Introduction
Idiopathic pulmonary hemosiderosis (IPH) is a rare
interstitial lung disease with an estimated incidence of 0.24
case/year/million in Swedish children and 1.23 cases/year/million
in Japanese children (1,2). IPH is characterized by diffuse
parenchymal infiltrates, hemoptysis and secondary iron deficiency
anemia (IDA) due to recurrent intrapulmonary capillary bleeding
(3). Rheumatoid arthritis (RA) has
at times been observed in patients with IPH (4,5), which
is of no known etiological significance. The present study reports
on the case of a patient with IPH who also presented with RA and
determined the possible association between IPH and RA, with the
aim of helping physicians to identify such patients early and
prevent illness and death.
Case report
A 21-year-old female patient was admitted to the
China-Japan Friendship Hospital (Beijing, China) in March 2019 for
confirmation of suspected IPH. From the age of 6 years, the patient
had required repeated blood transfusions for anemia, and for the
last 2 years, the patient had experienced exertional dyspnea,
presenting with diffuse alveolar infiltration on chest CT. The
patient also had occasional episodes of arthralgia and swelling of
the wrist joints, elbows, shoulders, ankles, cervical spine and
knees in the recent 2 months. In addition, the patient complained
of coughing up sputum occasionally but denied hemoptysis. The
patient reported a hospitalization 2 years previously due to
conscious disturbance of unknown causes, which subsided after a
short course of high-dose corticosteroids. However, the patient
failed to recall any further details with regard to the etiology
associated with her hospitalization. The patient was a non-smoker
and reported no exposure to cytotoxic drugs, environmental or
occupational allergens or toxic fumes, chemicals or dust.
On physical examination, the patient had pale skin.
Auscultation of the lungs revealed mild end-inspiratory crackles in
the bilateral lower lung zones. Cardiovascular, abdominal and
neurological system examinations were unremarkable. Finger-clubbing
was marked, but no skin lesions were noted.
The abnormal and most important clinically relevant
laboratory test results were summarized in Table I. Arterial blood gas analysis
revealed hypoxia. The hemoglobin level and hematocrit value were
decreased. The platelet count, prothrombin time and activated
partial thromboplastin time were normal. The reticulocyte count was
elevated. A test for occult blood in the stools was negative. The
direct and total serum bilirubin levels were normal. The serum iron
protein level was within the normal limits. Hemoglobin
electrophoresis, Coombs' test and test for cold agglutinins were
negative. A bone marrow biopsy indicated iron deficiency. The
erythrocyte sedimentation rate and C-reactive protein were normal.
Serum biochemistry, anticyclic-citrullinated protein (CCP) antibody
(466 U/ml; upper limit, <25 U/ml) and rheumatoid factor (RF)
(1,710 IU/ml; upper limit, <20 IU/ml) were highly increased and
severely positive. Anti-nuclear antibody, anti-dsDNA, anti-Scl-70
antibody, complement C3 and C4, as well as anti-neutrophil
cytoplasmic antibodies, were negative. A wide range of other
laboratory parameters associated with infection was assessed and
the results were negative or within normal limits.
 | Table IRelevant laboratory results of the
reported case. |
Table I
Relevant laboratory results of the
reported case.
| | Result | |
|---|
| Parameter | 03/21/2019 | 05/05/2019 | 07/05/2019 | 11/25/2019 | Reference/negative
range |
|---|
| Hematology | | | | | |
|
Hemoglobin
(g/l) | 56a | 119 | 132 | 138 | 115-150 |
|
HCT (%) | 21.2a | 41.1 | 41.6 | 40.9 | 35-45 |
|
MCV
(fl) | 65.8a | 73.3a | 80.3a | 80a | 82-100 |
|
MCH
(pg) | 17.4a | 21.2a | 25.5a | 27 | 27-34 |
|
Immunology/serology | | | | | |
|
CCP
(U/ml) | 466a | | | <25 | <25 |
|
RF
(IU/ml) | 1710a | | | 77.5a | <20 |
| Blood gas
analysis | | | | | |
|
PaO2
(mmHg) | 66a | 76a | 85 | 85 | 83-108 |
The results of the pulmonary function tests are
summarized in Table II and revealed
restrictive ventilation dysfunction and decreased diffusion
capacity; the forced vital capacity (FVC), forced expiratory volume
in 1 sec (FEV1), total lung capacity and diffusion
capacity of carbon monoxide were decreased. The ratio of
FEV1 to FVC was also slightly decreased. Serial chest CT
was performed using Aquilion 64-slice CT scanner (Toshiba Corp.)
with intervals of 1.25 mm at the time of hospital admission. The
scan was performed from the thoracic inlet to the lateral
costophrenic angle, revealing an interstitial pattern with
micronodules at the upper and middle zones of each lung lobe, as
well as diffuse bilateral ground-glass opacities and patchy
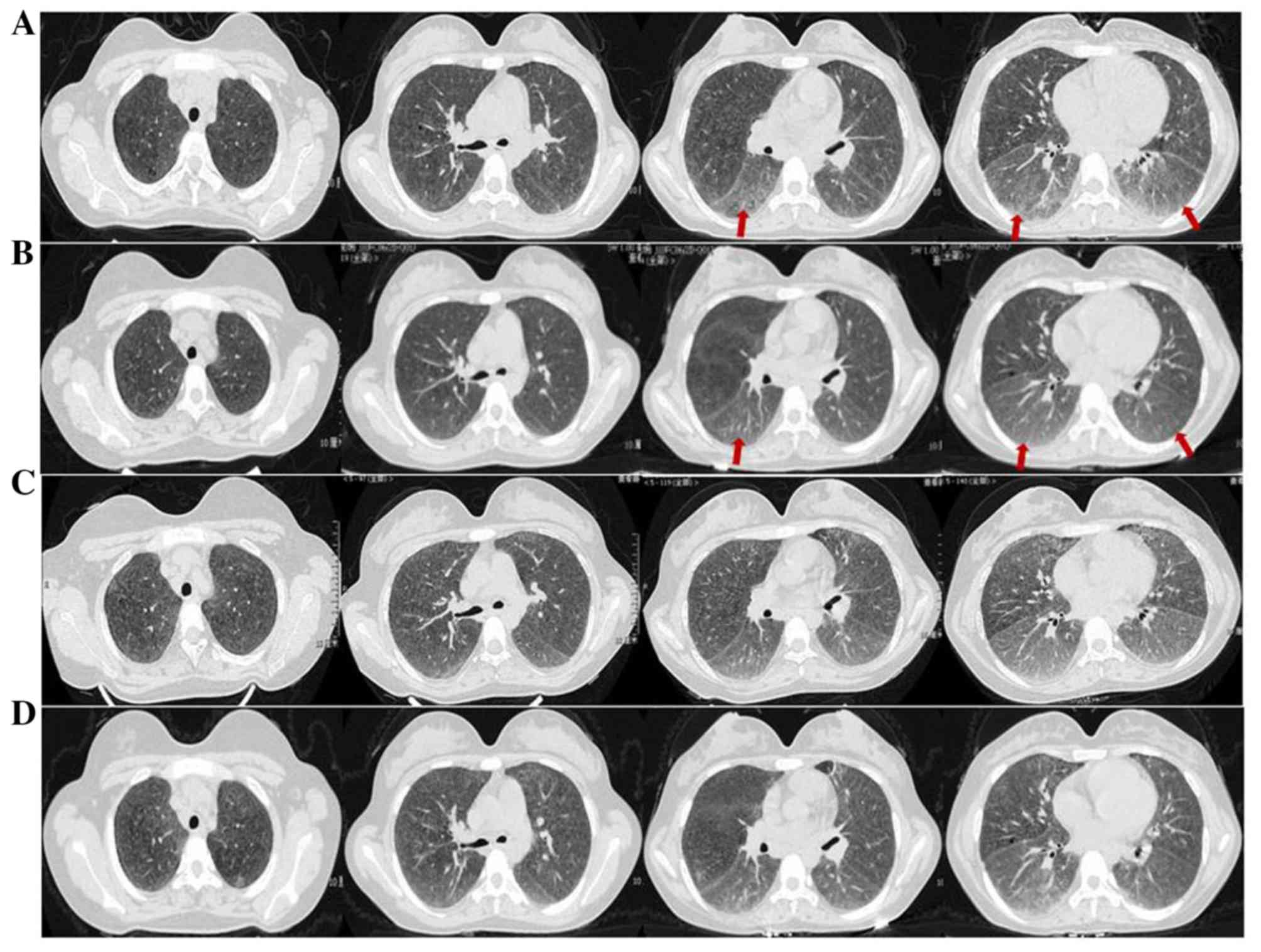
infiltrates at the lower lung lobes (Fig. 1A). The CT images were evaluated in
consensus by 2 radiologists experienced in chest radiology for
>20 years. Echocardiography did not reveal any significant
valvular lesions. X-ray scans of the hands, elbows and knees were
normal. Bronchoscopy was performed with a flexible bronchoscope
(Olympus Corp.). The patient received topical anesthetic with oral
mucosa (using lidocaine) and nasal passages (using oxybuprocaine
gel). Bronchoalveolar lavage fluid was obtained from the posterior
basal segment of the right lower lobe by administration followed by
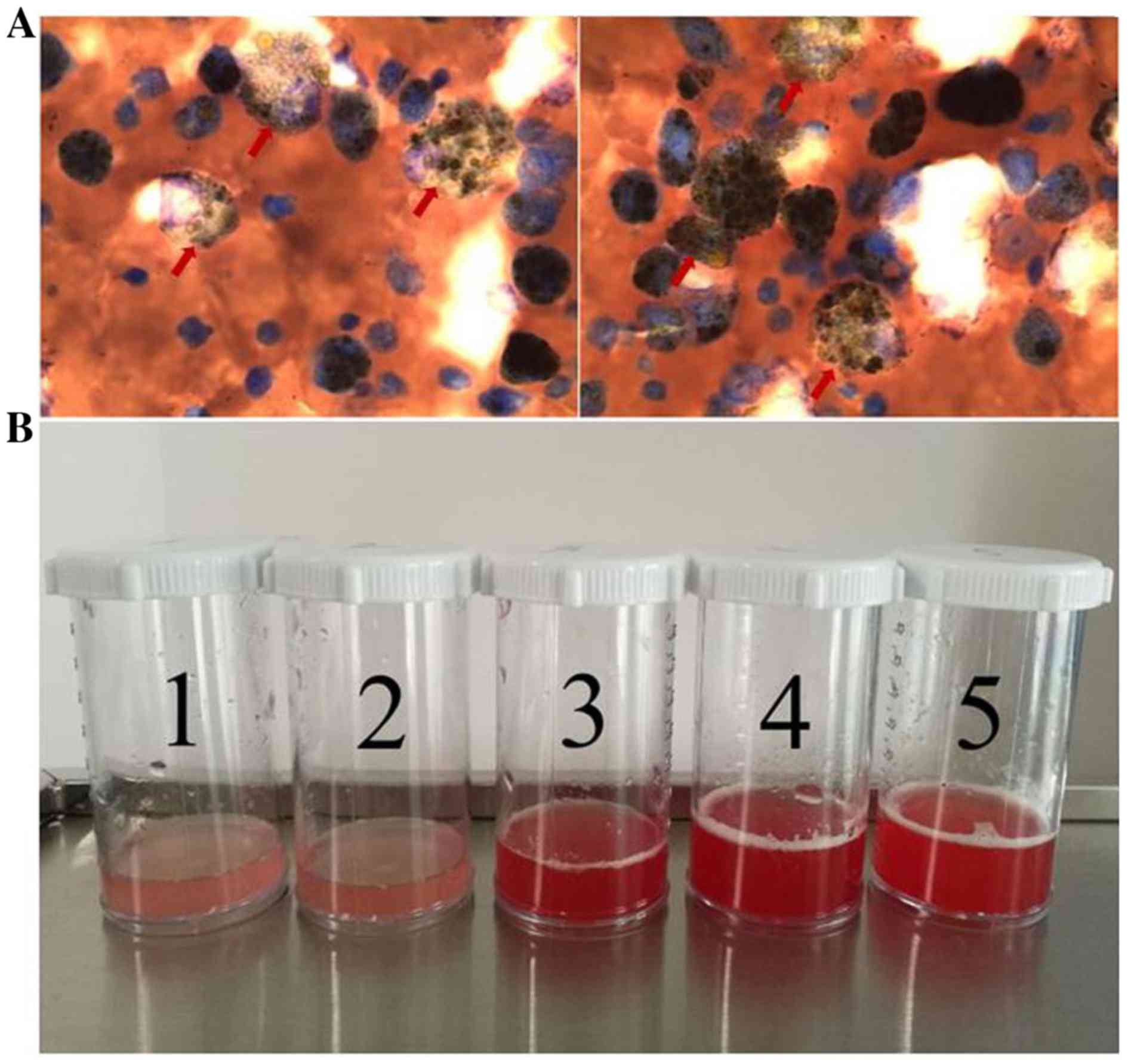
aspiration of 30 ml 0.9% NaCl and indicated the presence of
hemosiderin-laden macrophages (51.5%; Table III and Fig. 2A). There were five sequential
instillations performed at the same site at 1-2-min intervals, and
the volume used was five 30 ml aliquots. Observation of
progressively more hemorrhagic fluid with each successive lavage
suggested alveolar hemorrhage (Fig.
2B).
| Table IIResults of pulmonary function
tests. |
Table II
Results of pulmonary function
tests.
| | Result | | Result | |
|---|
| Tests | 03/20/2019 | 07/10/2019 | Reference/negative
range | 11/19/2019 | Reference/negative
range |
|---|
| FVC (l) | 2.34a | 2.52a | 3.46 | 2.44a | 3.50 |
| FVC%pred (%) | 67.60a | 72.90a | - | 69.60a | - |
| FEV1
(l) | 1.76a | 1.76a | 3.02 | 1.80a | 3.06 |
|
FEV1%pred (%) | 58.30a | 58.40a | - | 59.00a | - |
| FEV1/FVC
(%) | 75.18a | 69.85a | - | 73.93a | - |
| TLC (l) | 3.44a | 4.70 | 4.64 | 3.31a | 4.70 |
| TLC%pred (%) | 74.20a | 101.30 | - | 70.30a | - |
| DLCO%pred (%) | 43.00a | 50.30a | - | 44.20a | - |
| Table IIIResults of bronchoalveolar lavage
evaluation. |
Table III
Results of bronchoalveolar lavage
evaluation.
| Cell type | Percentage |
|---|
| Macrophages | 51.5 |
| Lymphocytes | 16.0 |
| Neutrophils | 31.5 |
| Eosinophils | 1.0 |
Cryobiopsy samples were obtained from the left lower
lobe by transbronchial lung cryobiopsy with a 1.9-mm probe
(freezing time, 5-8 sec) (6). A
total of 4 samples were taken. The frozen biopsy specimens were
thawed in saline and placed in formalin to obtain tissue for
histological analysis. Formalin-fixed lung tissues were
paraffin-embedded and cut into 5-µm sections for staining with
H&E (Sigma-Aldrich; Merck KGaA), Masson trichrome and Perls
(Prussian blue) staining (Nanjing Jiancheng Bioengineering
Institute) according to the manufacturer's protocols and examined
under a light microscope (Nikon Corp.). For confirmation of the
histological diagnosis, all of the available lung biopsy specimens
were reviewed by a pulmonary pathologist who was blinded to the
patient's clinical and radiological information.
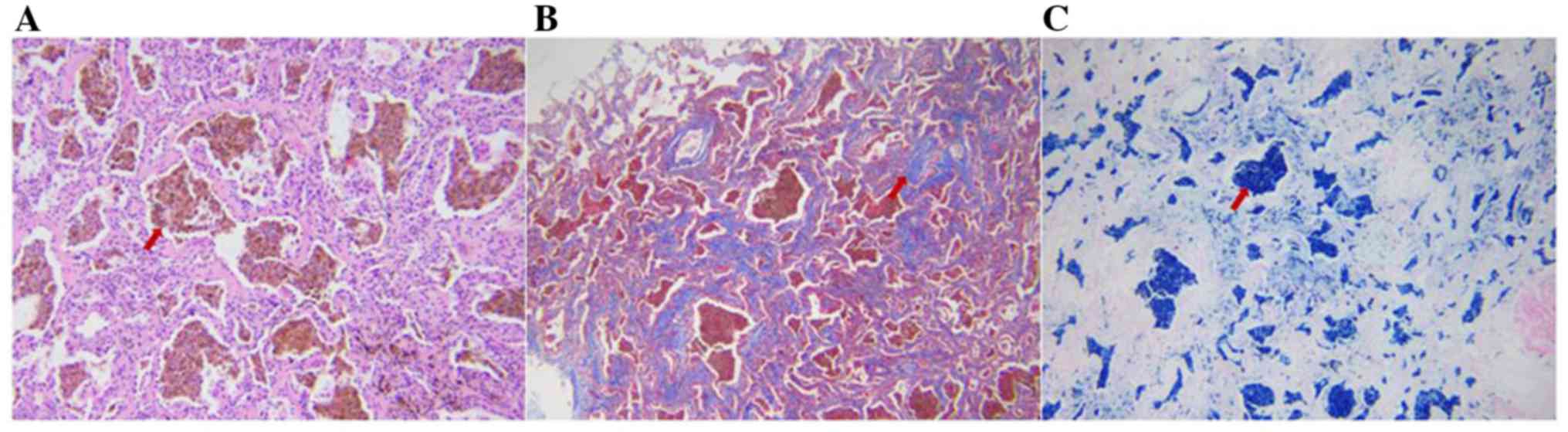
Histopathological examination revealed dense
accumulations of hemosiderin-laden macrophages and erythrocytes
invading into individual alveolar spaces, interalveolar septal
thickening and fibrosis (Fig. 3).
There was no evidence of concurrent vasculitis, granulomata or
organizing alveolitis on light microscopy, which was considered to
be consistent with the diagnosis of IPH (7,8).
Considering the results of the microscopic examination of the lung
tissue and the broad panel of tests employed, which did not reveal
the origin of the intra-alveolar bleeding, the diagnosis of primary
IPH was established.
High positivity for anti-CCP [a member of the family
of anti-citrullinated protein antibodies (ACPA)] was a significant
characteristic of this patient. It is well-known that there is a
close association between ACPA and bone destruction in RA (9). Previous studies support the
significance of ACPA in inflammation-associated bone diseases, not
confined to RA, but also in non-RA autoimmune conditions, and ACPA
may be detected in psoriatic arthritis, systemic lupus
erythematosus and juvenile idiopathic arthritis, as well as
scleroderma and CREST syndrome, followed by Sjögren's syndrome and
vasculitis (10-15).
However, the prevalence of ACPA positivity was higher in patients
with RA than in those mentioned above apart from RA (11,12).
Furthermore, similar pulmonary infiltrates detected
on the CT scan, as well as changes in pulmonary function, may be
observed in patients with systemic sclerosis. However, the patient
in the present study did not exhibit any typical clinical
manifestations of systemic sclerosis, including skin thickening on
the fingers, Raynaud's phenomenon and systemic sclerosis-associated
autoantibodies.
Thus, in the patient of the present study, the
combination of clinical presentation and laboratory results was
consistent with a diagnosis of RA according to the American College
of Rheumatology (ACR) criteria from 1987(16) (arthritis of 11 joint areas, arthritis
of hand joints, symmetric arthritis and serum RF) and the
ACR/European League Against Rheumatism 2010 classification criteria
(17) (joint swelling not better
explained by any other disease, joint involvement of >10
major/intermediate/small joints and at least 2 small joints, highly
positive RF and ACPA). In addition, the diagnosis of RA was
confirmed by an experienced rheumatologist. A final definitive
diagnosis of RA and IPH was then established.
After the diagnosis of IPH and RA had been made, the
patient began to receive high doses of oral prednisone (0.75 mg/kg)
for 8 weeks, which was gradually tapered to 0.5 mg/kg for another 6
weeks. The treatment led to a profound improvement of dyspnea and
arthralgia and a significant increase in hemoglobin levels, as well
as a decrease of the CCP and RF titers (Table I). A control high-resolution CT
(HRCT) obtained one month after steroid therapy revealed a marked
regression of the aforementioned lesions observed on previous HRCT
(Fig. 1B). Pulmonary function tests
and arterial blood gas analysis were also performed, indicating an
improvement during this period (Table
II). The patient was followed up at 3 and 6 months after
therapy by HRCT and no new lesions were observed (Fig. 1C and D). The patient has been closely monitored
due to the high relapse incidence of IPH (18) and remained in remission at the time
of the last follow-up in November 2019.
Discussion
IPH is a rare form of interstitial lung disease
characterized by recurrent intra-alveolar hemorrhage and secondary
IDA, usually occurring in young adults or children (1). Various theories have been proposed to
explain the etiology of IPH, including genetic, environmental and
auto-immune mechanisms (19-24).
The auto-immune theory provides the most acceptable explanation for
the pathophysiology of IPH, considering its frequent association
with auto-immune diseases, including RA (24).
The coexistence of IPH and RA was first reported by
Karlish (25), who described IPH
concurring with early RA in a 32-year-old male patient. A study by
Le Clainche et al (26)
followed 15 pediatric patients with IPH, revealing that ~25% of the
patients who survive with the condition for >10 years
subsequently develop autoimmune disorders, including RA. A total of
3 patients were reported to have developed RA at 6 months to 20
years after the initial diagnosis (26). In the patient of the present study,
RA developed with a marked delay from the onset of symptoms (15
years). The subsequent development of RA is of considerable
interest, since the association is insufficiently recognized due to
the delayed occurrence of RA associated with IPH (26).
RA is the most frequent systemic disease in the
general population (0.5-1.0%) and arthritis is at times associated
with respiratory symptoms, typically with diffuse parenchymal lung
disease (27). Macrophages are
central to the development of RA, and they may activate a wide
range of immune cells and secrete diverse tissue-degrading enzymes
mediating chronic pro-inflammatory, tissue-destructive and pain
responses in RA (28). In the
present case, the macrophages in the lung were abnormally activated
(hemosiderin-laden macrophages), which is necessary for
establishing the diagnosis of IPH by providing proof of chronic
intra-alveolar bleeding (29).
Furthermore, damage to pulmonary alveolar tissue by a variety of
agents may induce a macrophage-centered autoimmune process in
certain individuals, which may damage other non-pulmonary tissues,
e.g. in arthrosis (30). Thus, RA
may be caused by IPH through autoimmune responses, indicating IPH
may precede the development of RA; a multicenter cohort,
case-controlled study is required to confirm this.
Although IPH classically presents as a clinical
triad, including IDA, episodes of recurrent hemoptysis and
radiographic diffuse lung infiltrates, the clinical presentation
varies greatly, from silent attacks to a fulminant course with
rapidly worsening hypoxia (2,8,23), which may cause a significant delay in
the diagnosis of IPH. The quantity of hemoptysis may be variable
and is not a reliable index of the degree of pulmonary hemorrhage,
as alveolar bleeding does not readily reach the central airways
(2). Hemoptysis is also minimal and
goes unnoticed in children, as the sputum is mostly swallowed
(31). Thus, hemoptysis is only
detected in 50% of the patients (24), which leads to a delay between the
onset of symptoms and the diagnosis of IPH. There is frequently a
long delay of diagnosis, as the initial IDA usually precedes
pulmonary signs for several months (32). In the patient of the present study,
the delay in the diagnosis of IPH was by 15 years, since the onset
of anemia was at the age of 6 years. This delay may result in
pulmonary fibrosis due to iron overload within the lung, as free
ferric ions may induce the formation of toxic hydroxyl radicals
and/or stimulate fibrogenesis (33),
which is generally a poor prognostic factor (34). The delay in diagnosis may be due to
not only neglecting the cause of IDA, but also a lack of awareness
of clinicians regarding the condition.
Systemic corticosteroids have been reported as the
most effective treatment for IPH with favorable effects on alveolar
bleeding relapse and pulmonary fibrosis progression, as well as
with higher survival rates (35).
Early treatment with steroids has also been widely indicated to
shorten the duration of a hemorrhagic attack (36). Azathioprine in combination with
corticosteroids was proposed to be the best therapeutic regimen,
particularly for preventing IPH exacerbations (23). Considering the fertility of the case
of the present study, the patient treated with prednisone only for
a duration of 8 months and exhibited gradual improvement.
In conclusion, the current case provides supporting
evidence regarding the IPH-mediated pathology of RA; thus,
screening for autoimmune antibodies is recommended in patients with
IPH. In addition, through the present case, the authors intend to
emphasize that early IPH diagnosis and treatment are of grave
importance for obtaining a favorable outcome, which may delay or
even prevent the development of immune system diseases such as RA
in patients with IPH.
Acknowledgements
The authors would like to thank Dr Ling Zhao
(Department of Pathology, China-Japan Friendship Hospital, Beijing,
China) and Professor Min Liu (Department of Radiology, China-Japan
Friendship Hospital, Beijing, China) for their pathological and
radiological advice on the case.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81800036) and the National
Key Research and Development Program of China (grant no.
2016YFC0901102).
Availability of data and materials
All datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XR and TY analyzed and interpreted the patient data
and wrote the manuscript. JL and JZ collected original medical
records of the patient and drafted the manuscript. JG was in charge
of the follow-up of the patient, and acquisition and analysis of
these data. HD analyzed and interpreted the patient data and
revised the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Approval was obtained from the Medical Ethics
Committee of China-Japan Friendship Hospital (Beijing, China; no.
2018-148-K105). Written informed consent was obtained from the
patient regarding participation in the study. The sample collection
took place during hospitalization and the follow-up period.
Patient consent for publication
Informed consent was obtained from the patient
regarding the publication of data and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kjellman B, Elinder G, Garwicz S and Svan
H: Idiopathic pulmonary haemosiderosis in Swedish children. Acta
Paediatr Scand. 73:584–588. 1984.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ohga S, Takahashi K, Miyazaki S, Kato H
and Ueda K: Idiopathic pulmonary haemosiderosis in Japan: 39
possible cases from a survey questionnaire. Eur J Pediatr.
154:994–998. 1995.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Al Jassmi AM: Autoimmunity and delayed
diagnosis in pediatric idiopathic pulmonary hemosiderosis. J
Pediatr Hematol Oncol. 42:e240–e243. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Louie S, Russell LA and Richeson RB:
Circulating immune complexes with pulmonary hemorrhage during
pregnancy in idiopathic pulmonary hemosiderosis. Chest.
104:1907–1909. 1993.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lemley DE and Katz P: Rheumatoid-like
arthritis presenting as idiopathic pulmonary hemosiderosis: A
report and review of the literature. J Rheumatol. 13:954–957.
1986.PubMed/NCBI
|
|
6
|
Kania A, Misiaszek M, Vašáková M,
Szlubowski A, Bugalho A, Pankowski J, Szołkowska M, Roden AC,
Celejewska-Wójcik N, Nastałek P, et al: Cryobiopsy in the diagnosis
of idiopathic pulmonary hemosiderosis: A case report. J Thorac Dis.
11:3195–3201. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yao TC, Hung IJ, Jaing TH and Yang CP:
Pitfalls in the diagnosis of idiopathic pulmonary haemosiderosis.
Arch Dis Child. 86:436–438. 2002.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ioachimescu OC, Sieber S and Kotch A:
Idiopathic pulmonary haemosiderosis revisited. Eur Respir J.
24:162–170. 2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hecht C, Englbrecht M, Rech J, Schmidt S,
Araujo E, Engelke K, Finzel S and Schett G: Additive effect of
anti-citrullinated protein antibodies and rheumatoid factor on bone
erosions in patients with RA. Ann Rheum Dis. 74:2151–2156.
2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Saigal R, Bhakal SS, Goyal L and Goel AD:
Seroprevalence of anti-citrullinated protein antibodies (ACPA) in
patients with rheumatic diseases other than rheumatoid arthritis. J
Assoc Physicians India. 66:26–28. 2018.PubMed/NCBI
|
|
11
|
Yuan S, Chen D, Xiao Y, Lao M, Qiu Q,
Liang L and Yang X: Factors associated with erosive arthritis in
rheumatoid arthritis and other connective tissue diseases: A
retrospective study from a southern Chinese population. J Clin
Rheumatol. 22:22–29. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Conigliaro P, Chimenti MS, Triggianese P,
Sunzini F, Novelli L, Perricone C and Perricone R: Autoantibodies
in inflammatory arthritis. Autoimmun Rev. 15:673–683.
2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Pang SY, Liu HY, Huang YJ, Liu YF, Dai YM,
Zeng P and Zeng HS: Diagnostic performance of anti-citrullinated
protein/peptide antibodies in juvenile idiopathic arthritis. Genet
Mol Res. 15(gmr.15028641)2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wu R, Shovman O, Zhang Y, Gilburd B,
Zandman-Goddard G and Shoenfeld Y: Increased prevalence of
anti-third generation cyclic citrullinated peptide antibodies in
patients with rheumatoid arthritis and CREST syndrome. Clin Rev
Allergy Immunol. 32:47–56. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Cho SB, Lee JH, Ahn KJ, Bae BG, Kim T,
Park YB, Lee SK, Lee KH and Bang D: Anti-cyclic citrullinated
peptide antibodies and joint involvement in Behçet's disease.
Yonsei Med J. 53:759–764. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Arnett FC, Edworthy SM, Bloch DA, McShane
DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS,
et al: The American Rheumatism Association 1987 revised criteria
for the classification of rheumatoid arthritis. Arthritis Rheum.
31:315–324. 1988.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Aletaha D, Neogi T, Silman AJ, Funovits J,
Felson DT, Bingham CO III, Birnbaum NS, Burmester GR, Bykerk VP,
Cohen MD, et al: 2010 Rheumatoid arthritis classification criteria:
An American College of Rheumatology/European League Against
Rheumatism collaborative initiative. Arthritis Rheum. 62:2569–2581.
2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhang Y, Luo F, Wang N, Song Y and Tao Y:
Clinical characteristics and prognosis of idiopathic pulmonary
hemosiderosis in pediatric patients. J Int Med Res. 47:293–302.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gencer M, Ceylan E, Bitiren M and Koc A:
Two sisters with idiopathic pulmonary hemosiderosis. Can Respir J.
14:490–493. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Gerhardy B and Simpson G: Melioidosis and
idiopathic pulmonary hemosiderosis: A cast-iron case. Respirol Case
Rep. 1:46–47. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Cassimos CD, Chryssanthopoulos C and
Panagiotidou C: Epidemiologic observations in idiopathic pulmonary
hemosiderosis. J Pediatr. 102:698–702. 1983.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Nacaroglu HT, Sandal OS, Bag O, Erdem SB,
Bekem Soylu O, Diniz G, Ozturk A and Can D: Association of celiac
disease with idiopathic pulmonary hemosiderosis; Lane Hamilton
Syndrome. Iran J Pediatr. 25(e3312)2015.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Chen XY, Sun JM and Huang XJ: Idiopathic
pulmonary hemosiderosis in adults: review of cases reported in the
latest 15 years. Clin Respir J. 11:677–681. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Taytard J, Nathan N, de Blic J, Fayon M,
Epaud R, Deschildre A, Troussier F, Lubrano M, Chiron R, Reix P, et
al: New insights into pediatric idiopathic pulmonary hemosiderosis:
The French RespiRare(®) cohort. Orphanet J Rare Dis.
8(161)2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Karlish AJ: Idiopathic pulmonary
haemosiderosis with unusual features. Proc R Soc Med. 55:223–225.
1962.PubMed/NCBI
|
|
26
|
Le Clainche L, Le Bourgeois M, Fauroux B,
Forenza N, Dommergues JP, Desbois JC, Bellon G, Derelle J, Dutau G,
Marguet C, et al: Long-term outcome of idiopathic pulmonary
hemosiderosis in children. Medicine (Baltimore). 79:318–326.
2000.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Clement A, Nathan N, Epaud R, Fauroux B
and Corvol H: Interstitial lung diseases in children. Orphanet J
Rare Dis. 5(22)2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nagasawa Y, Takada T, Shimizu T, Narita J,
Moriyama H, Terada M, Suzuki E and Gejyo F: Inflammatory cells in
lung disease associated with rheumatoid arthritis. Intern Med.
48:1209–1217. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cohen S: Idiopathic pulmonary
hemosiderosis. Am J Med Sci. 317:67–74. 1999.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Smith BS: Idiopathic pulmonary
haemosiderosis and rheumatoid arthritis. Br Med J. 1:1403–1404.
1966.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Matsaniotis N, Karpouzas J, Apostolopoulou
E and Messaritakis J: Idiopathic pulmonary haemosiderosis in
children. Arch Dis Child. 43:307–309. 1968.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kuhn MJ: Idiopathic pulmonary
hemosiderosis: The importance of a chest radiograph in children
with unexplained anemia. Mt Sinai J Med. 52:358–362.
1985.PubMed/NCBI
|
|
33
|
Mateos F, Brock JH and Pérez-Arellano JL:
Iron metabolism in the lower respiratory tract. Thorax. 53:594–600.
1998.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Casian A and Jayne D: Current modalities
in the diagnosis of pulmonary vasculitis. Expert Opin Med Diagn.
6:499–516. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Bhatia S, Tullu MS, Vaideeswar P and
Lahiri KR: Idiopathic pulmonary hemosiderosis: Alveoli are an
answer to anemia. J Postgrad Med. 57:57–60. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Erkoçoğlu M, Civelek E and Kocabaş CN:
Unusual presentation: Concurrent IgA deficiency and idiopathic
pulmonary hemosiderosis. Pediatr Pulmonol. 51:E34–E36.
2016.PubMed/NCBI View Article : Google Scholar
|