Introduction
Vascular smooth muscle cells (VSMCs) are the
predominant cells in arteries (1).
Excessive VSMC proliferation and neo-intima formation are critical
processes in the development of atherosclerosis and restenosis
after percutaneous balloon angioplasty (2). In addition to VSMC growth, proper
regulation of apoptosis of VSMCs within vessel walls is also
important for preventing plaque rupture in atherosclerotic vessels
(3). For these reasons, many studies
have focused on the regulation of VSMC growth and apoptosis by
mechanistic target of rapamycin complex 1 (mTORC1) (4,5). It is
well established that mTORC1 contributes to vascular pathologies
such as intimal hyperplasia; accordingly, the use of the mTORC1
inhibitor rapamycin in drug-eluting stents has achieved a profound
reduction in the incidence of restenosis (5).
Sestrin2 is a stress-inducible protein that
regulates cell growth and survival, and can participate in cellular
responses to stress conditions (6).
Sestrin2 suppresses mTORC1 signaling via activation of
AMPK-activated protein kinase (AMPK) or inhibition of Rag GTPases
(7,8). Through these functions, Sestrin2
attenuates oxidative stress, fat accumulation, and insulin
resistance, thereby attenuating various age- and obesity-related
metabolic diseases (6). Sestrin2 is
also induced by a range of mitochondrial dysfunctions and promotes
mitochondrial biogenesis and mitohormesis (9). In the liver, Sestrin2 expression is
induced upon chronic hypernutrition and acts as an important
suppressor of hepatosteatosis by inhibiting mTORC1(10). Despite a great deal of evidence
supporting the idea that Sestrin2 inhibits mTORC1 in the context of
metabolic pathologies, the effect of Sestrin2/mTORC1 on VSMC growth
has not been previously studied.
Melatonin was first described as an endogenous
hormone that regulates circadian and seasonal rhythms. Several
lines of evidence have revealed that melatonin has many other
functions, including roles as an antioxidant, immune enhancer,
promoter of mitochondrial homeostasis, and tumor suppressor
(11). Multiple mechanisms,
including cell cycle arrest and epithelial-to-mesenchymal
transition, are thought to mediate the biologic effects of
melatonin on growth and invasion of cancer (12). In addition, melatonin blocks
TGF-β1-induced fibroblast proliferation, which leads to alleviated
pulmonary fibrogenesis (13).
However, it remains unclear whether melatonin regulates VSMC growth
and proliferation. In this study, we investigated whether melatonin
modulates VSMC growth and, if so, whether Sestrin2/mTORC1 is
implicated in this effect.
Materials and methods
Cell culture
Rat aortic smooth muscle cells were isolated from
4-week-old male Sprague-Dawley rats (90 to 100 g). Cervical
dislocation euthanasia was performed by well-trained individuals.
Trimmed aortas was washed with sterilized cold phosphate-buffered
saline (PBS) and sliced into pieces measuring 1-3 mm2.
The pieces of aorta were attached to dishes and cultured in
low-glucose Dulbecco's modified Eagle's medium (Hyclone)
supplemented with 20% fetal bovine serum (FBS; Hyclone) for 2 weeks
at 37˚C in 5% CO2. The medium was replaced every day.
Cells were maintained in low-glucose Dulbecco's modified Eagle's
medium supplemented with 20% FBS. Cells at passage 4-9 were used in
all experiments. All protocols for animal use and euthanasia were
reviewed and approved by the Animal Care and Use Committee of
Kyunpook National University School of Medicine and conducted
according to our institutional guideline (KNU-2011-0096-1).
Western blot analysis
Cells were lysed in lysis buffer (20 mM Tris-HCl [pH
7.4], 5 mM EDTA [pH 8.0], 10 mM
Na4P2O7, 100 mM NaF, 2 mM
Na3VO4, 1% NP-40) containing aprotinin,
leupeptin, PMSF, and phosphatase inhibitors cocktail 3 (Sigma).
Protein samples were separated on 10% SDS-PAGE gels and transferred
to PVDF membranes. Membranes were blocked with 5% skim milk in
Tris-buffered saline containing 0.1% Tween 20 (TBST) and incubated
with primary antibody overnight at 4˚C. Primary antibodies against
the following proteins were obtained from the indicated suppliers:
C/EBPβ, Sestrin2, phospho-p70S6K (T389), p70S6K, p-Rb (S807/811)
were from Cell Signaling Technology; Rb was from Santa Cruz
Biotechnology; and β-actin (1:5,000) from Sigma.-Membranes were
washed three times with TBST and incubated with HRP-conjugated
anti-mouse (Santa Cruz Biotechnology) or anti-rabbit secondary
antibody (Cell Signaling Technology). HRP was detected using the
ECL reagent (BioNote).
Small interfering RNA (siRNA)
transfection
For gene silencing, cells were transfected with 50
nM scrambled siRNA, siSestrin2, or siCEBP/β (Bioneer,) using
Lipofectamine RNAiMAX (Invitrogen).
Transfection of expression
vectors
For overexpression of Sestrin2, cells were
transfected with a FLAG-Sestrin2 expressing vector (a gift from Dr.
Jun Hee Lee, University of Michigan) or a control vector using
TransIT-LT1 Transfection Reagent (Mirus Bio) according to the
manufacturer's instructions.
Cell counting
Primary VSMCs transfected with scrambled small
interfering RNA (siRNA) or siRNA targeting Sestrin2 (siSestrin2)
for 24 h were serum-starved for 24 h and incubated with 10% FBS
with or without 2 mM melatonin (Sigma). Cells were trypsinized,
stained with Trypan blue solution, and counted with a
hemocytometer.
Cell viability assay
Cells were seeded at 3-5x103 cells per
well in 96-well plates. The cells were serum-starved for 24 h and
incubated with 10% FBS with or without 2 mM melatonin for 3 days,
and cell viability was measured using a CCK8 Solution Reagent
(CK04; Dojindo). The absorbance of each well at 495 nm was measured
on a VERSA MAX ELISA reader (Molecular Devices). The proportion of
viable cells in each treatment group was normalized against that of
control wells.
Flow cytometric analysis
Cell cycle distribution of VSMCs after treatment
with 10% FBS with or without 2 mM melatonin was detected by flow
cytometry. VSMCs were collected and fixed in 70% ethanol at 4˚C for
30 min. Before analysis, cells were centrifuged at 2000 rpm for 5
min to remove ethanol. Then, cells were suspended in 500 µl PBS
containing propidium iodide (33 µg/ml) and Ribonuclease A (1 mg/ml)
in darkness at 37˚C for 30 min. Fluorescence emitted by PI-DNA
complexes was measured on an Epics XL flow cytometer (BD
Bioscience).
Measurement of oxygen consumption
rate
Oxygen consumption rate (OCR) was measured in
24-well plates using a Seahorse XF-24 analyzer (Seahorse
Bioscience). VSMCs were serum-starved for 24 h and incubated with
10% FBS with or without melatonin (2 mM). On the day before the
experiment, the sensor cartridge was placed in calibration buffer
supplied by Seahorse Bioscience and incubated at 37˚C in a
non-CO2 incubator for 1 h. Oligomycin (Sigma), carbonyl
cyanide 3-chlorophenylhydrazone (CCCP, Sigma), and rotenone (Sigma)
were added at the indicated times during OCR measurement.
Reverse transcription-quantitative
PCR
Total RNA was prepared using QIAzol lysis reagent
(Qiagen), and complementary DNA (cDNA) was synthesized from total
RNA using the RevertAid First Strand cDNA Synthesis kit (Thermo
Fisher Scientific, Inc.). The resultant cDNA was amplified on a
7500 Fast Real-Time PCR System (Applied Biosystems). Relative
expression levels were calculated using the ∆∆Cq method (14); the levels of each mRNA were
normalized against the corresponding level of 36B4 mRNA
primer, and the sequences were as follows: CEBPβ forward,
ACGAGCGGCTGCAGAAGA, reverse, GGCAGCTGCTTGAACAAGTTC; SESN2
forward, ACCTTTCGTGCCCAGGATTAT, reverse, GGGTAGAGCCGCTGGATCA;
36B4 forward, TTCCCACTGGCTGAAAAGGT, and reverse,
GCCGCAGCCGCAAA.
MitoSox
Mitochondrial reactive oxygen species (ROS)
generation was assessed using MitoSOX Red Mitochondrial Superoxide
indicator (Invitrogen). VSMCs transfected with 50 nM scrambled
siRNA or siSestrin2 for 24 h on a confocal dish were serum-starved
for 24 h and incubated with 10% FBS with or without 2 mM melatonin.
Cells were treated with 5 µM MitoSOX reagent working solution and
incubated for 10 min at 37˚C in the dark. The cells were then
washed gently three times with warm HBSS buffer. Finally, cells
were counterstained with NucBlue Live Cell Stain ReadyProbes
(Invitrogen) and mounted in warm buffer for imaging. MitoSOX
fluorescence intensity were quantified using Image J software.
Immunocytochemistry
Cells were pretreated with or without 2 mM melatonin
for 24 h and then treated with 1 mM H2O2 for
6 h. Cells were fixed with 4% paraformaldehyde (Biosesang) and
washed with PBS. Cells were permeabilized with 0.1% Triton X-100
for 15 min and washed with PBS. Following 1 h of blocking in 5%
normal goat serum (Vector Laboratories) in PBS, cells were
incubated with primary anti-cleaved caspase-3 (1:400; Cell
Signaling Technology) antibody overnight at 4˚C. After washing with
PBS, the cells were incubated with Alexa Fluor® 568 goat
anti-rabbit (1:100; Thermo Fisher Scientific Inc.) secondary
antibodies for 2 h at room temperature. Nuclei were stained with
DAPI (Vector Laboratories). Immnofluorescence intensity of cleaved
caspase-3 was quantified using Image J software.
Statistical analysis
All values are presented as means ± SEM. ANOVA was
used for comparisons between multiple groups, followed by Tukey's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Melatonin inhibits VSMC
proliferation
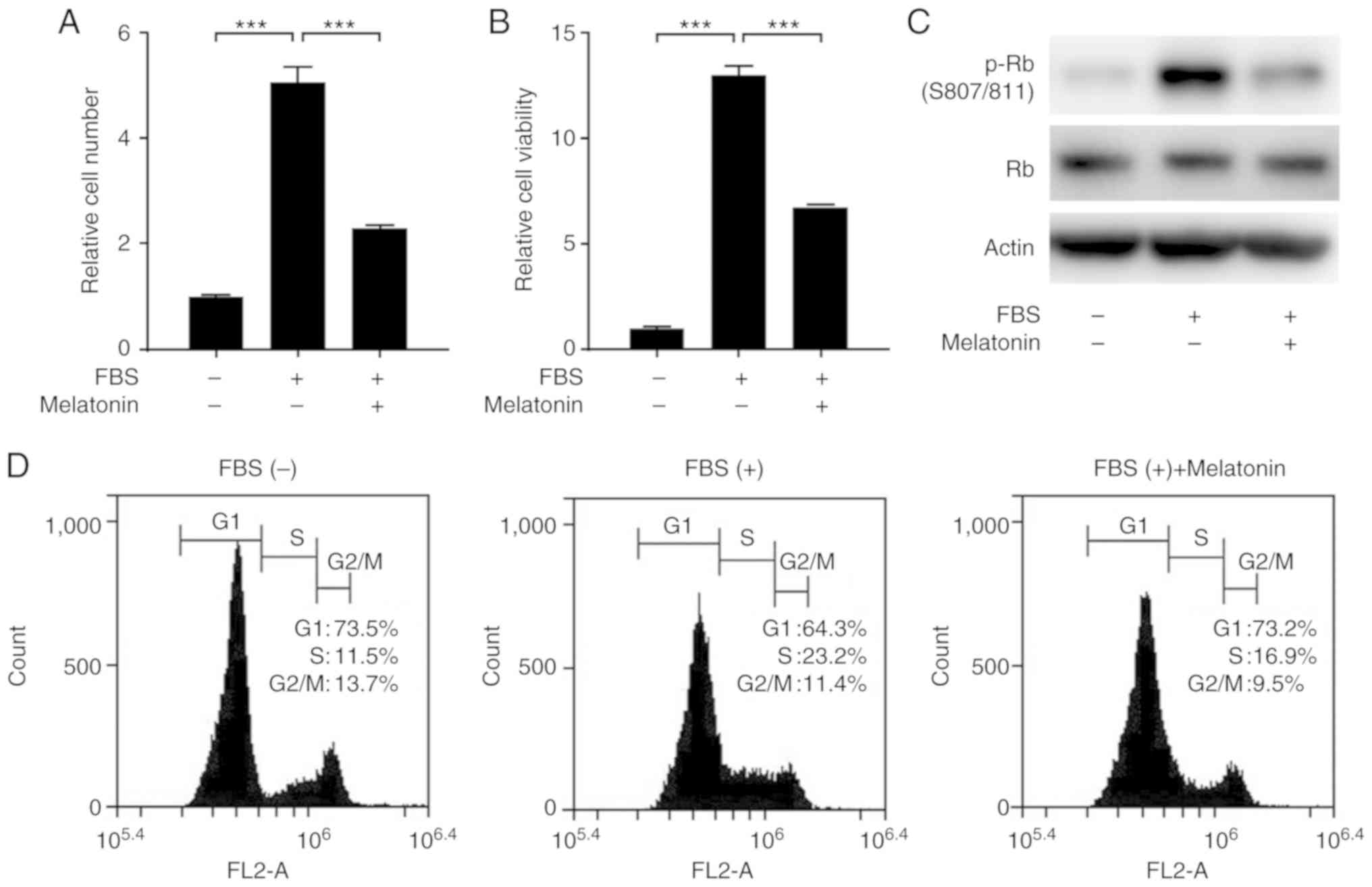
We first examined the effect of melatonin on
FBS-stimulated proliferation of VSMCs. Treatment of VSMCs with FBS
significantly increased the proliferation and viability of VSMCs,
but this effect was blocked by melatonin (Fig. 1A and B). Next, we explored whether melatonin
inhibits cell cycle progression in VSMCs. We found that melatonin
reduced the level of phosphorylated retinoblastoma protein (p-Rb)
(Fig. 1C). Flow cytometric analysis
of cell cycles showed that melatonin attenuated serum-stimulated
progression from G1 to S phase. In the melatonin-treated samples,
the cells accumulated in G1 phase (73.2% in melatonin-treated cells
vs. 64.3% in control cells) with a concomitant decrease in the
percentage of cells in S phase (16.9% in melatonin-treated cells
vs. 23.2% in control cells). Thus, melatonin arrests cells at the
G1 cell cycle phase, blocking proliferation (Fig. 1D).
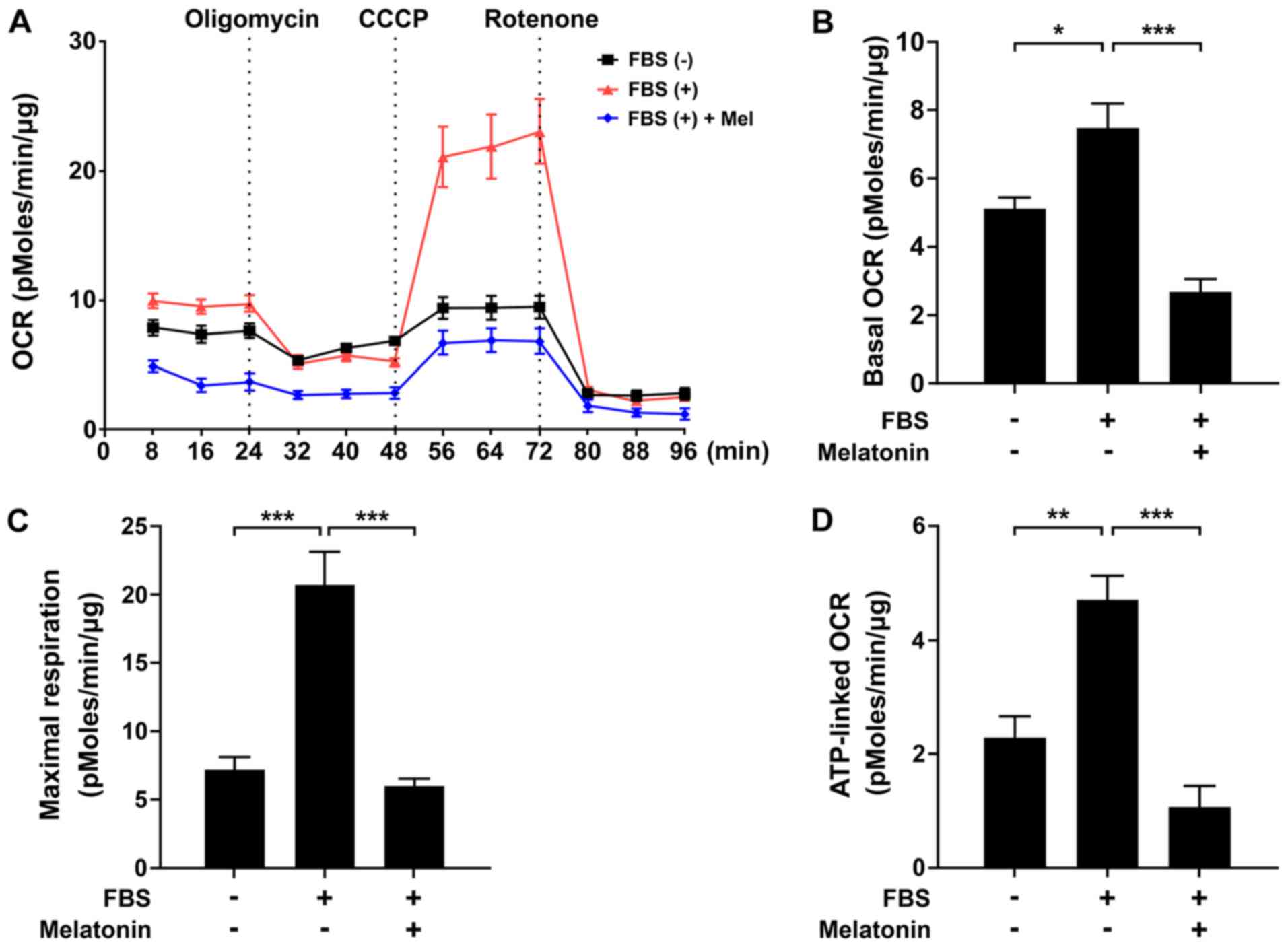
Melatonin induces mitochondrial
energetic stress and Sestrin2 expression in VSMCs
To determine the mechanism by which melatonin
inhibits VSMC proliferation, we evaluated the effect of melatonin
on mitochondrial function using an XF analyzer. We measured basal
OCR over time, and then observed the effects of the mitochondrial
inhibitors oligomycin, CCCP, and rotenone. Treatment of VSMCs with
melatonin caused a significant decrease in the major parameters of
mitochondrial function, including basal OCR and maximal
respiration, as well as ATP-linked respiration (Fig. 2A-D). After demonstrating that
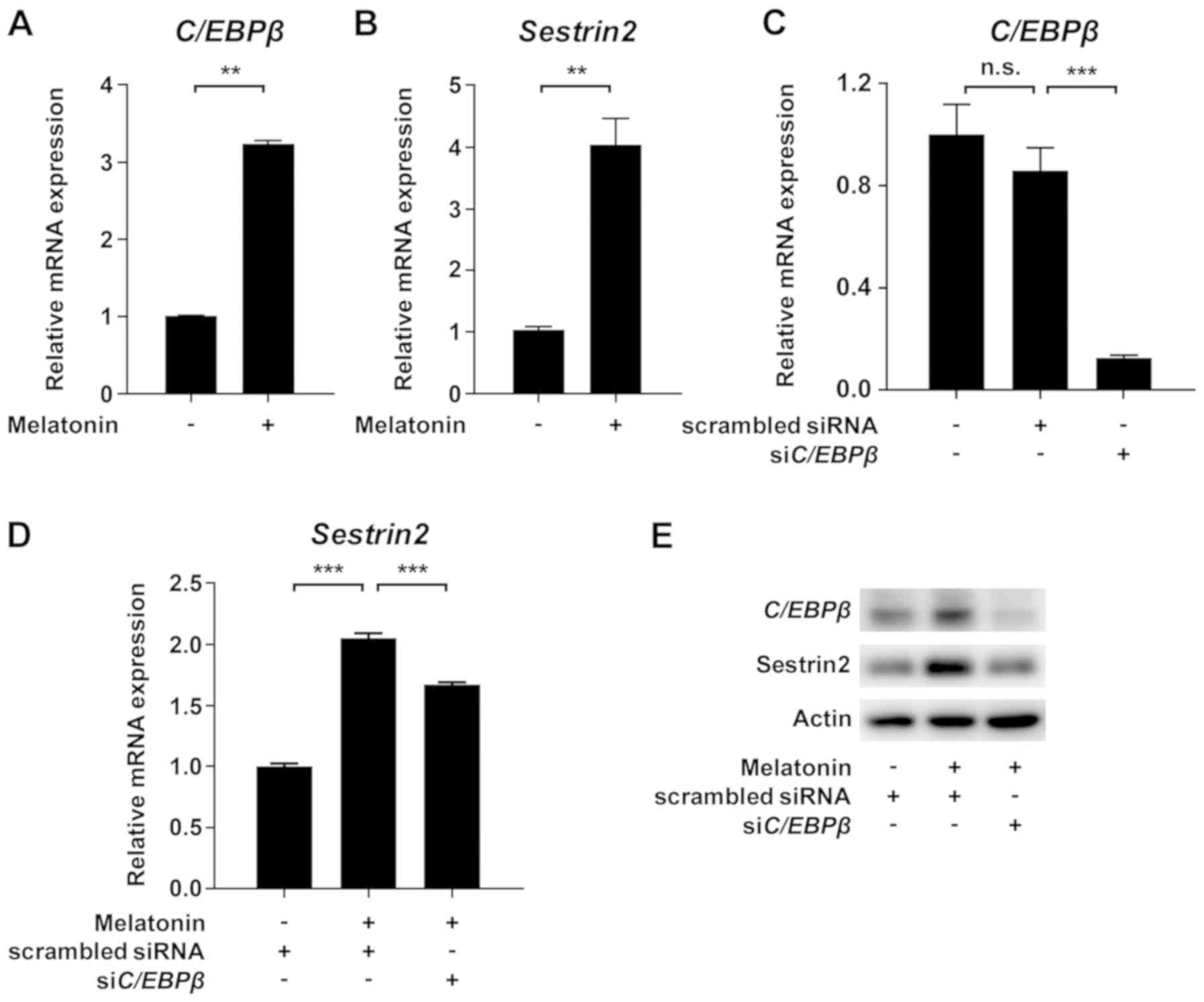
melatonin induced mitochondrial dysfunction, we evaluated
CCAAT/enhancer binding protein β (C/EBPβ) and Sestrin2, which
promote the response to mitochondrial stress (15,16). As
shown in Fig. 3A and B, treatment of VSMCs with melatonin
increased C/EBPβ and Sestrin2 protein levels. Furthermore,
siRNA-mediated knockdown of C/EBPβ inhibited melatonin-induced
upregulation of Sestrin2 mRNA and protein level (Fig. 3C-E). Taken together, these data
demonstrate that melatonin-induced mitochondrial energetic stress
results in upregulation of Sestrin2 via C/EBPβ.
Melatonin-induced Sestrin2 prevents
VSMC apoptosis
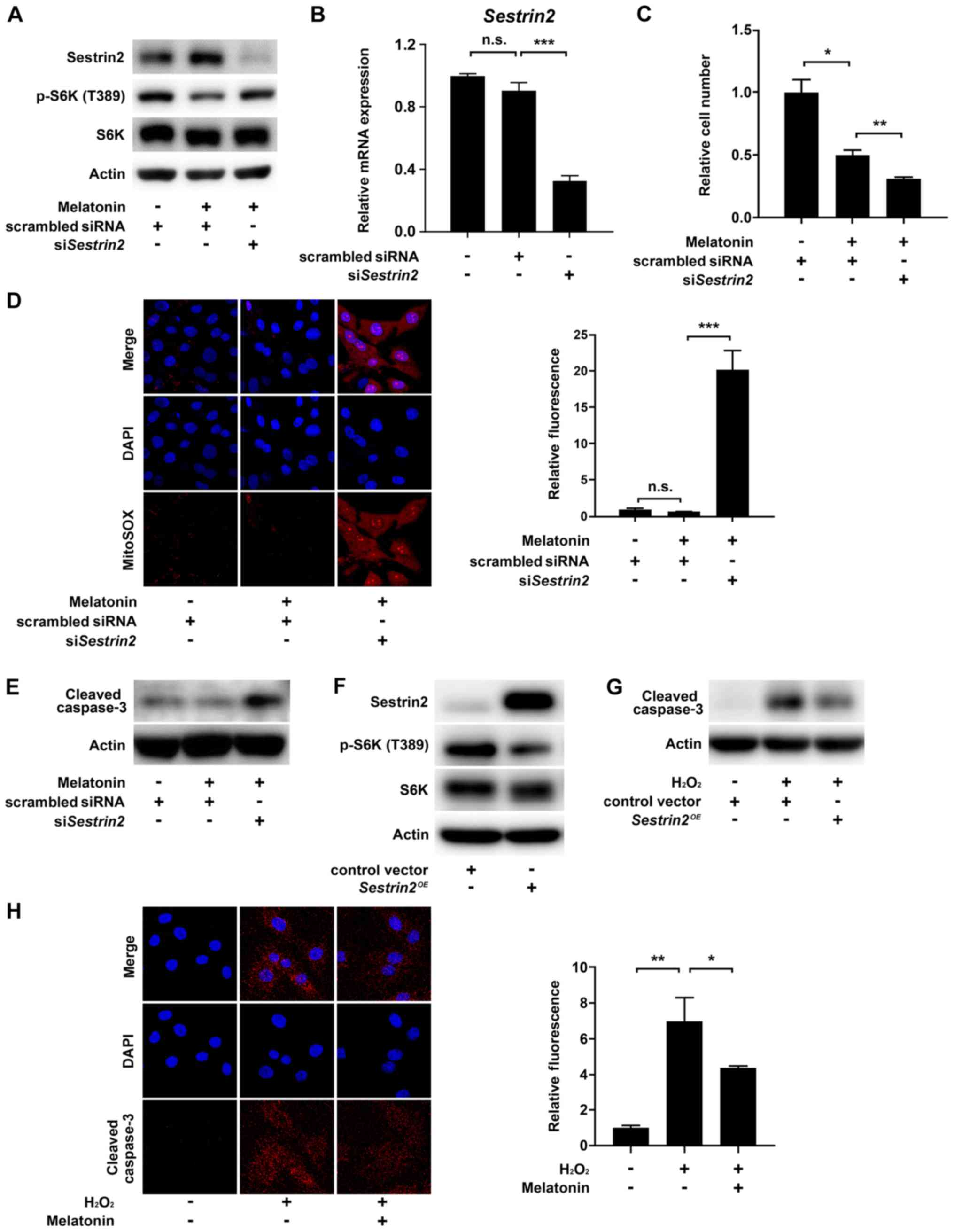
Next, we sought to confirm that Sestrin2 negatively
regulates mTORC1 activity in VSMCs, consistent with previous
results (7,8,17). As
mTORC1 controls cell growth via direct phosphorylation of Thr389 in
the hydrophobic motif site of p70S6 kinase 1 (S6K1), we determined
mTORC1 activity by measuring the levels of S6K (T389)
phosphorylation (18). The results
showed that melatonin increased Sestrin2 but decreased the levels
of S6K (T389) phosphorylation; this effect was abolished by
siRNA-mediated knockdown of Sestrin2 (siSESN2) in VSMCs (Fig. 4A). Successful knockdown of Sestrin2
was confirmed by quantitative PCR (Fig.
4B). Given that melatonin-induced Sestrin2 suppressed mTORC1
activity, we next asked whether inhibition of Sestrin2 would
restore melatonin-induced suppression of VSMC proliferation.
Unexpectedly, siSESN2 further decreased the number of VSMCs beyond
the reduction induced by melatonin treatment (Fig. 4C). Because Sestrin2 functions as a
ROS scavenger (19), we investigated
whether the siSESN2-induced decrease in cell number was caused by
cell death triggered by intracellular ROS production. Indeed,
siSESN2 significantly increased mitochondria-derived superoxide
production (stained by MitoSOX) and the level of cleaved caspase 3
in melatonin-treated VSMCs (Fig. 4D
and E). Moreover, Sestrin2
overexpression decreased mTORC1 activity in VSMCs and the level of
cleaved caspase 3 in H2O2-treated VSMCs,
further confirming the anti-proliferative and -apoptotic function
of Sestrin2 (Fig. 4F and G). Finally, immunofluorescence using a
cleaved caspase 3-specific antibody showed that melatonin reduced
H2O2-induced apoptosis of VSMCs (Fig. 4H). Collectively, these data indicated
that melatonin-induced Sestrin2 not only decreases VSMC
proliferation, but also blocks apoptosis of VSMCs by preventing
excessive ROS generation.
Discussion
The results of this study demonstrate that melatonin
induces mitochondrial energetic stress in VSMCs, leading to
induction of Sestrin2 via C/EBPβ. Melatonin-induced upregulation of
Sestrin2 suppressed mTORC1 activity in VSMCs, contributing to
suppression of VSMC proliferation. Furthermore, melatonin decreased
VSMC apoptosis through inhibition of ROS accumulation by
Sestrin2.
Although some previous studies reported that
melatonin improves mitochondrial respiratory function and enhances
ATP production, more recent studies showed that a supra-physiologic
dose of melatonin can be harmful to the kidney and other tissues
(11). Moreover, melatonin induces
mitochondrial depolarization and decreases oxidative
phosphorylation by inhibiting complex IV in some cancer cells
(20,21). In accordance with these findings, we
also showed that melatonin increased mitochondrial energetic stress
which was evident by decrease in basal OCR and maximal respiration,
as well as ATP-linked respiration. Consequently, mitochondrial
energetic stress by melatonin was responsible for induction of
Sestrin2 in VSMCs.
Sestrin2 is an important regulator of mitohormesis
in diverse tissues, including skeletal/cardiac muscle and brown
adipose tissue (9). Previous study
demonstrated that inhibitors of mitochondrial respiratory chain
complex I and III induce transcription of Sestrin2(22). Here, we showed that melatonin-induced
Sestrin2 upregulation is mediated by C/EBPβ, which is activated as
an aspect of mitochondrial retrograde signaling in response to
mitochondrial stress (23).
Consistent with previous studies reporting Sestrin2-mediated
inhibition of mTORC1 in various metabolic diseases, we also found
that melatonin plays a critical role in limiting VSMC proliferation
by the Sestrin2/mTORC1 axis.
It should be noted that knockdown of Sestrin2
promoted apoptosis in melatonin-treated VSMCs. Several lines of
evidence show that apoptosis of VSMCs in atherosclerosis is
sufficient to induce features of plaque vulnerability by thinning
the fibrous cap and enlarging the necrotic core, ultimately
resulting in plaque rupture (24).
Accordingly, decreasing VSMC apoptosis could retard plaque size and
instability, and thus has therapeutic potential (25). Sestrin2 promotes detoxification of
ROS in two ways: By recycling peroxiredoxin as a part of an
oxidoreductase enzyme, and by activating an antioxidant
transcriptional program by stabilizing Nrf2(6). In line with previous findings, we also
observed that melatonin-induced Sestrin2 prevents apoptosis of
VSMCs by limiting excessive ROS accumulation. Therefore, melatonin
represents a possible means to decrease both VSMC proliferation and
apoptosis. Although it will be necessary to perform animal
experiments to determine if these findings are valid in
vivo, collectively, our results support previous studies
showing that melatonin has anti-atherosclerotic effects in
preclinical models (26,27).
In summary, we showed here that melatonin suppresses
VSMC proliferation and apoptosis via Sestrin2-mediated inhibition
of mTORC1 and ROS scavenging, respectively. Given its ability to
control proliferation and apoptosis, melatonin represents a
potential lead compound for prevention of vessel lumen constriction
during the course of atherosclerosis and restenosis. Further
studies should investigate how melatonin causes mitochondrial
stress.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Research Foundation of Korea (NRF) grants funded by the Ministry of
Science and ICT (grant nos. NRF 2017M3A9G7073086 and
NRF-2018R1A2A1A05077703), an NRF grant funded by the Ministry of
Education (grant no. NRF-2017R1A6A3A04010231), and grants from the
Korea Health Technology R&D Project through the Korea Health
Industry Development Institute funded by the Ministry of Health and
Welfare, Republic of Korea (grant nos. HI16C1501 and
HI15C0001).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SHL and JKB conceptualized the study, designed the
research and performed the experiments. MP performed the
experiments. SWK, SWL, JGK, and IKL analyzed and interpreted the
data. YKC and KGP designed the experiments, analyzed and
interpreted the data, wrote and edited the manuscript, and
supervised the project. All authors reviewed the data and provided
feedback on the manuscript.
Ethics approval and consent to
participate
The present study was approved by The Animal Care
and Use Committee of Kyunpook National University School of
Medicine (approval no. KNU-2011-0096-1).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sehgel NL, Vatner SF and Meininger GA:
‘Smooth muscle cell stiffness syndrome’-Revisiting the structural
basis of arterial stiffness. Front Physiol. 6(335)2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Orford JL, Selwyn AP, Ganz P, Popma JJ and
Rogers C: The comparative pathobiology of atherosclerosis and
restenosis. Am J Cardiol. 86:6H–11H. 2000.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bennett MR, Sinha S and Owens GK: Vascular
smooth muscle cells in atherosclerosis. Circ Res. 118:692–702.
2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Rosner D, McCarthy N and Bennett M:
Rapamycin inhibits human in stent restenosis vascular smooth muscle
cells independently of pRB phosphorylation and p53. Cardiovasc Res.
66:601–610. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Martin KA, Rzucidlo EM, Merenick BL,
Fingar DC, Brown DJ, Wagner RJ and Powell RJ: The mTOR/p70 S6K1
pathway regulates vascular smooth muscle cell differentiation. Am J
Physiol Cell Physiol. 286:C507–C517. 2004.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lee JH, Budanov AV and Karin M: Sestrins
orchestrate cellular metabolism to attenuate aging. Cell Metab.
18:792–801. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Budanov AV and Karin M: p53 target genes
sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling.
Cell. 134:451–460. 2008.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Peng M, Yin N and Li MO: Sestrins function
as guanine nucleotide dissociation inhibitors for Rag GTPases to
control mTORC1 signaling. Cell. 159:122–133. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ro SH, Semple I, Ho A, Park HW and Lee JH:
Sestrin2, a regulator of thermogenesis and Mitohormesis in brown
adipose tissue. Front Endocrinol (Lausanne). 6(114)2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Lee JH, Budanov AV, Talukdar S, Park EJ,
Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, et al:
Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell
Metab. 16:311–321. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Luchetti F, Canonico B, Betti M,
Arcangeletti M, Pilolli F, Piroddi M, Canesi L, Papa S and Galli F:
Melatonin signaling and cell protection function. FASEB J.
24:3603–3624. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Favero G, Moretti E, Bonomini F, Reiter
RJ, Rodella LF and Rezzani R: Promising antineoplastic actions of
melatonin. Front Pharmacol. 9(1086)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhao X, Sun J, Su W, Shan H, Zhang B, Wang
Y, Shabanova A, Shan H and Liang H: Melatonin protects against lung
fibrosis by regulating the Hippo/YAP pathway. Int J Mol Sci.
19(pii: E1118)2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Seo K, Ki SH and Shin SM: Sestrin2-AMPK
activation protects mitochondrial function against glucose
deprivation-induced cytotoxicity. Cell Signal. 27:1533–1543.
2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhao Q, Wang J, Levichkin IV,
Stasinopoulos S, Ryan MT and Hoogenraad NJ: A mitochondrial
specific stress response in mammalian cells. EMBO J. 21:4411–4419.
2002.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Parmigiani A, Nourbakhsh A, Ding B, Wang
W, Kim YC, Akopiants K, Guan KL, Karin M and Budanov AV: Sestrins
inhibit mTORC1 kinase activation through the GATOR complex. Cell
Rep. 9:1281–1291. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Saxton RA and Sabatini DM: mTOR Signaling
in Growth, metabolism and disease. Cell. 168:960–976.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Budanov AV, Sablina AA, Feinstein E,
Koonin EV and Chumakov PM: Regeneration of peroxiredoxins by
p53-regulated sestrins, homologs of bacterial AhpD. Science.
304:596–600. 2004.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Loureiro R, Magalhaes-Novais S, Mesquita
KA, Baldeiras I, Sousa IS, Tavares LC, Barbosa IA, Oliveira PJ and
Vega-Naredo I: Melatonin antiproliferative effects require active
mitochondrial function in embryonal carcinoma cells. Oncotarget.
6:17081–17096. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sarti P, Magnifico MC, Altieri F,
Mastronicola D and Arese M: New evidence for cross talk between
melatonin and mitochondria mediated by a circadian-compatible
interaction with nitric oxide. Int J Mol Sci. 14:11259–11276.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Garaeva AA, Kovaleva IE, Chumakov PM and
Evstafieva AG: Mitochondrial dysfunction induces SESN2 gene
expression through activating transcription Factor 4. Cell Cycle.
15:64–71. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Biswas G, Guha M and Avadhani NG:
Mitochondria-to-nucleus stress signaling in mammalian cells: Nature
of nuclear gene targets, transcription regulation, and induced
resistance to apoptosis. Gene. 354:132–139. 2005.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Clarke MC, Figg N, Maguire JJ, Davenport
AP, Goddard M, Littlewood TD and Bennett MR: Apoptosis of vascular
smooth muscle cells induces features of plaque vulnerability in
atherosclerosis. Nat Med. 12:1075–1080. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
25
|
Lyon CA, Johnson JL, Williams H,
Sala-Newby GB and George SJ: Soluble N-cadherin overexpression
reduces features of atherosclerotic plaque instability.
Arterioscler Thromb Vasc Biol. 29:195–201. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Cheng X, Wan Y, Xu Y, Zhou Q, Wang Y and
Zhu H: Melatonin alleviates myosin light chain kinase expression
and activity via the mitogen-activated protein kinase pathway
during atherosclerosis in rabbits. Mol Med Rep. 11:99–104.
2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hu ZP, Fang XL, Fang N, Wang XB, Qian HY,
Cao Z, Cheng Y, Wang BN and Wang Y: Melatonin ameliorates vascular
endothelial dysfunction, inflammation, and atherosclerosis by
suppressing the TLR4/NF-κB system in high-fat-fed rabbits. J Pineal
Res. 55:388–398. 2013.PubMed/NCBI View Article : Google Scholar
|