Introduction
Chronic obstructive pulmonary disease (COPD) is
predicted to be the fourth leading cause of death worldwide by
2030, accounting for 5% of global mortalities (1-3).
The disease is characterized by an imbalance in proinflammatory and
anti-inflammatory factors, airway remodelling and an increased
production of reactive oxygen species (ROS). Cigarette smoke, the
primary cause of COPD, contains >7,000 chemicals that cause
bronchial epithelium damage, inflammatory cells infiltration and
tissue remodelling (4,5). Furthermore, a previous study revealed
that prolonged exposure to cigarette smoke resulted in irreversible
epithelial cell damage that increased the production of
pro-inflammatory chemokines and ROS, and induced apoptosis
(6). The increased production of the
pro-inflammatory chemokines interleukin (IL)-8 and tumor necrosis
factor α (TNF-α), the remodelling biomarkers matrix
metallopeptidase 9 (MMP9) and tissue inhibitor of metalloproteinase
1 (TIMP1), and ROS have been associated with the severity of COPD
(7-9).
Glucocorticoids (GCs) are used to treat numerous
chronic inflammatory diseases, such as asthma (10,11),
rheumatoid arthritis (12) and
inflammatory bowel disease (13-15).
GCs reverse histone acetylation by binding with glucocorticoid
receptors (GR) and recruiting histone deacetylase 2 (HDAC2) to the
activated transcription complex. At higher GC concentrations, the
GC-GR complex acts on the DNA recognition site to facilitate gene
transcription by enhancing histone acetylation (16). However, decreased GC sensitivity has
been reported in patients with COPD (17-21)
and in vitro and in vivo studies have provided
evidence for GC resistance in COPD (20-23).
Reversing GC resistance in COPD remains a clinical challenge and
novel therapeutic agents are required.
In the clinical treatment of COPD with traditional
Chinese medicine, several patients have noted symptom improvement
following administration of Epimedium brevicornum Maxim, the active
ingredient of which is icariin (24,25).
Icariin has been shown to exert anti-remodelling, anticancer and
cardiovascular protective effects, as well as to promote bone
formation (26-29).
Additionally, icariin exhibited anti-inflammatory and antioxidant
effects in cigarette smoke-induced inflammatory models in
vivo and in vitro, possibly by suppressing NF-κB
activation and altering the production of nitrous oxide, nitric
oxide synthase, superoxide dismutase and T lymphocytes (30-32).
The present study hypothesized that icariin could
alleviate cell injury in human bronchial epithelial cells (BEAS-2B)
exposed to cigarette smoke extract (CSE) by modulating
remodelling-related factors and restoring the balance of
pro-inflammatory and anti-inflammatory factors. Furthermore, the
present study investigated whether icariin reversed GC
resistance.
Materials and methods
BEAS-2B cell culture
Human BEAS-2B cells (American Type Tissue
Collection) were plated in six-well plates at a density of
1.5-2x106 cells/well, cultured in complete BEAS-2B
culture medium (BEBM) and incubated at 37˚C, 5% CO2.
Complete BEBM was formulated by supplementing BEBM basal medium
(Lonza Group Ltd.) with a BEGM SingleQuots kit (Lonza Group Ltd.),
which consisted of bovine pituitary extract, hydrocortisone, human
epidermal growth factor, epinephrine, transferrin, insulin,
retinoic acid, triiodothyronine, gentamicin and amphotericin-B.
CSE preparation and BEAS-2B cell
exposure
An unfiltered cigarette was combusted with the use
of a peristaltic pump. Cigarette smoke was slowly bubbled into 5 ml
complete BEBM from the start of the ignition to the end of the
cigarette burnout, which took ~5 min. The resulting medium was
adjusted to pH 7.4, sterile-filtered through a 0.22-µm Millex
filter (EMD Millipore) and defined as 100% CSE. BEAS-2B cells were
pre-treated with 20, 40 and 80 µM icariin (Shanghai Ronghe
Corporation) and 10 µM dexamethasone (DEX; Sigma-Aldrich, Merck
KGaA), or vehicle control for 24 h prior to exposure to 5% CSE for
4 h at 37˚C, 5% CO2 (32). The vehicle used was DMSO
(Sigma-Aldrich; Merck KGaA). The applied concentration of DMSO in
both control and treated groups was 1.6 µl/ml, lower than the
cytotoxic concentration (33). The
morphological changes to BEAS-2B cells were visualised using a
Zeiss AxioVert A1 fluorescence microscope (Carl Zeiss AG) at low
power (x10 magnification).
Cell proliferation assay
A total of 100 µl BEAS-2B cell suspension
(1x104 cells/ml) was plated per well in a 96-well plate
and cultured in complete BEBM overnight at 37˚C, 5% CO2.
Cell proliferation was determined using the Cell Counting Kit-8
(CCK-8; Dojindo Molecular Technologies, Inc.) according to the
manufacturer's protocol. The absorbance was measured at 450 nm
using a plate reader (TECAN Infinite 200 PRO; Tecan Group,
Ltd.).
Western blotting
Cellular proteins were extracted from the BEAS-2B
cells using a nuclear and cytoplasmic protein extraction kit
(Beyotime Institute of Biotechnology) according to the
manufacturer's protocol and phenylmethylsulfonyl fluoride. Total
protein concentration was determined using an enhanced
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology). Western blotting was subsequently performed as
previously described (34). The
following primary antibodies were used in the present study:
Anti-GR (Santa Cruz Biotechnology, Inc.; cat. no. sc-393232;
1:1,000), anti-HDAC2 (Cell Signaling Technology, Inc.; cat. no.
2540; 1:2,000), anti-NF-κB (Cell Signaling Technology, Inc.; cat.
no. 4764; 1:1,000) and anti-GAPDH (Cell Signaling Technology, Inc.;
cat. no. 5174; 1:5,000). Following primary antibody incubation, the
membranes were incubated with a horseradish peroxidase-labelled
secondary antibody (Santa Cruz Biotechnology, Inc.; cat. no. 2357;
1:50,000). Band intensities were quantified using ImageJ analysis
software (Version: 1.52t; National Institutes of Health), with
GAPDH as the loading control. All western blot experiments were
performed in triplicates.
ELISA analysis of IL-8, IL-10, TNF-α,
MMP9 and TIMP1 concentrations in BEAS-2B cell culture medium
The concentrations of IL-8 (Boatman Biotech, Co.,
Ltd.; cat. no. ETA05989), IL-10 (Shanghai ExCell Biology, Inc.;
cat. no. EM005), TNF-α (Shanghai ExCell Biology, Inc; cat. no.
EM008), MMP9 (R&D Systems, Inc.; cat. no. MMPT90), TIMP1
(R&D Systems, Inc.; cat. no. MTM100) in BEAS-2B cell culture
medium were quantified using ELISA kits (names, catalogue number
and supplier listed previously) according to the manufacturer's
protocol.
Immunofluorescence staining
The expression of nuclear factor erythroid-2-related
factor 2 (Nrf2) in BEAS-2B cells was analysed using
immunofluorescence staining. Complete BEBM was removed and the
cells were washed using phosphate-buffered saline (PBS). The cells
were subsequently fixed using ice cold 4% paraformaldehyde for 15
min at room temperature. The cells were washed 3 times in PBS for 5
min each. The cells were then blocked using Blocking Buffer (1x
PBS/5% normal goat serum; cat. no. ab7481; Abcam)/0.3% Triton X-100
(cat. no. T8787; Sigma Aldrich, Merck KGaA;) for 60 min at room
temperature. The cells were washed again and incubated with a
primary antibody against Nrf2 (Sigma-Aldrich, Merck KGaA; cat. no.
SAB4501984) overnight at 4˚C. The following day, the cells were
washed 4 times, 10 min each time, with PBS and incubated with a
donkey-anti-rabbit secondary antibody (Thermo Fisher Scientific,
Inc.; cat. no. A-21207; 1:200) for 1.5 h at room temperature. The
cells were subsequently counterstained with the nuclear stain DAPI
(Sigma-Aldrich, Merck KGaA; 0.6 mM in PBS) for 4 min at room
temperature. Stained cells were imaged using a Zeiss AxioVert A1
fluorescence microscope (Carl Zeiss AG; x40 magnification). A total
of 6 fields were analysed.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from BEAS-2B cells using
TRIzol™ reagent (Takara Bio, Inc.) following the manufacturer's
protocol. Total RNA was reverse transcribed into cDNA using the
iScriptDNA Synthesis kit (Bio-Rad Laboratories, Inc.; cat. no.
1708891) using the following temperature protocol: Priming at 25˚C
for 5 min, RT at 46˚C for 20 min, reverse transcriptase
inactivation at 95˚C for 1 min, and then holding at 4˚C. A total of
1 ml cDNA was subjected to qPCR using Power SYBR Green PCR Master
mix (Applied Biosystems, Thermo Fisher Scientific, Inc.; cat. no.
A25742) and the ABI 6500 fast Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling
conditions of the qPCR steps were as follows: Activation
(temperature: 50˚C; duration: 2 min; cycles: hold), Dual-LockTM DNA
polymerase (temperature: 95˚C; duration: 2 min; cycles: hold),
denaturation (temperature: 95˚C; duration: 15 sec; cycles: 40) and
annealing/extension (temperature: 60˚C; duration: 1 min; cycles:
40). The following primers were used for qPCR: GR forward,
5'-GGACCACCTCCCAAACTCTG-3' and reverse, 5'-GCTGTCCTTCCACTGCTCTT-3';
HDAC2 forward, 5'-CCATGGCGTACAGTCAAGGA3-' and reverse,
5'-TCATTTCTTCGGCAGTGGCT-3'; GAPDH forward,
5'-AGAAGGCTGGGGCTCATTTG-3' and reverse, 5'-AGGGGCCATCCACAGTCTTC-3';
NF-κB forward, 5'-CTGTCCTTTCTCATCCCATCTT-3' and reverse,
5'-TCCTCTTTCTGCACCTTGTC-3'; and IL-8 forward,
5'-TGGATTTCCCCCTTGCAACC-3' and reverse, 5'AAATCCTGACTGGGTCGCTG3'.
The relative expression level of each gene was determined against
the GAPDH level in the same sample. The fold-change of the target
genes was calculated by using the 2-∆∆Cq method
(35).
Cellular ROS analysis
ROS levels in BEAS-2B cell were detected using a
human intracellular ROS assay kit (Nanjing Jiancheng Bioengineering
Institute, Co., Ltd.) according to manufacturer's instructions.
Flow cytometry was also used to detect the levels of ROS in BEAS-2B
cells. BEAS-2B cells (0.5-1x106 cells/sample) were
incubated with dihydrogenrhodamine 123 (Sigma-Aldrich, Merck KGaA)
for 1 h at 37˚C and the fluorescence intensity of cellular oxidized
rhodamine 123 was detected using a flow cytometer (Attune NxT;
Thermo Fisher Scientific, Inc.) and analysed using using Attune NxT
Software (version 2.6; Thermo Fisher Scientific, Inc.).
Statistical analysis
Data are presented as the mean ± standard
deviation). All experiments were performed in duplicates at least
three separate times. The western blotting and immunofluorescence
staining experiments were performed three times. Statistical
analyses were performed using GraphPad Prism software (version
6.02; GraphPad Software, Inc.). The one-way ANOVA followed by the
Tukey's post hoc test was used to analyze the differences among
multiple groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Icariin increases the proliferation of
BEAS-2B cells exposed to CSE
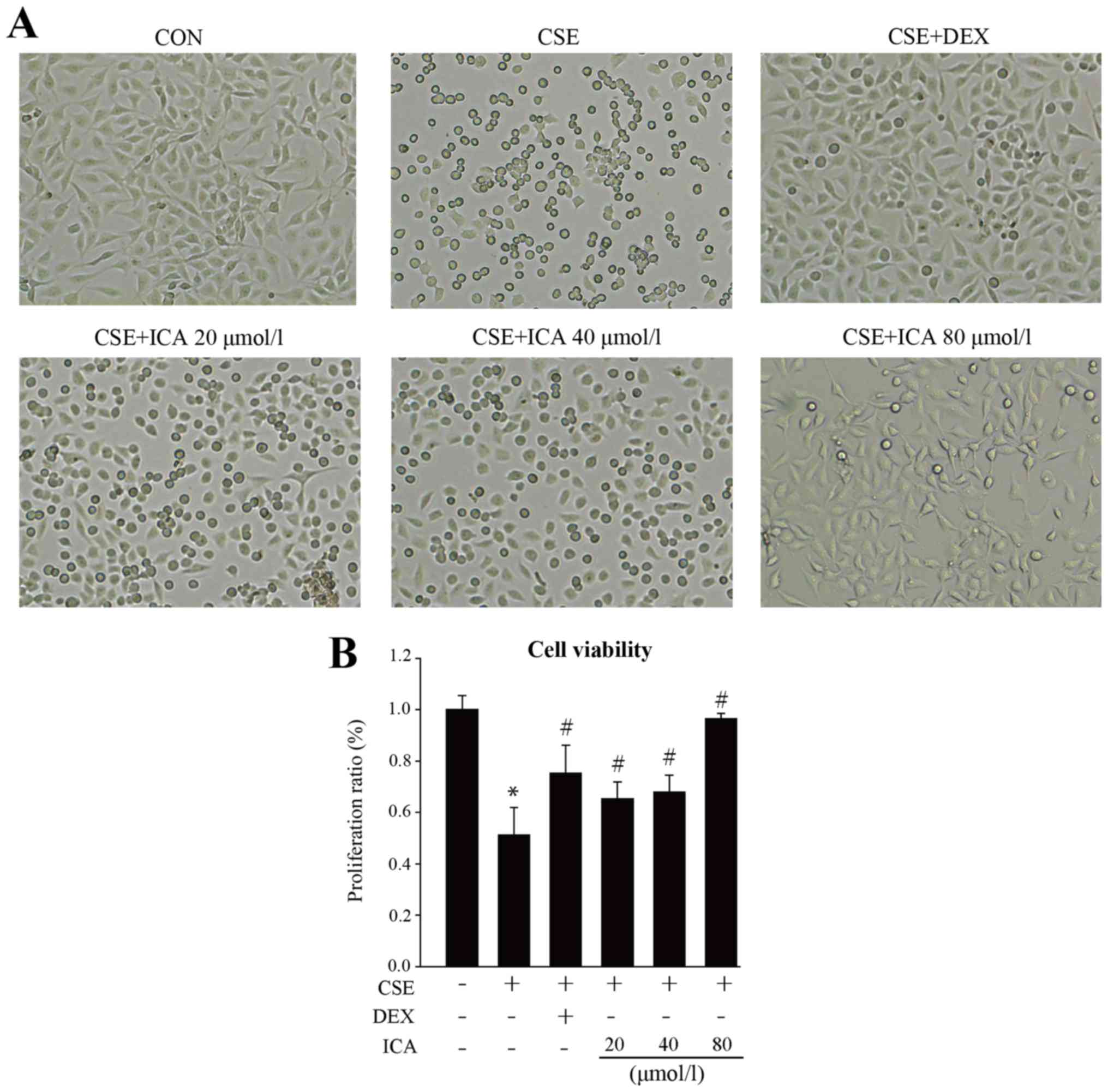
BEAS-2B cells exposed to CSE for 4 h exhibited
morphological changes, including retracted processes and flattening
and enlargement of cell bodies. These morphological changes were
less prominent in BEAS-2B cells treated with 20, 40 and 80 µM
icariin. The CCK-8 assay was used to determine the effect of
icariin on the proliferation of BEAS-2B cells exposed to CSE. The
proliferation of CSE-exposed BEAS-2B cells was significantly
reduced compared with the control group. Following treatment with
icariin, the proliferation of BEAS-2B cells increased in a
dose-dependent manner. Furthermore, cells treated with 80 µmol/l
icariin exhibited increased proliferation beyond the positive
control group (DEX; Fig. 1A-B).
Icariin balances the secretion of
pro-inflammatory and anti-inflammatory cytokines in BEAS-2B cells
exposed to CSE
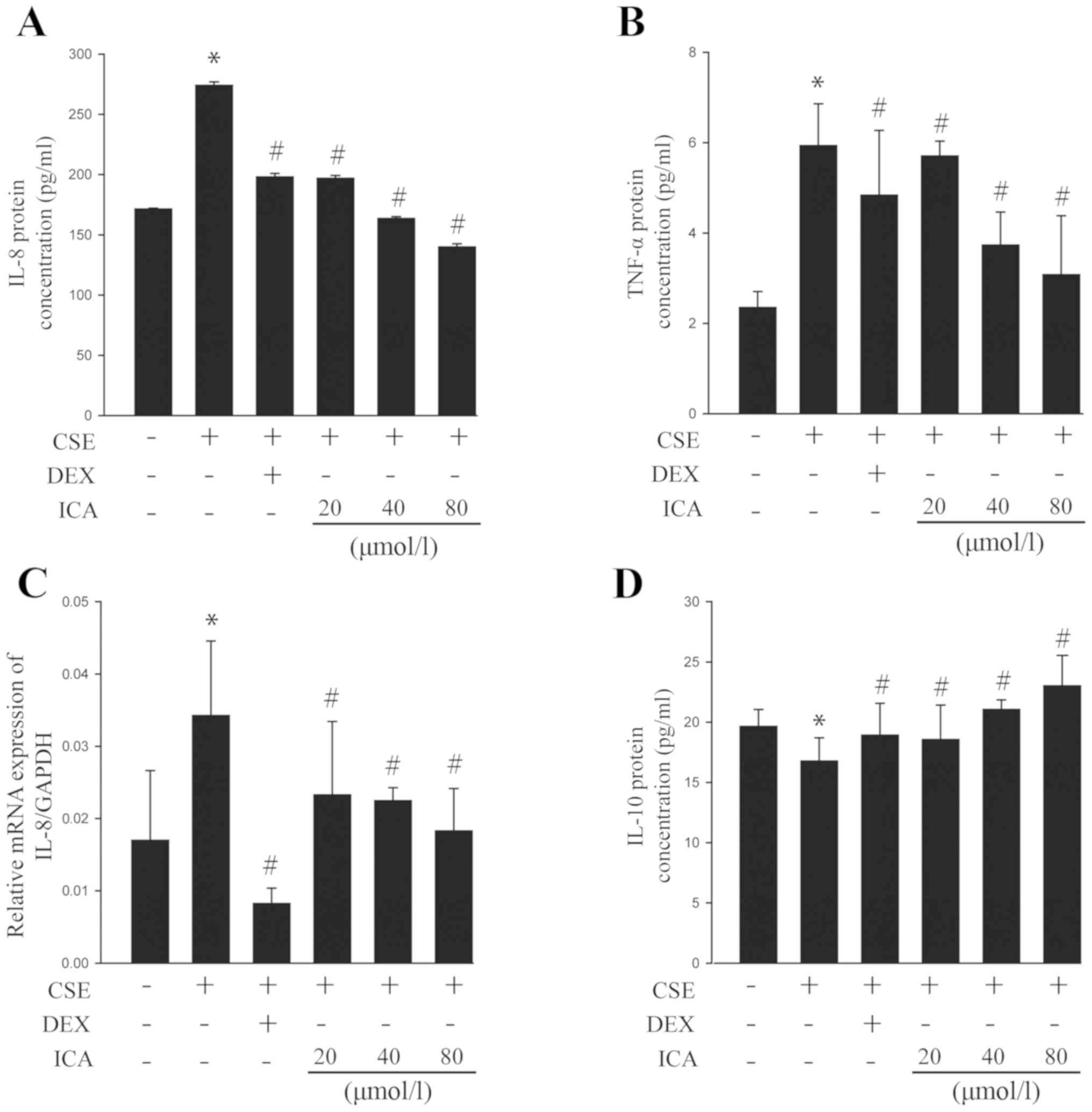
The levels of the pro-inflammatory cytokines IL-8
and TNF-α in the BEAS-2B cell culture medium were quantified using
ELISA and RT-qPCR. The results revealed that the expression of IL-8
and TNF-α increased in response to CSE exposure. However, icariin
exposure resulted in a significant reduction in this expression as
evidenced by protein (Fig. 2A and
B) and mRNA (Fig. 2C) levels of IL-8. Additionally,
exposure significantly increased the expression of the
anti-inflammatory cytokine IL-10 (Fig.
2D). Moreover, 40 and 80 µM icariin were superior to DEX for
the regulation of the pro-inflammatory and anti-inflammatory
balance. Collectively, the data suggested that icariin may play a
role in reducing CSE-induced cell injury by balancing the secretion
of pro-inflammatory and anti-inflammatory cytokines.
Icariin protects cells from damage
caused by the CSE-induced secretion of ROS and remodelling-related
factors
The level of ROS and DHR123 were significantly
increased in response to CSE exposure in BEAS-2B cells. Icariin (80
µM) inhibited ROS and DHR 123 expression to a similar extent as DEX
(Fig. 3A-C). These data suggested
that icariin may attenuate oxidative damage in cells induced by
CSE.
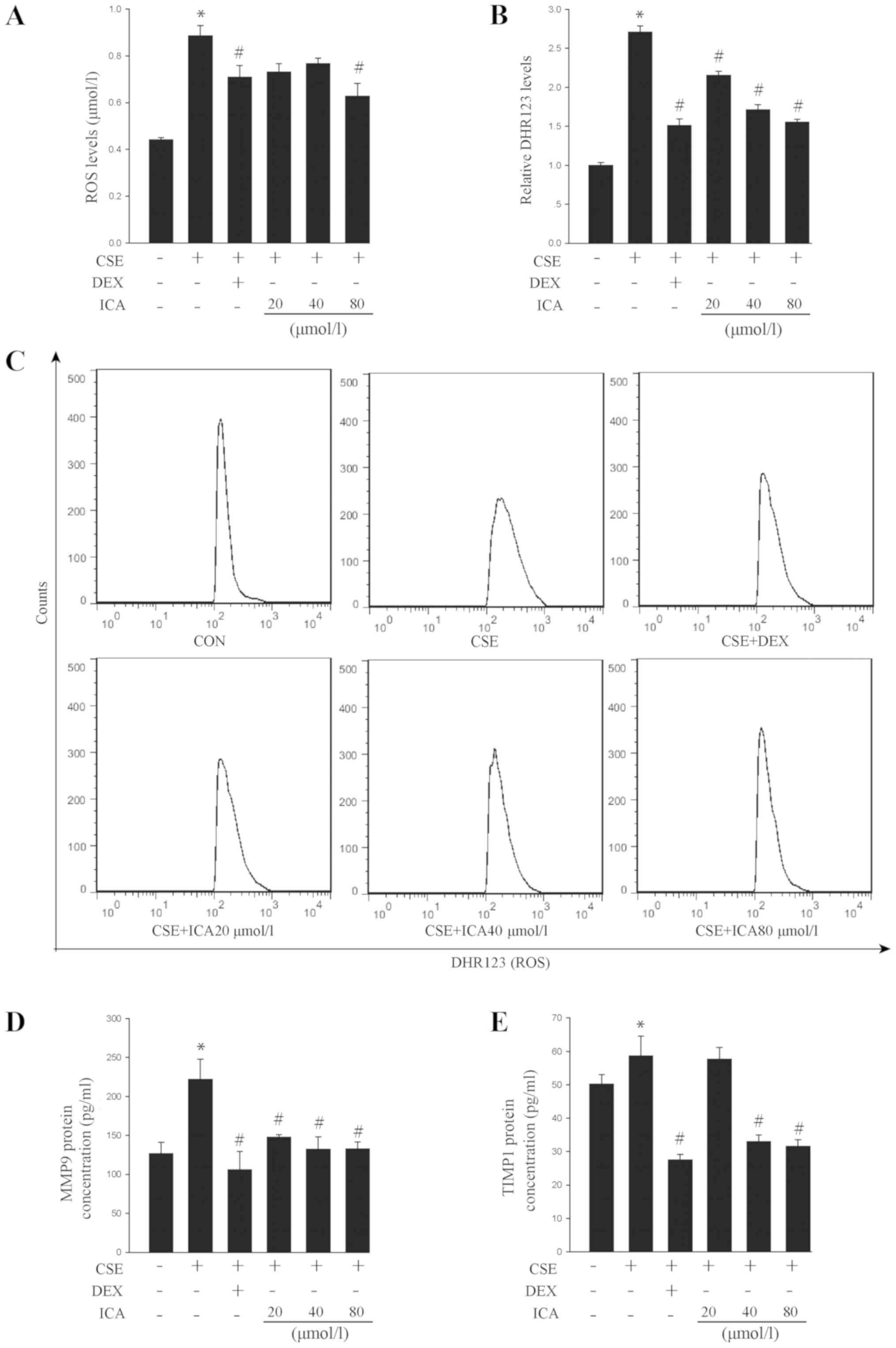 | Figure 3Effects of icariin on CSE-induced ROS
production and remodelling marker secretion. BEAS-2B cells were
pre-treated with 20, 40 and 80 µM icariin, 10 µM DEX or vehicle for
24 h, then treated with 5% CSE for 4 h. (A) ROS levels were
detected using a human intracellular ROS assay kit. (B) DHR123
levels were detected with flow cytometry. (C) representative flow
cytometry plots for the DHR123 analysis. ELISA analysis of the
remodelling-related factors (D) MMP9 and (E) TIMP1. Data are
expressed as the mean ± standard deviation (n=6).
*P<0.05 vs. vehicle and #P<0.05 vs.
CSE. CSE, cigarette smoke extract; ROS, reactive oxygen species;
DEX, dexamethasone; ICA, icariin; MMP9, matrix metalloprotease 9;
TIMP1, tissue inhibitor of metalloproteinase 1; DHR123,
dihydrogenrhodamine 123. |
MMP9 is associated with numerous pathological
processes, including remodelling of the respiratory tract (36). TIMP1, a multi-functional protein, is
an inhibitory molecule that alters cell viability and regulates
MMPs and cell-matrix renewal (37).
CSE significantly increased the levels of MMP9 and TIMP1 in the
BEAS-2B cell culture medium (Fig. 3D
and E). However, icariin treatment
decreased the levels of MMP9 and TIMP1, which may partially explain
its anti-remodelling effect.
Icariin attenuates CSE-induced GC
resistance
Due to primary GC resistance in COPD, GCs are unable
to effectively inhibit the inflammatory response. Studies have
shown that the factors associated with GC resistance include HDAC2,
Nrf2 and NF-κB (23,38,39). The
present study revealed that CSE significantly decreased the protein
and mRNA expression levels of GR, Nrf2 and HDAC2 and increased the
protein and mRNA levels of NF-κB. However, icariin treatment
reversed these changes (Figs. 4A-F
and S1A-D). Collectively, the
results suggested that icariin exerted a positive effect on GC
resistance.
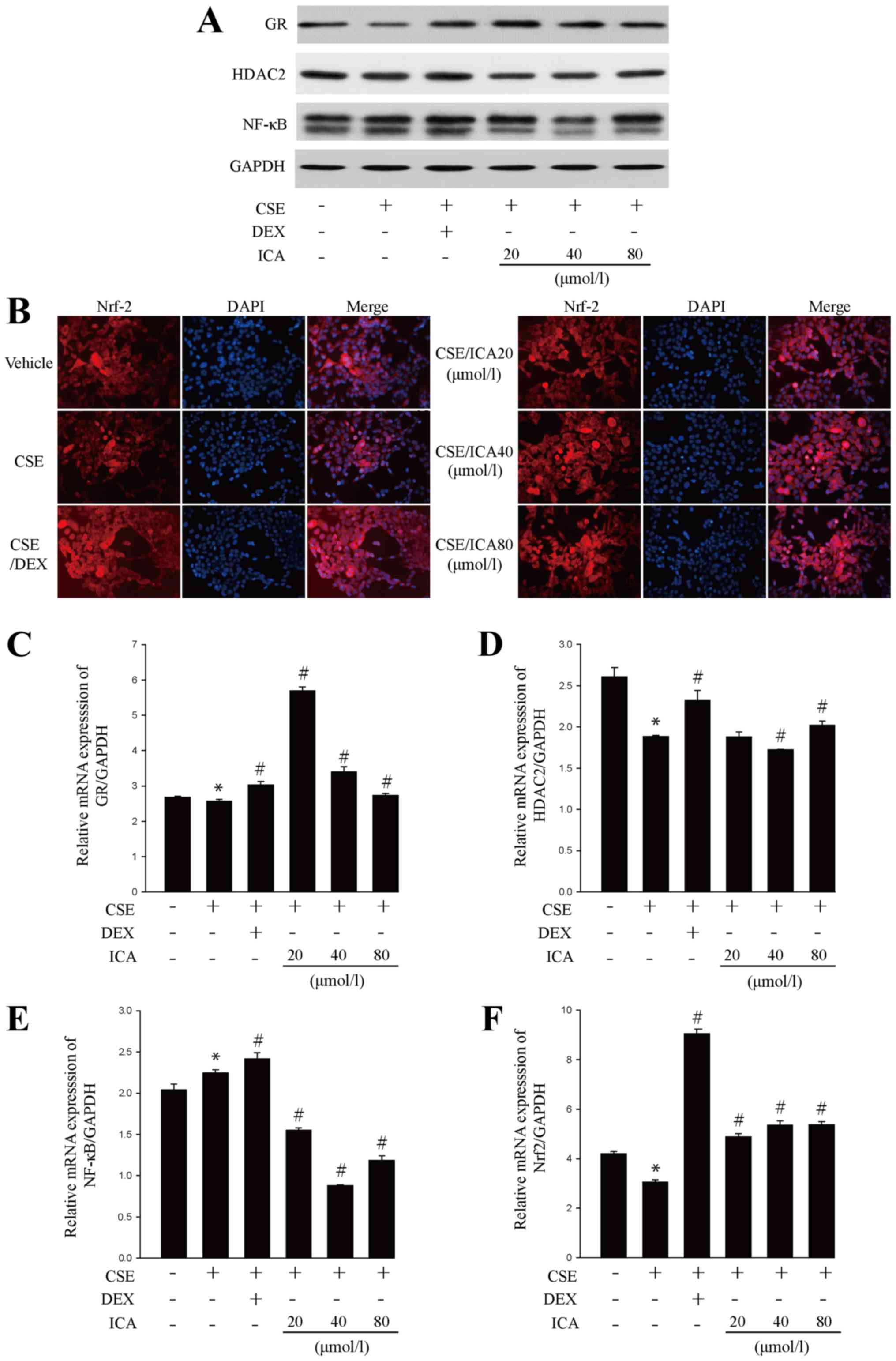 | Figure 4Effects of icariin treatment on
glucocorticoid resistance-related factors. BEAS-2B cells were
pre-treated with 20, 40 and 80 µM icariin, 10 µM DEX or vehicle for
24 h, then treated with 5% CSE for 4 h. (A) Western blotting
analysis of GR, HDAC2 and NF-κB protein expression. (B) Nrf2
immunofluorescence (red) in BEAS-2B cells (x40 magnification).
Reverse transcription-quantitative PCR analysis of (C) GR, (D)
HDAC2, (E) NF-κB and (F) Nrf2 mRNA expression. Data are expressed
as means ± SD (n=6). Western blotting and immunofluorescence
staining were performed 3 times. *P<0.05 vs. vehicle
and #P<0.05 vs. CSE. DEX, dexamethasone; CSE,
cigarette smoke extract; GR, glucocorticoid receptor; HDAC2,
histone deacetylase 2; Nrf2, erythroid 2 like 2; CSE, cigarette
smoke extract; DEX, dexamethasone; ICA, icariin. |
Discussion
The results of the present study revealed that
icariin significantly increased cell proliferation, regulated the
release of pro-inflammatory and anti-inflammatory factors,
inhibited ROS generation and decreased remodelling factor secretion
in an experimental model of CSE-exposed BEAS-2B cells. The
protective effects exhibited may be associated with the reversal of
GC resistance, increasing the levels of Nrf2, GR and HDAC2 and
decreasing NF-κB expression. These data support the unique
therapeutic value of icariin in the treatment of COPD.
Cigarette smoke contains thousands of toxic
chemicals and carcinogens and is regarded as a leading risk factor
for COPD, in which cigarette smoke triggers inflammatory responses,
induces the production of endogenous ROS and increases the
expression of remodelling-related factors (40-45).
The airway epithelium is the first barrier against environmental
insults such as cigarette smoke (46-48).
A previous study revealed that CSE decreases the cell viability of
mouse lung epithelial cells (49).
In the present study, the human bronchial epithelial cell line
BEAS-2B was exposed to CSE and the protective effects of icariin on
cell injury and proliferation were investigated. The CCK-8 assay
demonstrated that CSE decreased the proliferation of BEAS-2B cells
and icariin reversed this effect to a similar extent to DEX, which
indicated that icariin may protect against CSE-induced cell
injury.
COPD is a chronic non-specific disease in which
inflammation of the airways activates inflammatory epithelial and
smooth muscle cells and the release of inflammatory mediators such
as pro-inflammatory cytokines IL-8 and TNF-α, and the
anti-inflammatory cytokine IL-10(50). In patients with COPD, clinical
studies revealed that airway epithelial cells have higher baseline
levels of IL-8 and TNF-α expression and are correlated with the
prognosis and severity of disease (51-54).
Additionally, the specific inhibitors of IL-8 may alter the
progressive course of the disease (55). Acute exacerbations of the disease may
be due to increased mucus secretion as a result of increased
expression of mucin genes stimulated by high levels of IL-8
(7,56). TNF-α also has an important role in
the pathogenesis of COPD. TNF-α up-regulated the expression of
adhesion molecules causing a large influx of inflammatory cells,
and increased the production of matrix metalloproteinases and
tenascin promoting tissue damage and remodelling (57). IL-10 has also been implicated in the
development of COPD and its secretion is affected by COPD
progression and cigarette smoke (58). As an anti-inflammatory factor, IL-10
expression levels in sputum and serum were lower in patients with
COPD and smokers compared with healthy controls (59). A previous study revealed that IL-10
was positively correlated and IL-8 and TNF-α were negatively
correlated with the forced expiratory volume in one second (FEV1),
FEV1/predicted value ratio and FEV1/forced vital capacity. Plasma
levels of the inflammatory cytokines IL-8, IL-10 and TNF-α are
related to the severity of airway diseases and may serve as
prognostic biomarkers for COPD (7).
The results obtained in the present study revealed that icariin
prevented the CSE-induced upregulation of IL-8 and TNF-α and
increased IL-10 expression in BEAS-2B cells in vitro. Furthermore,
icariin (40 and 80 µM) were more effective than DEX.
The expression and activity of proteases and
anti-proteases are critical factors in the remodelling and
development of COPD (8,60,61).
MMP9, a zinc-dependent endopeptidase, is a biomarker for COPD as it
is not produced in normal lung tissues, but is produced in alveolar
type II, endothelial and epithelial cells in COPD (62-64).
MMPs form a group of neutral proteinases that can be divided into
three subgroups (65): collagenases
(MMP-1, MMP-8 and MMP-13); stromelysins and matrilysin (MMP-3,
MMP-10, MMP-11 and MMP-7); and type IV collagenases (MMP-2 and
MMP-9). MMP9 specifically degrade basement membrane type IV
collagen. Also, type V collagen as well as elastin can serve as
minor substrates for MMP9(66).
LeBert et al (67) found that
the depletion of MMP9 partially rescued the disordered collagen
fibres by using second-harmonic generation imaging technology. By
modifying collagen and elastin, MMP9 has a role in numerous
pathological processes such as remodelling, extracellular matrix
deposition and inflammation (62,63).
Serum and sputum MMP9 levels correlate with COPD severity and
significant clinical symptoms such as productive cough and a low
FEV1 (68,69). TIMPs are tissue inhibitors of MMPs
that act as multifunctional proteins to regulate cell matrix
renewal and cell activity (70-72).
Studies have shown that TIMP1 specifically inhibits the activity of
MMP9 (8,37,73). The
present study revealed a significant increase in MMP9 expression
and an adaptive decline in TIMP1 expression in CSE-exposed BEAS-2B
cells. Icariin significantly decreased MMP9 expression and
increased TIMP1 expression, suggesting that icariin may serve a
protective role in CSE-induced remodelling.
Preclinical studies and clinical trials have
revealed that an imbalance in oxidant/antioxidant factors in
patients with COPD is due to long-term exposure to cigarette smoke,
which results in the production of high concentrations of ROS
(74,75). This imbalance plays a vital role in
promoting airway remodelling and inflammation (76). ROS are implicated in the progression
of COPD and increased ROS generation has been documented in
patients with COPD (75,77). Increased ROS may lead to epithelial
cell injury and death, protease/antiprotease activity imbalance and
mucus hypersecretion (75,77). The present study revealed that CSE
exposure significantly increased the level of ROS in BEAS-2B cells,
which was then decreased following icariin treatment. Therefore,
the protective effects of icariin against CSE-induced damage may be
partly due to a decrease in the production of ROS.
Taken together, the results obtained in the present
study revealed that icariin protected BEAS-2B against CSE-induced
cell damage by decreasing the pro-inflammation/anti-inflammation
imbalance, oxidative damage and airway remodelling. Furthermore,
the effects of icariin on GC resistance were investigated as GCs
exert a significant anti-inflammatory effect, but this effect is
reduced in patients with COPD due to GC resistance (19,20,78). GCs
enter the cytoplasm to form a complex with GRs, which is then
transferred to the nucleus and acetylated. The complex subsequently
binds to the GR response element and leads to the transcription of
hormone-sensitive genes. HDAC2 deacetylates the acetylated GC-GR
complex and competitively binds to NF-κB to reduce acetylation of
NF-κB, thereby decreasing the release of inflammatory factors such
as IL-8 and TNF-α. The dynamic transformation of acetylation and
deacetylation of GRs in the nucleus is closely associated with
transcription of inflammatory factors. Therefore, primary GC
resistance in patients with COPD may be attributed to the lack of
HDAC2 in cells (19,79,80).
Additionally, HDAC2 is involved in GR-mediated gene transcriptional
repression through the deacetylation of the GR-GC complex (21,78).
Numerous studies have shown that activation of Nrf2 may be involved
in GC resistance by enhancing the activity of HDAC2 (81,82). The
present study revealed that CSE-exposed BEAS-2B cells expressed low
levels of GR mRNA and protein, while pre-treatment with icariin
rescued GR expression. Furthermore, HDAC2 expression, which drives
the GR to compete with Nrf2 following GR deacetylation, was also
increased by icariin, and Nrf2 expression, which regulates HDAC2
activity, was correspondingly elevated. By contrast, icariin
decreased the protein and mRNA expression of NF-κB. These data
suggested that the mechanism by which icariin protected against
CSE-induced cell injury may be attributed to the reversal of GC
resistance.
In conclusion, the present study revealed that
icariin exerted anti-inflammatory, antioxidative and
anti-remodelling effects in CSE-induced cell damage, potentially by
reversing GC resistance. The results are consistent with those
reported in a previous study, in which icariin alleviated
CSE-induced inflammatory responses by normalizing GR expression and
decreasing NF-κB expression in vivo and in vitro
(83). Furthermore, icariin was
revealed to serve a protective role against CSE-mediated oxidative
stress in the human lung epithelial cell line A549 by quenching ROS
and upregulating glutathione via a PI3K/AKT/Nrf2-dependent
mechanism (32). However, the
clinical application of icariin in COPD and other GC-resistant
diseases requires further investigation.
Supplementary Material
Quantification of glucocorticoid
resistance-related factors after icariin treatment. BEAS-2B cells
were pre-treated with 20, 40 and 80 μM icariin, 10 μM DEX or
vehicle for 24 h, then treated with 5% CSE for 4 h. Western
blotting quantification of (A) GR, (B) HDAC2 and (C) NF-κB protein
expression. (D) The relative density of Nrf2 immunofluorescence.
Data are expressed as means ± SD (n=3). Western blotting and
immunofluorescence staining were performed 3 times.
*P<0.05 vs. vehicle and #P<0.05 vs.
CSE. DEX, dexamethasone; CSE, cigarette smoke extract; GR,
glucocorticoid receptor; HDAC2, histone deacetylase 2; Nrf2,
erythroid 2 like 2; CSE, cigarette smoke extract; DEX,
dexamethasone; ICA, icariin.
Acknowledgements
Not applicable.
Funding
This project was supported by funding from the
National Natural Science Foundation of China (grant no. 81302931 to
HYZ, grant no. 81703829 to JS, grant no. 81573758 to JCD and grant
no. 81603406 to FL).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HZ and JS designed the study. LH, FL, LL, LZ, CY,
QL, JQ and JD performed the laboratory experiments. LH and FL
performed the statistical analysis and drafted the manuscript. HZ,
JS and JD made significant conceptual contributions to the
manuscript. HZ, JS and JD reviewed the final version of the paper.
All the authors provided intellectual content and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by The Affiliated
Huashan Hospital of Fudan University (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mathers CD and Loncar D: Projections of
global mortality and burden of disease from 2002 to 2030. PLoS Med.
3(e442)2006.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hanania NA and Marciniuk DD: A unified
front against COPD: Clinical practice guidelines from the American
College of Physicians, the American College of Chest Physicians,
the American Thoracic Society, and the European Respiratory
Society. Chest. 140:565–566. 2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rabe KF, Hurd S, Anzueto A, Barnes PJ,
Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R,
van Weel C, et al: Global strategy for the diagnosis, management,
and prevention of chronic obstructive pulmonary disease: GOLD
executive summary. Am J Respir Crit Care Med. 176:532–555.
2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Aghapour M, Raee P, Moghaddam SJ, Hiemstra
PS and Heijink IH: Airway epithelial barrier dysfunction in chronic
obstructive pulmonary disease: Role of cigarette smoke exposure. Am
J Respir Cell Mol Biol. 58:157–169. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ng DS, Liao W, Tan WS, Chan TK, Loh XY and
Wong WS: Anti-malarial drug artesunate protects against cigarette
smoke-induced lung injury in mice. Phytomedicine. 21:1638–1644.
2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Murray LA, Dunmore R, Camelo A, Da Silva
CA, Gustavsson MJ, Habiel DM, Hackett TL, Hogaboam CM, Sleeman MA
and Knight DA: Acute cigarette smoke exposure activates apoptotic
and inflammatory programs but a second stimulus is required to
induce epithelial to mesenchymal transition in COPD epithelium.
Respir Res. 18(82)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Huang AX, Lu LW, Liu WJ and Huang M:
Plasma inflammatory cytokine IL-4, IL-8, IL-10, and TNF-α levels
correlate with pulmonary function in patients with asthma-chronic
obstructive pulmonary disease (COPD) overlap syndrome. Med Sci
Monit. 22:2800–2808. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wang C, Li Z, Liu X, Peng Q, Li F, Li D
and Wang C: Effect of Liuweibuqi capsule, a Chinese patent
medicine, on the JAK1/STAT3 pathway and MMP9/TIMP1 in a chronic
obstructive pulmonary disease rat model. J Tradit Chin Med.
35:54–62. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Marushchak M, Maksiv K and Krynytska I:
The specific features of free radical oxidation in patients with
chronic obstructive pulmonary disease and arterial hypertension.
Pol Merkur Lekarski. 47:95–98. 2019.PubMed/NCBI
|
|
10
|
Bateman ED, Hurd SS, Barnes PJ, Bousquet
J, Drazen JM, FitzGerald JM, Gibson P, Ohta K, O'Byrne P, Pedersen
SE, et al: Global strategy for asthma management and prevention:
GINA executive summary. Eur Respir J. 31:143–178. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Barnes PJ, Greening AP and Crompton GK:
Glucocorticoid resistance in asthma. Am J Respir Crit Care Med. 152
(Suppl):S125–S140. 1995.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Burmester GR and Pope JE: Novel treatment
strategies in rheumatoid arthritis. Lancet. 389:2338–2348.
2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Baumgart DC and Sandborn WJ: Inflammatory
bowel disease: Clinical aspects and established and evolving
therapies. Lancet. 369:1641–1657. 2007.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Torres J, Mehandru S, Colombel JF and
Peyrin-Biroulet L: Crohn's disease. Lancet. 389:1741–1755.
2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Dignass A, Van Assche G, Lindsay JO,
Lémann M, Söderholm J, Colombel JF, Danese S, D'Hoore A, Gassull M,
Gomollón F, et al: The second European evidence-based Consensus on
the diagnosis and management of Crohn's disease: Current
management. J Crohn's Colitis. 4:28–62. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rhen T and Cidlowski JA: Antiinflammatory
action of glucocorticoids-new mechanisms for old drugs. N Engl J
Med. 353:1711–1723. 2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Barnes PJ: New therapies for chronic
obstructive pulmonary disease. Med Princ Pract. 19:330–338.
2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Morjaria JB, Malerba M and Polosa R:
Biologic and pharmacologic therapies in clinical development for
the inflammatory response in COPD. Drug Discov Today. 15:396–405.
2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Barnes PJ: How corticosteroids control
inflammation: Quintiles Prize Lecture 2005. Br J Pharmacol.
148:245–254. 2006.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Rodriguez JM, Monsalves-Alvarez M,
Henriquez S, Llanos MN and Troncoso R: Glucocorticoid resistance in
chronic diseases. Steroids. 115:182–192. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Barnes PJ and Adcock IM: Glucocorticoid
resistance in inflammatory diseases. Lancet. 373:1905–1917.
2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Cosio BG, Tsaprouni L, Ito K, Jazrawi E,
Adcock IM and Barnes PJ: Theophylline restores histone deacetylase
activity and steroid responses in COPD macrophages. J Exp Med.
200:689–695. 2004.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Barnes PJ, Ito K and Adcock IM:
Corticosteroid resistance in chronic obstructive pulmonary disease:
Inactivation of histone deacetylase. Lancet. 363:731–733.
2004.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhou Z, Zheng W, Liang T, Yan Q, Zhang C,
Huang H, Liu X and Ye X: Efficacy and safety of Chuankezhi
injection in patients with chronic obstructive pulmonary disease: A
systematic review and meta-analysis protocol. Medicine (Baltimore).
99(e18620)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhao YL, Song HR, Fei JX, Liang Y, Zhang
BH, Liu QP, Wang J and Hu P: The effects of Chinese yam-epimedium
mixture on respiratory function and quality of life in patients
with chronic obstructive pulmonary disease. J Tradit Chin Med.
32:203–207. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
He W, Sun H, Yang B, Zhang D and Kabelitz
D: Immunoregulatory effects of the herba Epimediia glycoside
icariin. Arzneimittelforschung. 45:910–913. 1995.PubMed/NCBI
|
|
27
|
Shi Y, Yan WH, Lin QY and Wang WM: Icariin
influences cardiac remodeling following myocardial infarction by
regulating the CD147/MMP-9 pathway. J Int Med Res. 46:2371–2385.
2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Dong H, Ming S, Fang J, Li Y and Liu L:
Icariin ameliorates angiotensin II-induced cerebrovascular
remodeling by inhibiting Nox2-containing NADPH oxidase activation.
Hum Cell. 32:22–30. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ming LG, Chen KM and Xian CJ: Functions
and action mechanisms of flavonoids genistein and icariin in
regulating bone remodeling. J Cell Physiol. 228:513–521.
2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Xu CQ, Liu BJ, Wu JF, Xu YC, Duan XH, Cao
YX and Dong JC: Icariin attenuates LPS-induced acute inflammatory
responses: Involvement of PI3K/Akt and NF-kappaB signaling pathway.
Eur J Pharmacol. 642:146–153. 2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chen SR, Xu XZ, Wang YH, Chen JW, Xu SW,
Gu LQ and Liu PQ: Icariin derivative inhibits inflammation through
suppression of p38 mitogen-activated protein kinase and nuclear
factor-kappaB pathways. Biol Pharm Bull. 33:1307–1313.
2010.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wu J, Xu H, Wong PF, Xia S, Xu J and Dong
J: Icaritin attenuates cigarette smoke-mediated oxidative stress in
human lung epithelial cells via activation of PI3K-AKT and Nrf2
signaling. Food Chem Toxicol. 64:307–313. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Krystosek A and Sachs L: Control of
lysozyme induction in the differentiation of myeloid leukemic
cells. Cell. 9:675–684. 1976.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hu L, Yu Y, Huang H, Fan H, Hu L, Yin C,
Li K, Fulton DJ and Chen F: Epigenetic regulation of interleukin 6
by histone acetylation in macrophages and its role in
paraquat-induced pulmonary Fibrosis. Front Immunol.
7(696)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Papakonstantinou E, Karakiulakis G,
Batzios S, Savic S, Roth M, Tamm M and Stolz D: Acute exacerbations
of COPD are associated with significant activation of matrix
metalloproteinase 9 irrespectively of airway obstruction, emphysema
and infection. Respir Res. 16(78)2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Navratilova Z, Kolek V and Petrek M:
Matrix metalloproteinases and their inhibitors in chronic
obstructive pulmonary disease. Arch Immunol Ther Exp (Warsz).
64:177–193. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Vandewalle J, Luypaert A, De Bosscher K
and Libert C: Therapeutic mechanisms of glucocorticoids. Trends
Endocrinol Metab. 29:42–54. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Li J, Liu D, Wu J, Zhang D, Cheng B, Zhang
Y, Yin Z, Wang Y, Du J and Ling C: Ginsenoside Rg1 attenuates
ultraviolet B-induced glucocortisides resistance in keratinocytes
via Nrf2/HDAC2 signalling. Sci Rep. 6(39336)2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Stabile AM, Marinucci L, Balloni S,
Giuliani A, Pistilli A, Bodo M and Rende M: Long term effects of
cigarette smoke extract or nicotine on nerve growth factor and its
receptors in a bronchial epithelial cell line. Toxicol In Vitro.
53:29–36. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Munakata S, Ishimori K, Kitamura N,
Ishikawa S, Takanami Y and Ito S: Oxidative stress responses in
human bronchial epithelial cells exposed to cigarette smoke and
vapor from tobacco- and nicotine-containing products. Regul Toxicol
Pharmacol. 99:122–128. 2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Eisner MD, Anthonisen N, Coultas D,
Kuenzli N, Perez-Padilla R, Postma D, Romieu I, Silverman EK and
Balmes JR: Committee on Nonsmoking COPD Environmental and
Occupational Health Assembly. An official American Thoracic Society
public policy statement: Novel risk factors and the global burden
of chronic obstructive pulmonary disease. Am J Respir Crit Care
Med. 182:693–718. 2010.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lee H, Jung KH, Park S, Kil YS, Chung EY,
Jang YP, Seo EK and Bae H: Inhibitory effects of Stemona tuberosa
on lung inflammation in a subacute cigarette smoke-induced mouse
model. BMC Complement Altern Med. 14(513)2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Ramos CO, Campos KKD, Costa GP, Cangussú
SD, Talvani A and Bezerra FS: Taurine treatment decreases
inflammation and oxidative stress in lungs of adult mice exposed to
cigarette smoke. Regul Toxicol Pharmacol. 98:50–57. 2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Chen L, Ge Q, Tjin G, Alkhouri H, Deng L,
Brandsma CA, Adcock I, Timens W, Postma D, Burgess JK, et al:
Effects of cigarette smoke extract on human airway smooth muscle
cells in COPD. Eur Respir J. 44:634–646. 2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
BéruBé K, Aufderheide M, Breheny D,
Clothier R, Combes R, Duffin R, Forbes B, Gaça M, Gray A, Hall I,
et al: In vitro models of inhalation toxicity and disease The
report of a FRAME workshop. Altern Lab Anim. 37:89–141.
2009.PubMed/NCBI
|
|
47
|
López-Rodríguez JC, Benedé S, Barderas R,
Villalba M and Batanero E: Airway epithelium plays a leading role
in the complex framework underlying respiratory allergy. J Investig
Allergol Clin Immunol. 27:346–355. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Gras D, Chanez P, Vachier I, Petit A and
Bourdin A: Bronchial epithelium as a target for innovative
treatments in asthma. Pharmacol Ther. 140:290–305. 2013.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Pouli AE, Hatzinikolaou DG, Piperi C,
Stavridou A, Psallidopoulos MC and Stavrides JC: The cytotoxic
effect of volatile organic compounds of the gas phase of cigarette
smoke on lung epithelial cells. Free Radic Biol Med. 34:345–355.
2003.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Singh S, Verma SK, Kumar S, Ahmad MK,
Nischal A, Singh SK and Dixit RK: Correlation of severity of
chronic obstructive pulmonary disease with potential biomarkers.
Immunol Lett. 196:1–10. 2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Zhang X, Zheng H, Zhang H, Ma W, Wang F,
Liu C and He S: Increased interleukin (IL)-8 and decreased IL-17
production in chronic obstructive pulmonary disease (COPD) provoked
by cigarette smoke. Cytokine. 56:717–725. 2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Schneider D, Ganesan S, Comstock AT,
Meldrum CA, Mahidhara R, Goldsmith AM, Curtis JL, Martinez FJ,
Hershenson MB and Sajjan U: Increased cytokine response of
rhinovirus-infected airway epithelial cells in chronic obstructive
pulmonary disease. Am J Respir Crit Care Med. 182:332–340.
2010.PubMed/NCBI View Article : Google Scholar
|
|
53
|
de Godoy I, Donahoe M, Calhoun WJ, Mancino
J and Rogers RM: Elevated TNF-alpha production by peripheral blood
monocytes of weight-losing COPD patients. Am J Respir Crit Care
Med. 153:633–637. 1996.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Xie J, Yang XY, Shi JD, Deng XQ and Long
W: A new inflammation marker of chronic obstructive pulmonary
disease-adiponectin. World J Emerg Med. 1:190–195. 2010.PubMed/NCBI
|
|
55
|
Barnes PJ: Therapy of chronic obstructive
pulmonary disease. Pharmacol Ther. 97:87–94. 2003.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Wu H, Yang S, Wu X, Zhao J, Zhao J, Ning
Q, Xu Y and Xie J: Interleukin-33/ST2 signaling promotes production
of interleukin-6 and interleukin-8 in systemic inflammation in
cigarette smoke-induced chronic obstructive pulmonary disease mice.
Biochem Biophys Res Commun. 450:110–116. 2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Malaviya R, Laskin JD and Laskin DL:
Anti-TNFα therapy in inflammatory lung diseases. Pharmacol Ther.
180:90–98. 2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Zhang L, Cheng Z, Liu W and Wu K:
Expression of interleukin (IL)-10, IL-17A and IL-22 in serum and
sputum of stable chronic obstructive pulmonary disease patients.
COPD. 10:459–465. 2013.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Pelegrino NR, Tanni SE, Amaral RA,
Angeleli AY, Correa C and Godoy I: Effects of active smoking on
airway and systemic inflammation profiles in patients with chronic
obstructive pulmonary disease. Am J Med Sci. 345:440–445.
2013.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Mercer PF, Shute JK, Bhowmik A, Donaldson
GC, Wedzicha JA and Warner JA: MMP-9, TIMP-1 and inflammatory cells
in sputum from COPD patients during exacerbation. Respir Res.
6(151)2005.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Yao H, Hwang JW, Sundar IK, Friedman AE,
McBurney MW, Guarente L, Gu W, Kinnula VL and Rahman I: SIRT1
redresses the imbalance of tissue inhibitor of matrix
metalloproteinase-1 and matrix metalloproteinase-9 in the
development of mouse emphysema and human COPD. Am J Physiol Lung
Cell Mol Physiol. 305:L615–L624. 2013.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Greenlee KJ, Werb Z and Kheradmand F:
Matrix metalloproteinases in lung: Multiple, multifarious, and
multifaceted. Physiol Rev. 87:69–98. 2007.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Tiede SL, Wassenberg M, Christ K,
Schermuly RT, Seeger W, Grimminger F, Ghofrani HA and Gall H:
Biomarkers of tissue remodeling predict survival in patients with
pulmonary hypertension. Int J Cardiol. 223:821–826. 2016.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Vogel ER, Britt RD Jr, Faksh A, Kuipers I,
Pandya H, Prakash YS, Martin RJ and Pabelick CM: Moderate hyperoxia
induces extracellular matrix remodeling by human fetal airway
smooth muscle cells. Pediatr Res. 81:376–383. 2017.PubMed/NCBI View Article : Google Scholar
|
|
65
|
MacDougall JR and Matrisian LM:
Contributions of tumor and stromal matrix metalloproteinases to
tumor progression, invasion and metastasis. Cancer Metastasis Rev.
14:351–362. 1995.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Yao PM, Buhler JM, d'Ortho MP, Lebargy F,
Delclaux C, Harf A and Lafuma C: Expression of matrix
metalloproteinase gelatinases A and B by cultured epithelial cells
from human bronchial explants. J Biol Chem. 271:15580–15589.
1996.PubMed/NCBI View Article : Google Scholar
|
|
67
|
LeBert DC, Squirrell JM, Rindy J,
Broadbridge E, Lui Y, Zakrzewska A, Eliceiri KW, Meijer AH and
Huttenlocher A: Matrix metalloproteinase 9 modulates collagen
matrices and wound repair. Development. 142:2136–2146.
2015.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Linder R, Rönmark E, Pourazar J, Behndig
A, Blomberg A and Lindberg A: Serum metalloproteinase-9 is related
to COPD severity and symptoms-cross-sectional data from a
population based cohort-study. Respir Res. 16(28)2015.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Chaudhuri R, McSharry C, Spears M, Brady
J, Grierson C, Messow CM, Miele G, Nocka K, MacNee W, Connell M, et
al: Sputum matrix metalloproteinase-9 is associated with the degree
of emphysema on computed tomography in COPD. Transl Respir Med.
1(11)2013.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Lambert E, Dassé E, Haye B and Petitfrère
E: TIMPs as multifacial proteins. Crit Rev Oncol Hematol.
49:187–198. 2004.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Toricelli M, Melo FH, Peres GB, Silva DC
and Jasiulionis MG: Timp1 interacts with beta-1 integrin and CD63
along melanoma genesis and confers anoikis resistance by activating
PI3-K signaling pathway independently of Akt phosphorylation. Mol
Cancer. 12(22)2013.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Arpino V, Brock M and Gill SE: The role of
TIMPs in regulation of extracellular matrix proteolysis. Matrix
Biol. 44-46:247–254. 2015.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Mocchegiani E, Giacconi R and Costarelli
L: Metalloproteases/anti-metalloproteases imbalance in chronic
obstructive pulmonary disease: Genetic factors and treatment
implications. Curr Opin Pulm Med. 17 (Suppl 1):S11–S19.
2011.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Pryor WA and Stone K: Oxidants in
cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and
peroxynitrite. Ann N Y Acad Sci. 686:12–27; discussion 27-28.
1993.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Boukhenouna S, Wilson MA, Bahmed K and
Kosmider B: Reactive oxygen species in chronic obstructive
pulmonary disease. Oxid Med Cell Longev.
2018(5730395)2018.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Jiang Y, Wang X and Hu D: Mitochondrial
alterations during oxidative stress in chronic obstructive
pulmonary disease. Int J Chron Obstruct Pulmon Dis. 12:1153–1162.
2017.PubMed/NCBI View Article : Google Scholar
|
|
77
|
MacNee W: Pulmonary and systemic
oxidant/antioxidant imbalance in chronic obstructive pulmonary
disease. Proc Am Thorac Soc. 2:50–60. 2005.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Barnes PJ: Mechanisms and resistance in
glucocorticoid control of inflammation. J Steroid Biochem Mol Biol.
120:76–85. 2010.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Ingawale DK, Mandlik SK and Patel SS: An
emphasis on molecular mechanisms of anti-inflammatory effects and
glucocorticoid resistance. J Complement Integr Med. 12:1–13.
2015.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Ito K, Yamamura S, Essilfie-Quaye S, Cosio
B, Ito M, Barnes PJ and Adcock IM: Histone deacetylase 2-mediated
deacetylation of the glucocorticoid receptor enables NF-kappaB
suppression. J Exp Med. 203:7–13. 2006.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Payne DN and Adcock IM: Molecular
mechanisms of corticosteroid actions. Paediatr Respir Rev.
2:145–150. 2001.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Ogawa S, Lozach J, Benner C, Pascual G,
Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M,
Rosenfeld MG and Glass CK: Molecular determinants of crosstalk
between nuclear receptors and toll-like receptors. Cell.
122:707–721. 2005.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Li L, Sun J, Xu C, Zhang H, Wu J, Liu B
and Dong J: Icariin ameliorates cigarette smoke induced
inflammatory responses via suppression of NF-κB and modulation of
GR in vivo and in vitro. PLoS One. 9(e102345)2014.PubMed/NCBI View Article : Google Scholar
|