Introduction
Rosai-Dorfman disease (RDD) is a benign histiocyte
disorder that was originally described by Rosai and Dorfman in 1969
as sinus histiocytosis with massive lymphadenopathy. It mostly
affects young adults. Patients typically present with a fever,
leukocytosis, and non-painful cervical lymphadenopathy (1,2). RDD
sometimes involves extranodal organs, including the skin, soft
tissue, and central nervous system (1). Bone involvement occurs in less than 10%
of cases. Primary bone RDD, in the absence of lymphadenopathy,
accounts for less than 1% of all cases (2). RDD is usually self-limiting disease,
making systemic therapy rarely required (3). The imaging of osseous RDD typically
shows a lytic lesion on radiography and CT. The differential
diagnosis is broad and includes osteomyelitis, Langerhans cell
histiocytosis, lymphoma, primary bone sarcoma and metastatic bone
tumor (4). Therefore accurate
diagnosis depends on histological examination.
We herein report a rare case of primary bone RDD
that occurred in the pelvic bone of a two-year-old boy and we
present a literature review of the management and clinical course
of this type of disease.
Case report
A two-year-old boy was admitted to our hospital due
to limping of the right lower extremity, which persisted for two
months without obvious pain. He had no perinatal medical problems.
He had no history of the infection, such as upper respiratory tract
infection or viral enteritis.
A physical examination showed slight limitation in
the range of motion of his right hip joint without a leg length
discrepancy. There was no swelling, redness, local heat, or
percussion pain around his right hip joint. There was no cervical,
axillary, popliteal, or inguinal lymphadenopathy. A laboratory
examination revealed no abnormalities; his white blood cell count
and hemoglobin and serum C-reactive protein (CRP) levels were
normal. Tests for tumor markers, including AFP, CEA, CA19-9, CA125,
NSE, HCG-β and sIL-2R, were negative.
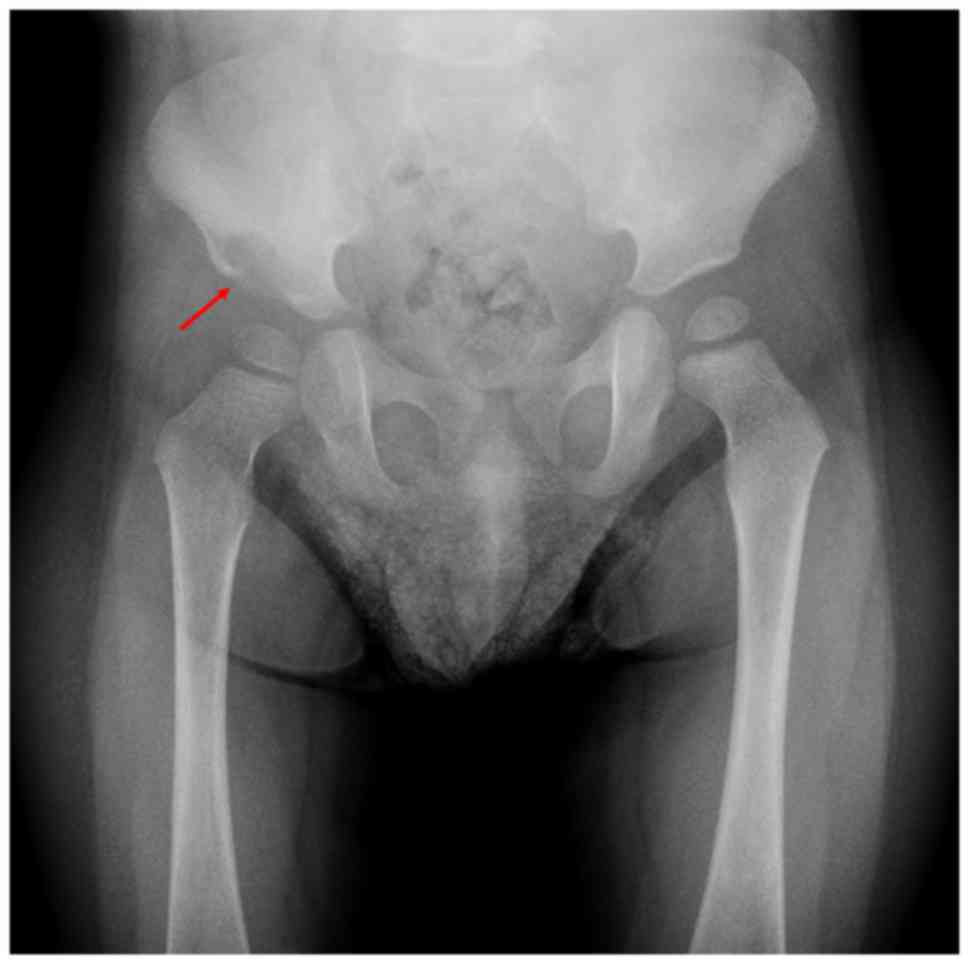
Plain radiography showed an osteolytic lesion at the
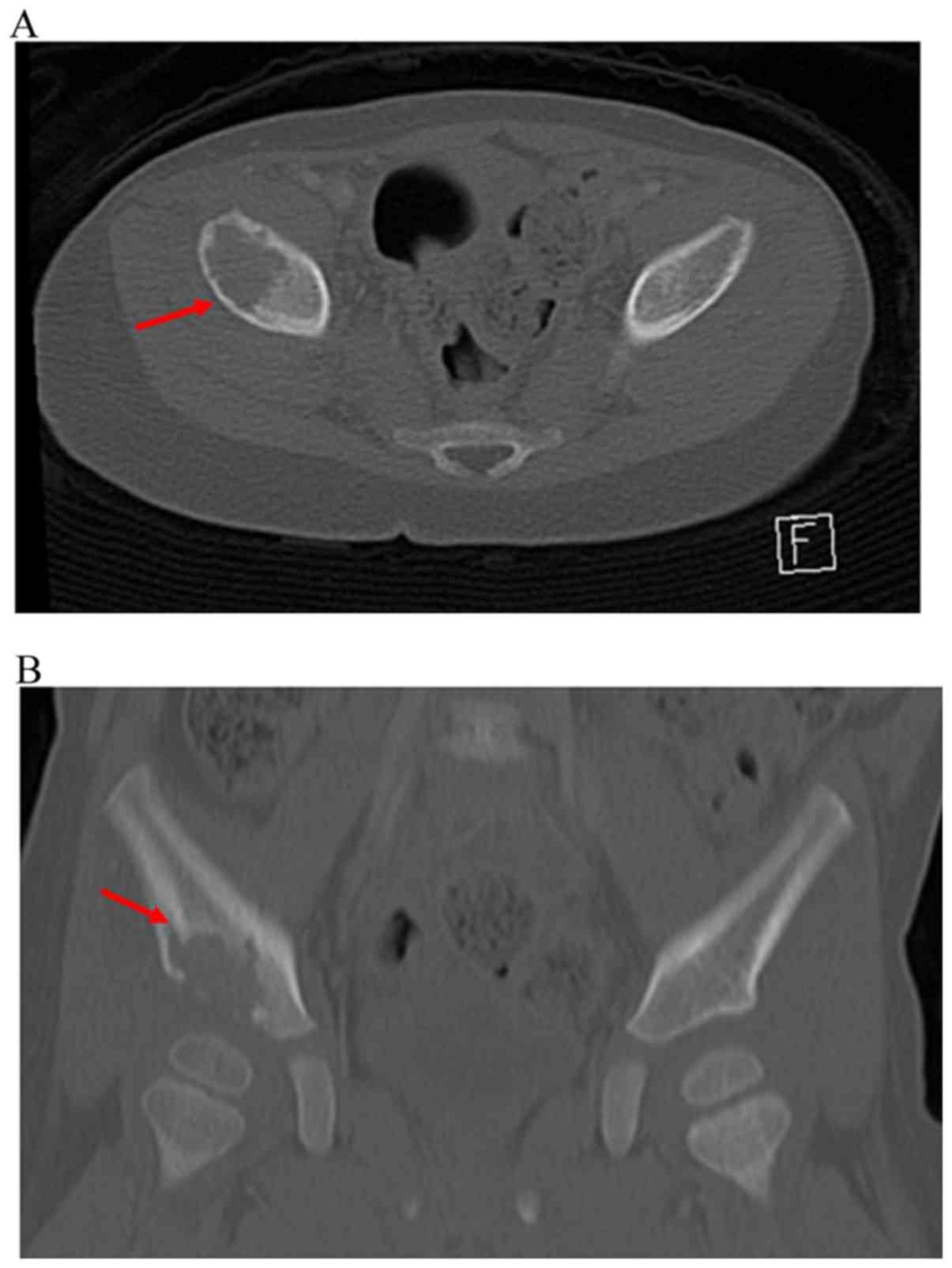
peri-acetabular region of his right ilium (Fig. 1). Computed tomography (CT) showed a
purely osteolytic lesion of the right ilium with slight
discontinuity of the thin cortical bone and minor cortical fracture
(Fig. 2). New bone formation was not
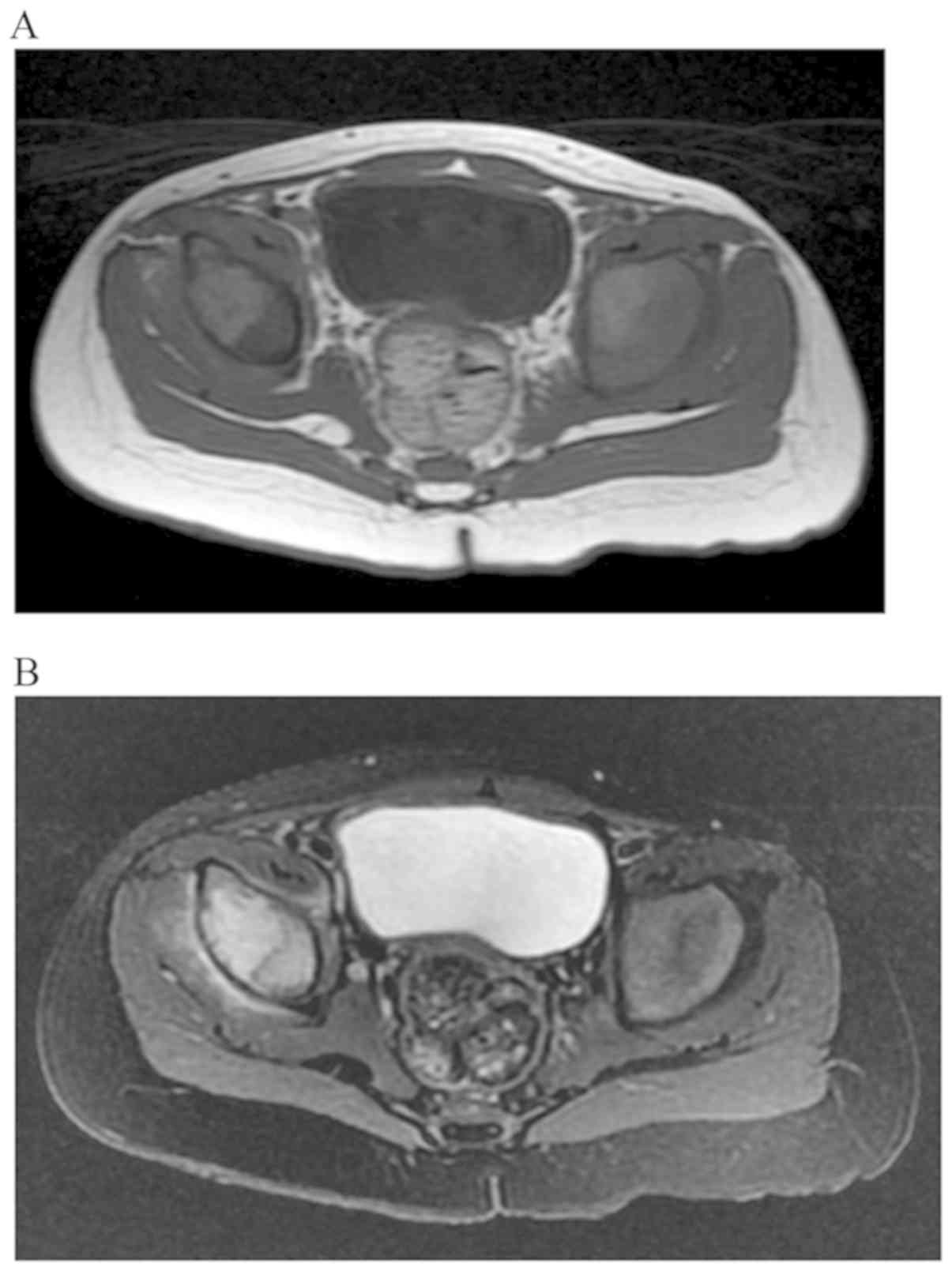
observed. Magnetic resonance imaging of the pelvis showed a
peri-acetabular lesion at the ilium with an iso- to high
heterogeneous signal intensity on T1-weighted imaging and a high
heterogeneous signal intensity on T2-weighted imaging (Fig. 3). Fluorodeoxyglucose positron
emission tomography (FDG-PET) showed an abnormal accumulation in
the right peri-acetabular lesion.
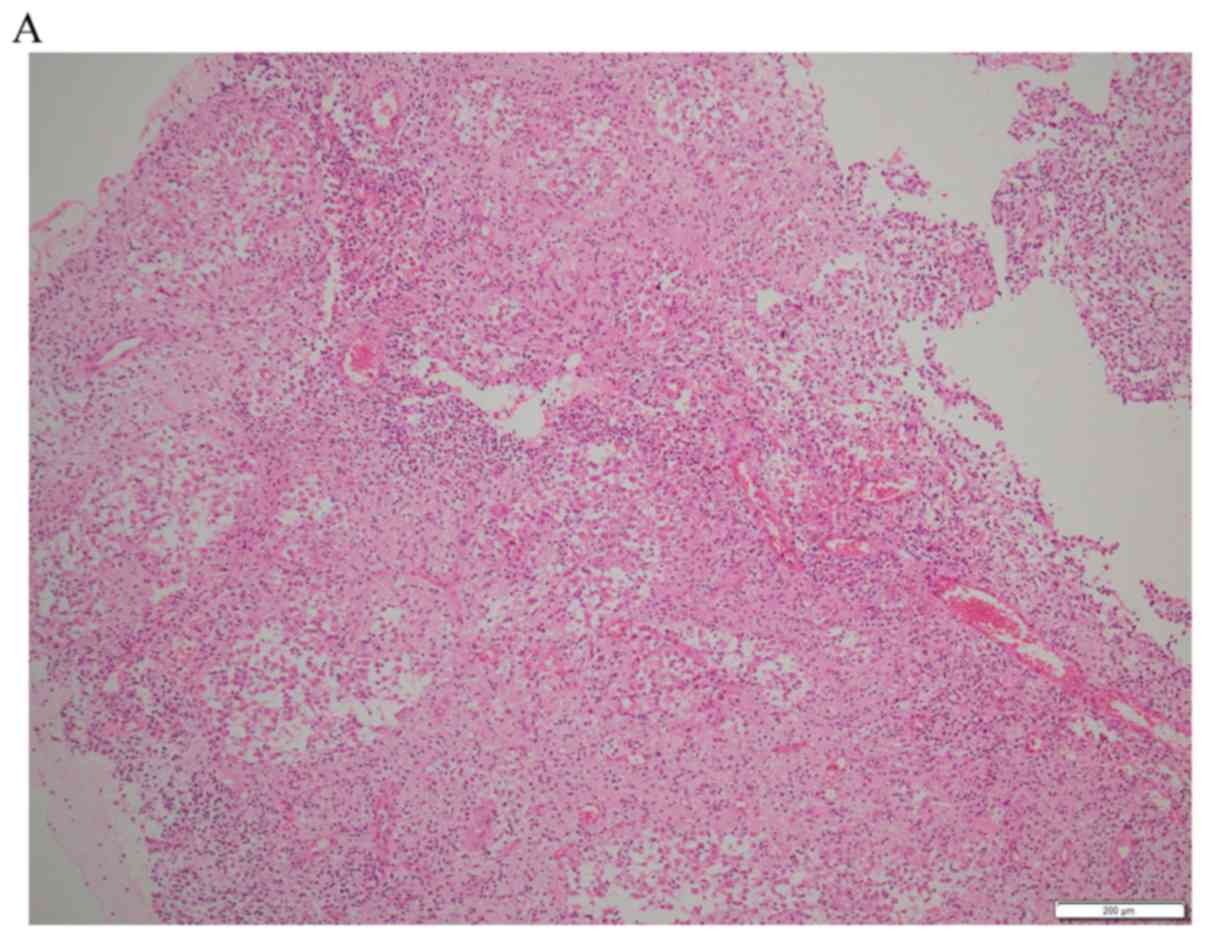
An incisional biopsy was performed to obtain a
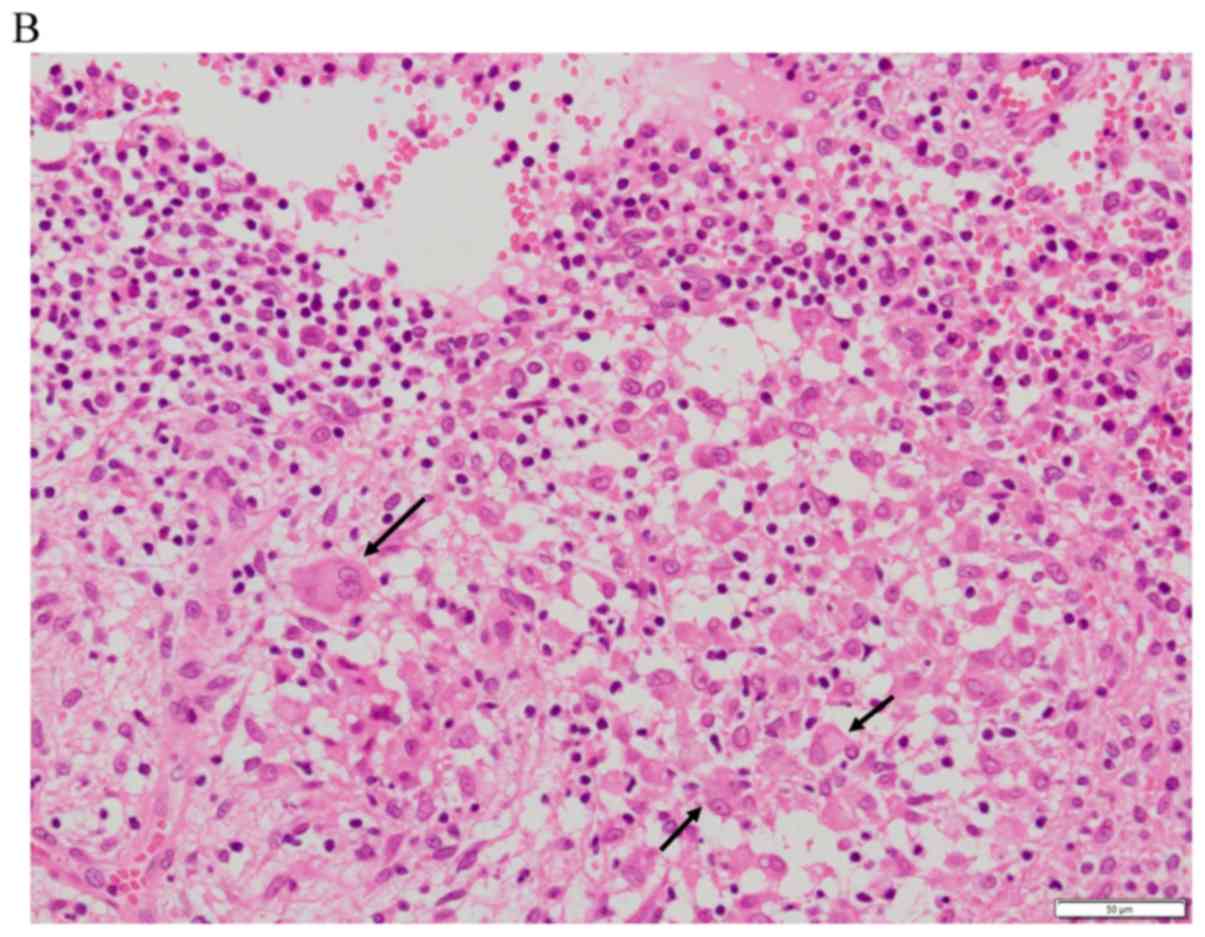
definite pathological diagnosis. Microscopy showed numerous large
histiocytes interspersed with different amounts of lymphocytes,
neutrophils, and plasma cells. Emperipolesis was observed in the
cytoplasm of the large histiocytes. Immunohistochemistry was
positive for CD68, CD163 and S-100 and negative for CD1a (Fig. 4). The patient was diagnosed with
primary RDD of bone in the ilium. Because the osteolytic lesion
gradually diminished without any progression of clinical symptoms
after the excisional biopsy, we carefully observed the patient
without additional treatment. After 18 months of follow-up, the
bone lesion on radiography and CT had disappeared completely, and
no joint deformity was observed (Fig.
5).
We are concerned about the risk of recurrence,
growth failure, and osteoarthritis. We are planning to follow him
until he reaches adulthood.
Discussion
RDD is a rare histiocytic disorder initially
described as a separate entity in 1969 by Rosai and Dorfman under
the term sinus histiocytosis with massive lymphadenopathy (1). The majority of patients are adolescents
and young adults, and the mean age at the onset is 20 years old.
The analysis of a registry of 423 worldwide cases of RDD showed
that the mean age of the onset was 20.6 years, with 58% of cases
occurring in men and 42% in women (2).
The pathological findings of RDD are characterized
by the proliferation of numerous large histiocytes containing
abundant eosinophilic cytoplasm, enmeshed in a variably cellular,
mixed inflammatory infiltration composed of plasma cells,
lymphocytes, neutrophils, foamy macrophages and rare eosinophils.
The characteristic feature of the large histiocytes in RDD is
conspicuous emperipolesis-namely lymphocytophagocytosis-with
intracytoplasmic lymphocytes, plasma cells, or neutrophils
(3,5). The histiocytes of RDD are
immunoreactive for CD68, CD163 and S100 protein and lack reactivity
for CD1a (3). This
immunohistochemical feature of RDD differentiates it from LCH, in
which the histiocytes are CD1a-positive and do not display
emperipolesis. ECD can be differentially diagnosed from RDD based
on the lack of emperipolesis, negative staining for S100 protein,
and a characteristic imaging appearance that includes diaphyseal
osteosclerosis of the long bone (4).
In the present case, microscopic findings showed
emperipolesis within the histiocyte cytoplasm. Immunohistochemistry
was positive for CD45, CD68, CD163, and lysozome and negative for
CD1a. The patient had no clinical or laboratory evidence to support
diagnoses of osteomyelitis, osteosarcoma, Ewing's sarcoma, or
metastatic bone tumor, such as metastatic neuroblastoma or
lymphoma. Thus, the diagnosis of primary bone RDD of the ilium was
made.
Extranodal disease occurs in the upper respiratory
tract, salivary glands, eyelids, and skin in approximately 28% of
cases. Bone involvement in association with nodal disease is seen
in <10% of cases (2). Mosheimer
et al (6) reviewed 108 RDD
patients with bone involvement and reported that primary RDD of the
bone was observed in 67 (74.4%) cases. Typical symptoms include
pain and swelling, but bone lesions may be an incidental finding.
Few reports have described the details of primary bone RDD
(4,7-31).
The largest series describes 15 cases of primary intraosseous RDD
(31).
The clinical information of 25 previously reported
cases of primary bone RDD are summarized in Table I. Of the 25 patients, 9 patients were
male (36%), and 16 were female (64%). The mean age was 28.7 (range:
1.5-60) years old. The two-year-old boy in the present case is the
second youngest patient to have a solitary bone lesion without
lymphadenopathy. Treatment in most patients consisted of curettage
(n=12) or resection (n=8). One patient underwent curettage followed
by radiotherapy. Four patients were managed conservatively, and
their condition was classified as stable disease. One patient (n=9)
received prednisone because his bone lesion was unresectable.
Paryani reported a patient (n=21) who underwent radiotherapy for
recurrent disease after curettage for the primary lesion. Overall,
the clinical outcome of primary intraosseous RDD is good. Curettage
and resection are effective for achieving local control.
 | Table IPrevious reports of primary bone
Rosai-Dorfman disease. |
Table I
Previous reports of primary bone
Rosai-Dorfman disease.
| No. | Author, year | Age, years | Sex | Site | Symptom | Treatment | Outcome (follow-up
duration) | (Refs.) |
|---|
| 1 | Hamels et al,
1985 | 1.5 | M | Radius | Pain, swelling | Curettage | NED (36 months) | (7) |
| 2 | Lewin et al,
1985 | 7 | M | Metacarpal | Pain for 1 year | Amputation, RT | NED (24 months) | (8) |
| 3 | Nawroz and
Wilson-Storey, 1989 | 11 | M | Radius | Pain, swelling | Curettage | NED (48 months) | (9) |
| 4 | Allegranza et
al, 1991 | 14 | F | Parietemporal
bone | Pain | Curettage | NED (17 months) | (10) |
| 5 | Kademani et
al, 2002 | 44 | F | Maxilla | Pain, swelling | Resection | NED (14 months) | (11) |
| 6 | George et al,
2003 | 41 | F | Radius | Pain | Curettage | NED (14 months) | (12) |
| 7 | Loh et al,
2004 | 57 | F | Triquetrum | Pain for 1 year | Curettage, RT | NED (12 months) | (13) |
| 8 | Mota Gamboa et
al, 2004 | 19 | F | Tibia | Swelling pain | Resection | NED (10 months) | (14) |
| 9 | Rodriguez-Galindo
et al, 2004 | 9 | F | Frontal bone | Pain | Curettage | SD (12 months) | (15) |
| 10 | Al-Saad et al,
2005 | 17 | M | T9 vertebra | Pain | Prednisolone | CR (7 weeks) | (16) |
| 11 | Miyake et al,
2005 | 38 | F | Femur | Pain | Observation | SD (6 months) | (17) |
| 12 | Sundaram et
al, 2005 | 60 | F | Femur, fibula | Pathological
fracture | Curettage | Femoral lesion:
NED, Fibular lesion: SD (30 months) | (18) |
| 13 | Tubbs et al,
2005 | 13 | M | Mastoid bone | Neck pain for 2
months | Mastoidectomy | Developed new
cervical lesion (4 months) | (19) |
| 14 | Robert et
al, 2006 | 23 | F | Sacrum | Pain | Embolization,
resection | NED (12
months) | (20) |
| 15 | Keskin et
al, 2007 | 29 | F | Maxilla | Pain/swelling for 2
year | Resection | NED (16
months) | (21) |
| 16 | Kang et al,
2011 | 25 | F | Right talus | Pain for 2
months | Curettage | NED (11
months) | (22) |
| 17 | Walczak et
al, 2011 | 50 | F | Right distal
femur | Pain for 7
months | Curettage | NED (22
months) | (23) |
| 18 | Hsu et al,
2011 | 16 | M | Right glenoid | Pain for 1
week | Observation | NED (9 months) | (24) |
| 19 | Tripathy et
al, 2012 | 52 | F | Carpal bone | Pain for 2
years | Observation | NED (9 months) | (25) |
| 20 | Dean et al,
2012 | 16 | M | Distal radius,
carpal Bones: Multifocal | Pain for 8
months | Observation | SD (6 months) | (26) |
| 21 | Paryani et
al, 2014 | 49 | F | Right distal
femur | Pain | Curettage, RT | Recurrence (1 year)
and SD for 15 months after RT | (27) |
| 22 | Kim et al,
2014 | 15 | M | T6 and T12 vertebra
body | Back pain for 6
months | Resection | NED (1 year)) | (28) |
| 23 | Xu et al,
2015 | 56 | F | Right proximal
tibia | Progressive pain
for 1 year | Curettage | NED (4 years) | (29) |
| 24 | Hartenstine et
al, 2016 | 38 | F | Right 9th rib | Back pain for 2
weeks | Excisional
biopsy | NED (15
months) | (30) |
| 25 | Baker et al,
2017 | 19 | M | Left distal
femur | Thigh pain lasting
several months | Curettage | NED (23
months) | (4) |
In the present case, the osteolytic lesion of the
RDD showed spontaneous remission without residual bone deformity
after curettage of the lesion at the incisional biopsy. However,
previous reports indicated various treatment strategies, including
corticosteroids, chemotherapy, radiotherapy, surgical curettage,
and resection. Among all RDD patients, 20% show spontaneous
remission without therapy (3).
Mosheimer et al (6) reported
that additional nodal manifestations of osseous RDD led to a more
systemic treatment approach. Mostly intensive treatment was related
to disease manifestations of problematic organs, including the
central nervous system, vessels, orbit, and nasal cavity. Demicco
et al (31) reported the
clinical course of RDD of bone. Of 12 patients that were available
for follow-up, 5 eventually developed additional extraosseous
manifestations, including testicular, lymph node, and subcutaneous
lesions. One patient developed additional multiple lesions of bone
without extraosseous disease. These additional lesions developed
from three months to three years after initial treatment. Thus, at
least three years of follow-up may be necessary to detect the
development of additional lesions.
RDD arising from bone has been reported in the
literature. However, primary bone RDD without lymphadenopathy is
extremely rare. Furthermore, the majority of patients are
adolescents and young adults, and the mean age at the onset is 20
years old. There was only one report describe a patient under five
years old. Before the biopsy, we suspected Langerhans cell
histiocytosis as the differential diagnosis. It is important to
consider primary RDD of bone as a differential diagnosis when
osteolytic lesions are observed, even if the patient is under five
years old.
Acknowledgements
The authors would like to thank Dr Oda Yoshinao,
Professor of the Department of Anatomic Pathology, Pathological
Sciences, Kyushu University (Fukuoka, Japan), for his advice
regarding pathological diagnosis.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Author's contributions
YIz, KS, HK, YO and AM examined clinical findings,
including laboratory data and radiographic images, and discussed
the rsults. YIm pathologically diagnosed the patient. HK provided
helpful advice due to their knowledge of bone diseases in
childhood. YIz drafted the manuscript. AM takes full responsibility
for the work as a whole, including the study design, access to data
and the decision to submit and publish the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Clinical information was obtained by reviewing
medical records. The study protocol was approved by the Ethics
Committee of University of Fukui.
Patient consent for publication
Written informed consent for the publication of
patient data/images was obtained from the patient's parents.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rosai J and Dorfman RF: Sinus
histiocytosis with massive lymphadenopathy: A newly recognized
benign clinicopathological entity. Arch Pathol. 87:63–70.
1969.PubMed/NCBI
|
|
2
|
Foucar E, Rosai J and Dorfman R: Sinus
histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease):
Review of the entity. Semin Diagn Pathol. 7:19–73. 1990.PubMed/NCBI
|
|
3
|
Dalia S, Sagatys E, Sokol L and Kubal T:
Rosai-Dorfman disease: Tumor biology, clinical features, pathology,
and treatment. Cancer Control. 21:322–327. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Baker JC, Kyriakos M, McDonald DJ and
Rubin DA: Primary Rosai-Dorfman disease of the femur. Skeletal
Radiol. 46:129–135. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fletcher CD, Bridge JA, Hogendoorn PW and
Mertens F (eds): WHO Classification of Tumours of the Soft Tissue
and Bone. IARCPress, Lyon, p468, 2013.
|
|
6
|
Mosheimer BA, Oppl B, Zandieh S, Fillitz
M, Keil F, Klaushofer K, Weiss G and Zwerina J: Bone involvement in
Rosai-Dorfman disease (RDD): A case report and systematic
literature review. Curr Rheumatol Rep. 19(29)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hamels J, Fiasse L and Thiery J: Atypical
lymphohistiocytic bone tumor (osseous variant of Rosai-Dorfman
disease?). Virchows Arch A Pathol Anat Histopatthol. 408:183–189.
1985.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lewin JR, Das SK, Blumenthal BI, D'Cruz C,
Patel RB and Howell GE: Osseous pseudotumor. Thesole manifestation
of sinus histiocytosis with massive lymphadenopathy. Am J Clin
Pathol. 84:547–550. 1985.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nawoz IM and Wilson-Storey D: Sinus
histiocytosis with massive lumphadenopathy (Rosai-Dorfman disease).
Histopathology. 14:91–99. 1989.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Allegranza A, Barbareschi M, Solero CL,
Fornari M and Lasio G: Primary lymphohistiocytic tumour of bone: A
primary osseous localization of Rosai-Dorfman disease.
Histpathology. 18:83–86. 1991.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kademani D, Patel SG, Prasad ML, Huvos AG
and Shah JP: Intraoral presentation of Rosai-Dorfman disease: A
case report and review of the literature. Oral Surg Oral Med Oral
Pathol Radiol Endod. 93:699–704. 2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
George J, Stacy G, Peabody T and Montag A:
Rosai-Dorfman disease manifesting as a solitary lesion of the
radius in a 41-year-old woman. Skeletal Radiol. 32:236–239.
2003.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Loh SY, Tan KB Wong YS and Lee YS:
Rosai-Dorfman disease of the triquetrum without lymphadenopathy. A
case report. J Bone Joint Surg Am. 86:595–598. 2004.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mota Gamboa JD, Caleiras E and Rosas-Uribe
A: Extranodal Rosai-Dorfman disease. Clinical and pathological
characteristics in a patient with a pseudotumor of bone. Pathol Res
Pract. 200:423–428. 2004.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Rodriguez-Galindo C, Helton KJ, Sánchez
ND, Rieman M, Jeng M and Wang W: Extranodal Rosai-Dorfman disease
in children. J Pediatr Hematol Oncol. 26:19–24. 2004.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Al-Saad K, Thorner P, Ngan BY, Gerstle JT,
Kulkarni AV, Babyn P, Grant RM, Read S, Laxer RM and Chan HS:
Extranodal Rosai-Dorfman disease with multifocal bone and epidural
involvement causing recurrent spinal cord compression. Pediatr Dev
Pathol. 8:593–598. 2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Miyake M, Tateisi U, Maeda T, Arai Y,
Sugimura K and Hasegawa T: Extranodal Rosai-Dorfman disease: A
solitary lesion with soft tissue reaction. Radiat Med. 23:439–442.
2005.PubMed/NCBI
|
|
18
|
Sundaram C, Uppin Shantveer G,
Chandrashekar P, Prasad VB and Umadevi M: Multifocal osseous
involvement as the sole manifestation of Rosai-Dorfman disease.
Skeletal Radiol. 34:658–664. 2005.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tubbs RS, Kelly DR, Mroczek-Musulman EC,
Hammers YA, Berkow RL, Oakes WJ and Grabb PA: Spinal cord
compression as a result of Rosai-Dorfman disease of the upper
cervical spine in a child. Childs Nerv Syst. 21:951–954.
2005.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Robert EG, Fallon KB and Tender GC:
Isolated Rosai-Dorfman disease of the sacrum. Case illustration. J
Neurosurg Spine. 4(425)2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Keskin A, Genç F and Günhan O:
Rosai-Dorfman disease involving maxilla: A case report. J Oral
Maxillofac Surg. 65:2563–2568. 2007.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kang RW, McGill KC, Lin J and Gitelis S:
Chronic Ankle pain and Swelling in a 25-year-old woman: An unusual
case. Clin Orthop Relat Res. 469:1517–1521. 2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Walczak BE, Halperin DM, Bdeir RW and
Irwin RB: Orthopaedic case of the month A 50-year-old woman with
persistent knee pain. Clin Orthop Relat Res. 469:3527–3532.
2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Hsu AR, Bhatia S, Kang RW, Arvanitis L,
Nicholson GP and Virkus WW: Extranodal Rosai-Dorfman disease
presenting as an isolated glenoid lesion in a high school athlete.
J Shoulder Elbow Surg. 21:e6–e11. 2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tripathy K, Misra A, Sahu AK and Patnaik
K: Extranodal Rosai-Dorfamn disease in a carpal bone. Indian J
Ortthop. 46:487–489. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Dean EM, Wittig JC, Vilalobos C and Garcia
RA: A 16-year-old boy with multifocal, painless osseous lesions.
Clin Orthop Relat Res. 470:2640–2645. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Paryani NN, Daugherty LC, O'Connor MI and
Jiang L: Extranodal Rosai-Dorfman disease of the bone treated with
surgery and radiotherapy. Rare Tumors. 6(5531)2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kim DY, Park JH, Shin DA, Yi S, Ha Y, Yoon
DH and Kim KN: Rosai-Dorfman disease in thoracic spine: A rare case
of compression fracture. Korean J Spine. 11:198–201.
2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Xu J, Liu CH, Wang YS and Chen CX:
Extranodal Rosai-Dorfaman disease as isolated lesion of the tibia
diagnosed by fine-needle aspiration cytology. Medicine (Baltimore).
94(e2038)2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hartenstine J, Jackson H and Mortman K: A
38-year-old woman with an osteolytic rib lesion. Chest.
149:e79–e85. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Demicco EG, Rosenberg AE, Bjornsson J,
Rybak LD, Unni KK and Nielsen GP: Primary Rosai-Dorfman disease of
bone: A clinicopathologic study of 15 cases. Am J Surg Pathol.
34:1324–1333. 2010.PubMed/NCBI View Article : Google Scholar
|