C). These data demonstrated that miR-489 inhibited the
viability and differentiation of ISO-treated CFs.
HDAC2 is a direct target of
miR-489
Computational predictions of miR-489 target genes
were performed using TargetScan software. As shown in Fig. 3A, the 3′-untranslated region (UTR) of
HDAC2 was complementary to miR-489, suggesting that HDAC2 may be a
direct downstream target of miR-489. In order to validate this
result, WT or MUT HDAC2 sequences were cloned into the 3′-UTR of
the firefly luciferase gene. The results revealed that the miR-489
mimic transfection reduced the luciferase activity in 293T cells
that were co-transfected with WT HDAC2, but not with MUT HDAC2
(Fig. 3B). Furthermore, RT-qPCR was
used to determine the expression level of HDAC2 in ISO-treated CFs
transfected with miR-489 mimic, miR-489 inhibitor or the
corresponding NC. Compared with the NC group, HDAC2 expression was
downregulated in miR-489 mimic-transfected cells and upregulated in
miR-489 inhibitor-transfected cells (Fig. 3C).
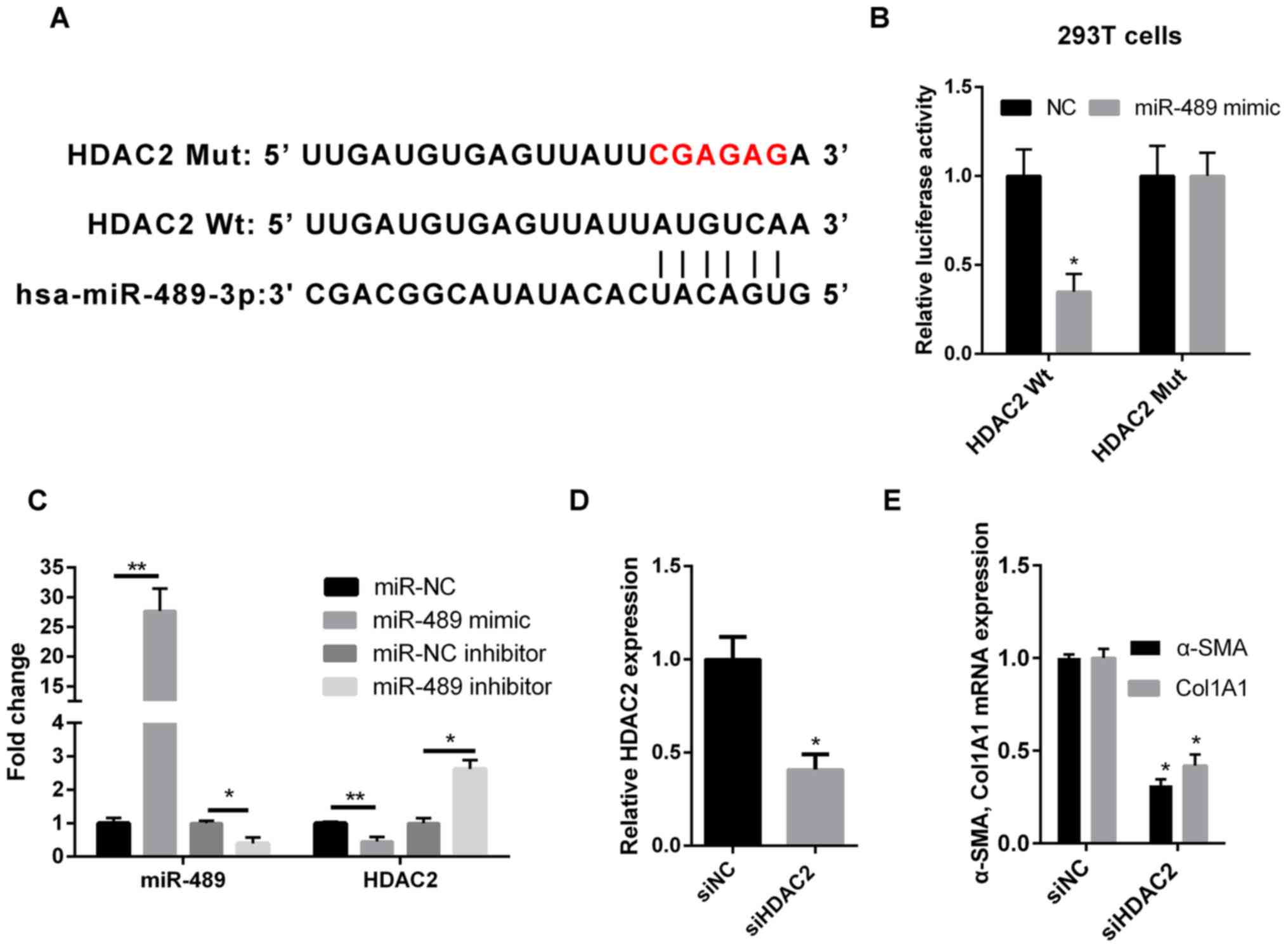 | Figure 3.miR-489 directly targets HDAC2. (A)
Bioinformatics analysis suggested that the 3′-untranslated region
of HDAC2 was complementary to miR-489. (B) The luciferase reporter
assay revealed that miR-489 binds to WT HDAC2, but not MUT HDAC2 in
293T cells. (C) RT-qPCR was used to determine the expression levels
of miR-489 and HDAC2 mRNA in isoproterenol-treated CFs transfected
with NC, miR-489 mimic or miR-489 inhibitor. (D) HDAC2 expression,
and (E) Col1A1 and α-SMA mRNA levels in CFs transfected with siNC
and siHDAC2 were determined by RT-qPCR. The data are presented as
the mean ± standard deviation. *P<0.05 and **P<0.01, vs.
corresponding NC group. miR, microRNA; HDAC2, histone deacetylase
2; WT, wild-type; MUT, mutant; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; CFs, cardiac
fibroblasts; NC, negative control; Col1A1, collagen I; α-SMA,
α-smooth muscle actin; si, small interfering RNA. |
Silencing of HDAC2 decreases the
expression levels of Col1A1 and α-SMA in CFs
To further investigate the role of HDAC2 in cardiac
fibrosis, HDAC2 expression was knocked down using siRNA followed by
treatment with ISO. The transfection efficiency was confirmed by
RT-qPCR. Relative HDAC2 expression was significantly reduced in
cells transfected with siHDAC2. (Fig.
3D). As shown in Fig. 3E, the
expression levels of Col1A1 and α-SMA were markedly reduced in the
siHDAC2-transfected group, as compared with those in the siNC
group.
miR-489 suppresses cardiac fibrosis
via HDAC2
The present study earlier revealed that HDAC2 is a
downstream target of miR-489 and was involved in the expression of
fibrosis-associated markers. In order to determine whether the
biological function of miR-489 in fibrogenesis was mediated by
HDAC2, ISO-treated CFs were transfected with NC or miR-489 mimic,
or co-transfected with HDAC2 overexpression plasmid and miR-489
mimic. RTqPCR demonstrated that the level of HDAC2 was notably
increased in CFs transfected with the HDAC2 overexpression plasmid
(Fig. 4A), while HDAC2 markedly
reversed the inhibitory effect of miR-489 on HDAC2 expression in
co-transfected CFs (Fig. 4B). As
shown in Fig. 4C and D, cells
co-transfected with HDAC2 overexpression plasmid and miR-489 mimic
exhibited significantly increased expression levels of α-SMA and
Col1A1, as well as enhanced cell viability, compared with the cells
transfected with the miR-489 mimic alone. These results further
suggested that HDAC2 serves a role in attenuating the viability and
differentiation of ISO-treated CFs.
Discussion
The present study revealed that miR-489 served an
important role in ISO-induced cardiac fibrosis. Specifically,
miR-489 attenuated cardiac fibrosis and decreased the expression
levels of Col1A1 and α-SMA by downregulating HDAC2 expression.
Emerging evidence suggests that miRNAs are strongly
associated with the development of cardiac fibrosis. For instance,
Zhang et al (30) reported
that miR-29b inhibited angiotensin II-induced cardiac fibrosis by
targeting transforming growth factor (TGF)-β1, thereby inhibiting
the TGF-β/SMAD3 signaling pathway. Wang et al (20) further revealed that cardiac
hypertrophy-related factor, a long non-coding RNA, regulated
cardiac hypertrophy by serving as an endogenous sponge for miR-489,
thereby decreasing its expression level. However, the underlying
mechanism of miR-489 in cardiac fibrosis has not been fully
elucidated. In the present study, Sprague-Dawley rats were treated
with ISO to induce cardiac fibrosis, and H&E and Masson
staining were used to confirm that the model of cardiac fibrosis
was successfully established. Additionally, miR-489 expression was
found to be significantly downregulated in ISO-treated cardiac
tissues and CFs, whereas the mRNA expression levels of Col1A1 and
α-SMA were increased in ISO-treated cardiac tissues and CFs.
miR-489 overexpression significantly reduced cell viability and
Col1A1 and α-SMA mRNA expression in ISO-treated CFs. Taken
together, these findings demonstrated that miR-489 mimic suppressed
cell viability and differentiation of ISO-treated CFs.
HDAC inhibitors have been demonstrated to inhibit
fibrosis following injury in a number of organs (31,32).
Furthermore, the involvement of HDAC2 in heart disease has been
reported (24). Trivedi et al
(33) also indicated that HDAC2
regulated the cardiac hypertrophy response by modulating glycogen
synthase kinase 3β activity. The present study revealed that
miR-489 expression was decreased following ISO treatment in
vivo or in vitro. Bioinformatics analysis and
dual-luciferase reporter assay demonstrated that HDAC2 mRNA
directly interacted with miR-489. RT-qPCR analysis found that the
overexpression of miR-489 inhibited HDAC2 expression Subsequently,
it was observed that HDAC2 knockdown decreased Col1A1 and α-SMA
expression levels in CFs, whereas HDAC2 overexpression reversed the
inhibitory effects of miR-489b on ISO-treated CFs. Therefore, the
data suggested that miR-489 suppressed ISO-induced cardiac fibrosis
by downregulating HDAC2. However, other possible targets of miR-489
may exist, whilst HDAC2 could also be subject to the regulation by
other miRNAs. Therefore, further experiments of miR-489 on CFs are
required to elucidate the mechanism in cardiac fibrosis further and
the application of miR-489 for the treatment of cardiovascular
diseases.
In conclusion, the results obtained in the present
study indicated that miR-489 served an important role in the
development of ISO-induced cardiac fibrosis by regulating HDAC2.
The present study provided new insight into the mechanisms
underlying cardiac fibrogenesis and suggested that miR-489 may
serve as a potential therapeutic target for the treatment of
cardiac fibrosis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XY and TY designed the study. TY and SZ analyzed the
data and prepared the figures. XY drafted the manuscript. All
authors approved this manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
the Third Affiliated Hospital of Soochow University (Changzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sci.
71:549–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dzeshka MS, Lip GY, Snezhitskiy V and
Shantsila E: Cardiac fibrosis in patients with atrial fibrillation:
Mechanisms and clinical implications. J Am Coll Cardiol.
66:943–959. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Segura AM, Frazier OH and Buja LM:
Fibrosis and heart failure. Heart Fail Rev. 19:173–185. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Edgley AJ, Krum H and Kelly DJ: Targeting
fibrosis for the treatment of heart failure: A role for
transforming growth factor-β. Cardiovasc Ther. 30:e30–e40. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Espira L and Czubryt MP: Emerging concepts
in cardiac matrix biology. Can J Physiol Pharmacol. 87:996–1008.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fan D, Takawale A, Lee J and Kassiri Z:
Cardiac fibroblasts, fibrosis and extracellular matrix remodeling
in heart disease. Fibrogenesis Tissue Repair. 5:152012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barnes JL and Gorin Y: Myofibroblast
differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney
Int. 79:944–956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weber KT, Sun Y, Tyagi SC and Cleutjens
JP: Collagen network of the myocardium: Function, structural
remodeling and regulatory mechanisms. J Mol Cell Cardiol.
26:279–292. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Swynghedauw B: Molecular mechanisms of
myocardial remodeling. Physiol Rev. 79:215–262. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nature reviews. Cancer. 6:857–866.
2006.PubMed/NCBI
|
|
11
|
Feng H, Wang Y, Su J, Liang H, Zhang CY,
Chen X and Yao W: MicroRNA-148a suppresses the proliferation and
migration of pancreatic cancer cells by down-regulating ErbB3.
Pancreas. 45:1263–1271. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Jiaqi C, Zhaoying C and Huimin C:
MicroRNA-506-3p regulates neural stem cell proliferation and
differentiation through targeting TCF3. Gene. 593:193–200. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vienberg S, Geiger J, Madsen S and
Dalgaard LT: MicroRNAs in metabolism. Acta Physiol (Oxf).
219:346–361. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thum T, Catalucci D and Bauersachs J:
MicroRNAs: Novel regulators in cardiac development and disease.
Cardiovasc Res. 79:562–570. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van Rooij E, Sutherland LB, Thatcher JE,
DiMaio JM, Naseem RH, Marshall WS, Hill JA and Olson EN:
Dysregulation of microRNAs after myocardial infarction reveals a
role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA.
105:13027–13032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wei C, Kim IK, Kumar S, Jayasinghe S, Hong
N, Castoldi G, Catalucci D, Jones WK and Gupta S: NF-κB mediated
miR-26a regulation in cardiac fibrosis. J Cell Physiol.
228:1433–1442. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jiang X, Tsitsiou E, Herrick SE and
Lindsay MA: MicroRNAs and the regulation of fibrosis. FEBS J.
277:2015–2021. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang X, Wang HX, Li YL, Zhang CC, Zhou CY,
Wang L, Xia YL, Du J and Li HH: MicroRNA Let-7i negatively
regulates cardiac inflammation and fibrosis. Hypertension.
66:776–785. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu Q, Han L, Yan W, Ji X, Han R, Yang J,
Yuan J and Ni C: miR-489 inhibits silica-induced pulmonary fibrosis
by targeting MyD88 and Smad3 and is negatively regulated by lncRNA
CHRF. Sci Rep. 6:309212016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang K, Liu F, Zhou LY, Long B, Yuan SM,
Wang Y, Liu CY, Sun T, Zhang XJ and Li PF: The long noncoding RNA
CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res.
114:1377–1388. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yoon S and Eom GH: HDAC and HDAC
inhibitor: From cancer to cardiovascular diseases. Chonnam Med J.
52:1–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li M, Zheng Y, Yuan H, Liu Y and Wen X:
Effects of dynamic changes in histone acetylation and deacetylase
activity on pulmonary fibrosis. Int Immunopharmacol. 52:272–280.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li X, Wu XQ, Xu T, Li XF, Yang Y, Li WX,
Huang C, Meng XM and Li J: Role of histone deacetylases(HDACs) in
progression and reversal of liver fibrosis. Toxicol Appl Pharmacol.
306:58–68. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eom GH, Cho YK, Ko JH, Shin S, Choe N, Kim
Y, Joung H, Kim HS, Nam KI, Kee HJ and Kook H: Casein kinase-2α1
induces hypertrophic response by phosphorylation of histone
deacetylase 2 S394 and its activation in the heart. Circulation.
123:2392–2403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Katwa LC, Guarda E and Weber KT:
Endothelin receptors in cultured adult rat cardiac fibroblasts.
Cardiovascular research. 27:2125–2129. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, Wang B, Zhou C and Bi Y: Matrine
induces apoptosis in angiotensin II-stimulated hyperplasia of
cardiac fibroblasts: Effects on Bcl-2/Bax expression and caspase-3
activation. Basic Clin Pharmacol Toxicol. 101:1–8. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zaitone SA and Abo-Gresha NM: Rosuvastatin
promotes angiogenesis and reverses isoproterenol-induced acute
myocardial infarction in rats: Role of iNOS and VEGF. Eur J
Pharmacol. 691:134–142. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li M, Jiang Y, Jing W, Sun B, Miao C and
Ren L: Quercetin provides greater cardioprotective effect than its
glycoside derivative rutin on isoproterenol-induced cardiac
fibrosis in the rat. Can J Physiol Pharmacol. 91:951–959. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM
and Lan HY: miR-29b as a therapeutic agent for angiotensin
II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol
Ther. 22:974–985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR,
Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, et al: Inhibition of
histone deacetylation blocks cardiac hypertrophy induced by
angiotensin II infusion and aortic banding. Circulation. 113:51–59.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kong Y, Tannous P, Lu G, Berenji K,
Rothermel BA, Olson EN and Hill JA: Suppression of class I and II
histone deacetylases blunts pressure-overload cardiac hypertrophy.
Circulation. 113:2579–2588. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W,
Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, et al: Hdac2
regulates the cardiac hypertrophic response by modulating Gsk3 beta
activity. Nat Med. 13:324–331. 2007. View
Article : Google Scholar : PubMed/NCBI
|