Introduction
Adrenocortical carcinoma (ACC) is a rare urological
tumor with an annual incidence of 0.7-2/million (1). ACC is highly invasive and metastatic.
Meanwhile, the prognosis of ACC is poor and most patients survive
only 4-30 months. The 5-year overall survival rate is 16-47% and
only 5-10% for advanced patients (2). In addition, diagnosis of ACC is
difficult. Indeed, more than one-half of the patients display
metastatic symptoms as the first clinical manifestation and many
cases remain difficult to diagnose even after pathological
diagnosis. Therefore, there is great interest in determining the
molecular mechanisms of ACC onset and progression and in developing
diagnostic and therapeutic strategies.
Microarray technologies and bioinformatics analysis
have made high-throughput genome-wide sequencing and measurement of
gene expression possible. Thus, key signaling pathways can be
elucidated comprehensively and systematically, thereby revealing
the molecular mechanisms of disease development and progression. In
the present study, three mRNA microarray datasets from the Gene
Expression Omnibus (GEO) database were screened for obtaining
differentially expressed genes (DEGs) and ACC hub genes were chosen
from the most significant module. Subsequently, a protein-protein
interaction (PPI) network was established and gene enrichment,
survival, co-expression and cluster analysis were performed for the
hub genes. These analyses may help clarify the mechanisms of
carcinogenesis and progression of ACC and identify new targets for
treatment.
Materials and methods
Research process
In the present study, three microarray datasets from
the GEO database (www.ncbi.nlm.nih.gov/geo/) were screened according to
specific inclusion and exclusion criteria. A total of 96 DEGs were
chosen to analyze. The PPI network of DEGs was constructed and
corresponding enrichment analysis was performed. From these
analyses, hub genes were identified from the most significant
module (degree cutoff=2; node score cutoff=0.2; K-core=2) in the
PPI network. Subsequently, enrichment, survival and cluster
analysis were performed on these hub genes. Finally, the Oncomine
online database (www.oncomine.org/) was used to further verify the
differential expression of hub genes between ACC and normal tissue,
and to analyze the relationships between clinical phenotypes and
gene expression. Fig. 1 summarizes
this research process.
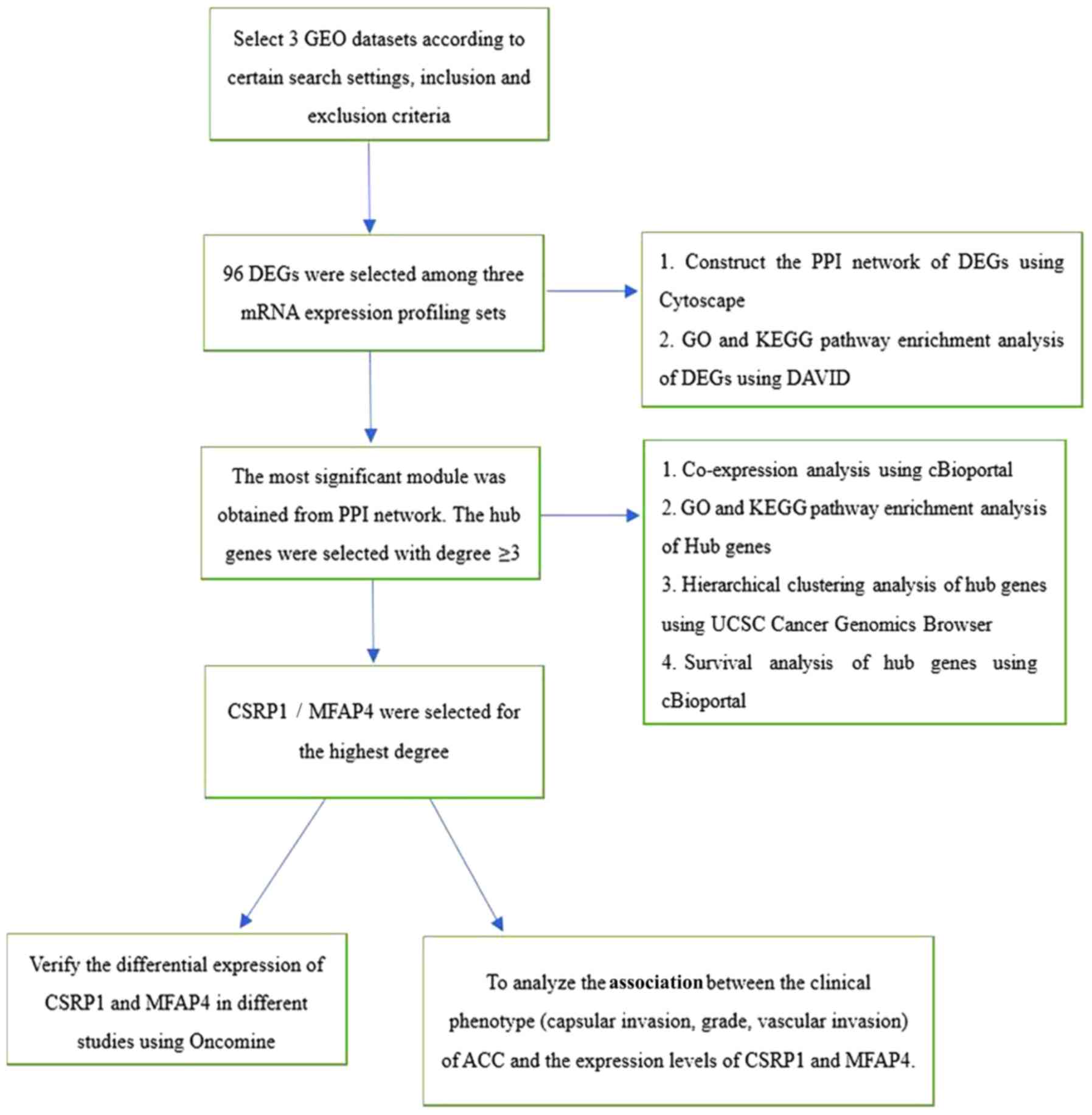 | Figure 1Research process. DEG, differentially
expressed gene; PPI, protein-protein interaction; GO, Gene
Ontology; CSRP1, cysteine and glycine rich protein 1; MFAP4,
microfibril associated protein 4; GEO, Gene Expression Omnibus;
KEGG, Kyoto Encyclopedia of Genes and Genomes; DAVID, Database for
Annotation, Visualization and Integrated Discovery; UCSC,
University of California Santa Cruz. |
Dataset screening
Relevant datasets were obtained from the GEO
database using the key words ‘Adrenal cortical carcinoma’ OR
‘Adrenocortical carcinoma’ OR ‘Adrenal carcinoma’. The research
type was set to ‘Expression profiling by array’ and the organism
was selected as ‘Homo sapiens’. In total, 28 relevant
datasets were initially identified. Datasets GSE19750(3), GSE12368(4) and GSE14922(5) were ultimately selected according to the
following inclusion criteria: i) Achievable comparison of ACC with
normal adrenal tissue; and ii) original data can be downloaded in
CEL format. In addition, the following exclusion criteria were
applied: i) Childhood ACC; and ii) use of molecular targeted drugs
for ACC before surgical treatment.
DEG identification
Using GEO 2R online analysis software (www.ncbi.nlm.nih.gov/geo/geo2r/), each
dataset was divided into ACC group and normal tissue group. The
TOP250 option was then used to obtain a genomic profile of DEGs
between the tumor and normal groups in each dataset. A P-value
<0.01 and LogFC absolute value ≥0.5 were used as initial
screening conditions, where FC indicates fold change. DEGs which
are shared between datasets are presented in Venn diagrams.
KEGG and GO enrichment analyses
DEGs were subjected to gene enrichment analysis to
obtain the main biological functions and signaling pathways in
which they were involved. The Gene Ontology (GO) Consortium
(geneontology.org/) is a database of new semantics
vocabulary standards that are applicable to various species that
can define and describe gene and protein functions (6). GO genetic annotations fall into three
broad categories: i) Molecular function (MF); ii) biological
process (BP); and iii) cellular component (CC). Gene function was
defined and described according to these categories.
Kyoto Encyclopedia of Genes and Genomes (www.kegg.jp; version 94.0; KEGG) is a comprehensive
database that integrates information on genomic, chemical and
system functions (7). Using the KEGG
database, information on the signaling pathways of genes can be
obtained to deeply excavate the molecular mechanisms of the
genes.
Database for Annotation, Visualization and
Integrated Discovery (DAVID) is an online bioinformatics analysis
and integration tool (david.ncifcrf.gov) for Functional Annotation, Gene
Functional Classification, Gene ID Conversion and other analyses
(8). DAVID (version 6.8) was used to
complete the GO and KEGG enrichment analyses of DEGs and hub genes
to obtain information on their molecular functions, biological
processes, cytogenetics and signaling pathways.
PPI network construction and module
analysis
Functional links among proteins often reflect the
genetic association among their genes. A PPI network can be used to
describe the interactions among proteins and identify hub
regulatory genes of disease. The STRING database (version 11.0;
string-db.org) can search for interactions
between known and predicted proteins, which can be used to analyze
and establish the PPI network of DEGs (9). Cytoscape (version 3.4.0; Cytoscape User
Support, Education and New Initiatives are supported by the
National Resource for Network Biology; award no. P41 GM103504) is
an open source bioinformatics software platform for visualizing
molecular interaction networks (10). The Cytoscape plugin MCODE is an
application for cluster analysis (11). With Cytoscape, a visualization of the
molecular functions of DEGs can be obtained. Using the clustering
analytic function of MCODE, the most significant module in a PPI
network of DEGs was obtained; the hub genes were derived from this
module.
Hub gene selection and analysis
After obtaining the most significant module in the
PPI network of DEGs, genes with a score ≥3 were selected as hub
genes. PubMed Gene was employed to perform functional description
of the hub genes (www.ncbi.nlm.nih.gov/gene/). The cBioPortal
(www.cbioportal.org) platform was used to
establish a network relationship between the hub genes and their
co-expressed genes. The Cytoscape plugin BiNGO was used to
visualize the BP of hub genes (12).
The University of California Santa Cruz (UCSC) Cancer Genomics
Browser (https://genome-cancer.ucsc.edu/) is a genomic database
containing >22,700 shares of sample information (13). Users can explore the relationships
between genomic changes and clinical phenotypes using visualized
clinical data and phenotypic characteristics, such as age, tissue
grade and pathology subtypes. Hierarchical clustering analysis of
hub genes by the USCS Cancer Genomics Browser can identify the
differential expression of hub genes between tumors and normal
samples. The analysis can evaluate whether hub genes could be used
as diagnostic markers.
To assess the potential function of hub genes in
clinical progression of ACC, the prognostic analysis and clinical
correlation analysis were performed. Overall survival rate and
disease-free survival rate in ACC were analyzed using cBioPortal.
Oncomine (www.oncomine.org) was used to further
verify whether the expression of hub genes between ACC and normal
tissues was significant different (P<0.05) and to evaluate the
relationships between expression of hub genes and clinical
phenotypes, including capsular invasion, grade and vascular
invasion. The clinical correlation analysis is based on the
Kolmogorov–Smirnov test. During the verification of Oncomine
database, we set the following parameters: i) Analysis type, cancer
vs. normal analysis; ii) cancer type, adrenal cortex carcinoma; and
iii) data type, mRNA.
Results
Identification of DEGs in ACC
In the present study, ‘Adrenocortical carcinoma’,
‘Adrenal cortical carcinoma’ and ‘Adrenal carcinoma’ were used as
the search terms for the GEO database. Initially, 815 studies were
obtained. Subsequently, 29 studies were obtained through study type
filter (set as expression profiling by array), of which only nine
were of human tissue origin and the rest were animal or cytological
experiments. In the residual nine studies, GSE90713 involved
metastatic ACC samples and GSE73417 involved a neoplastic
transplant model. GSE19776, GSE19775, GSE28476 and GSE15918 did not
include compared normal tissues. Thus, only three datasets were
ultimately selected.
Finally, three datasets from the GEO database were
selected according to the aforementioned criteria: i) GSE19750 (44
ACC; 4 normal); ii) GSE12368 (28 ACC; 6 normal); and iii) GSE14922
(4 ACC; 4 normal; 4 non-functioning adenomas; 4 secretory type).
DEGs were identified in each dataset. In total, 1,464, 764 and
1,088 DEGs were identified in GSE19750, GSE12368 and GSE14922,
respectively. As a result, 96 DEGs were shared across all three
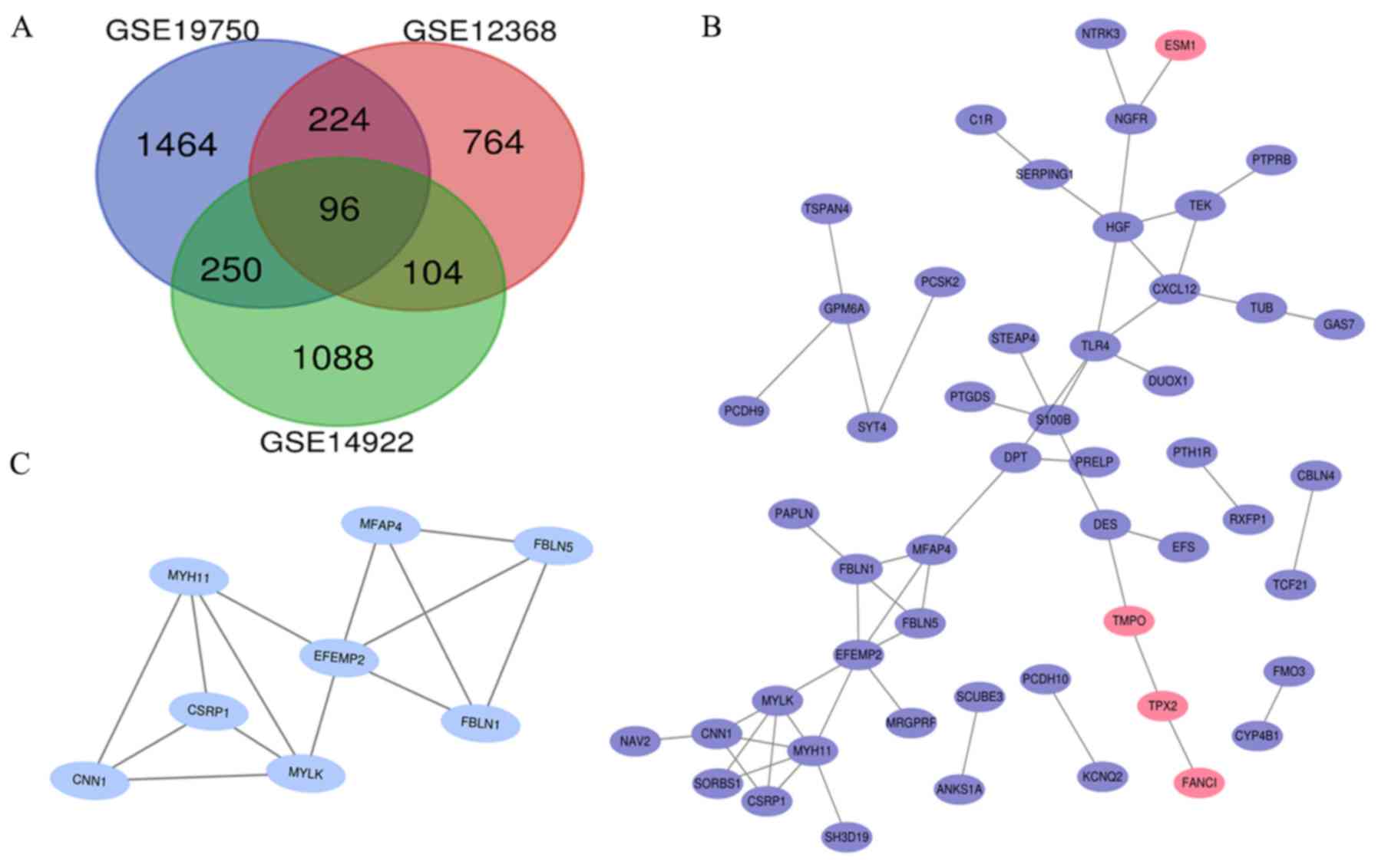
datasets (Fig. 2A).
GO and KEGG enrichment analysis of
DEGs
DAVID ver. 6.8 was used to perform GO and KEGG
enrichment analyses for all identified DEGs. The pathways with
P<0.05 and the highest enrichment, based on the number of
enriched genes, are presented in Table
I. The CCs associated with the DEGs in the present study were
mainly extracellular structures, including ‘extracellular exosome’,
‘extracellular region’ and ‘extracellular space’. The MFs of these
DEGs were predominantly associated with functional binding,
including ‘actin binding’ and ‘integrin binding’. Moreover, DEGs
were found to be related with some tumor biological process, such
as ‘cell adhesion’, ‘muscle contraction’ and ‘negative regulation
of inflammatory response’. According to the KEGG signaling pathway
analysis, DEGs were significantly enriched in ‘drug
metabolism-cytochrome P450’ and the ‘pertussis’ pathways.
 | Table IGO and KEGG enrichment analysis of
differentially expressed genes. |
Table I
GO and KEGG enrichment analysis of
differentially expressed genes.
| A, Cellular
component |
|---|
| Term | Description | Count in gene
set | P-value |
|---|
| GO:0070062 | Extracellular
exosome | 26 | 0.0019 |
| GO:0005576 | Extracellular
region | 20 | 0.0003 |
| GO:0005615 | Extracellular
space | 17 | 0.0008 |
| B, Molecular
function |
| Term | Description | Count in gene
set | P-value |
| GO:0005509 | Calcium ion
binding | 12 | 0.0007 |
| GO:0003779 | Actin binding | 6 | 0.010 |
| GO:0005178 | Integrin
binding | 4 | 0.0150 |
| C, Biological
process |
| Term | Description | Count in gene
set | P-value |
| GO:0007155 | Cell adhesion | 8 | 0.0092 |
| GO:0006936 | Muscle
contraction | 5 | 0.0022 |
| GO:0050728 | Negative regulation
of inflammatory response | 4 | 0.0078 |
| D, KEGG
pathway | | | |
| Term | Description | Count in gene
set | P-value |
| Hsa00982 | Drug
metabolism-cytochrome P450 | 3 | 0.0395 |
| Hsa05133 | Pertussis | 3 | 0.0471 |
PPI network and module analysis
Cytoscape (version 3.4.0) was used to construct a
PPI network of DEGs (Fig. 2B). MCODE
was used to extract the most significant module from the PPI
network (Fig. 2C). The MCODE
parameters were the following: i) Degree cut-off=2; ii) node score
cut-off=0.2; iii) max depth=100; and iv) k-score=2(11). The most prominent module had 8 nodes
and 14 edges. DAVID was used to perform GO and KEGG enrichment
analyses of the module (Table II).
The genes in this most prominent module were not significantly
enriched in KEGG pathway analysis (P>0.05). In the GO analysis,
the module was mainly enriched in some extracellular functions and
structures, such as the ‘extracellular exosome’, ‘extracellular
region’, ‘elastic fiber’ and ‘elastic fiber assembly’.
| Table IIGO enrichment analysis of
differentially expressed genes in the most significant module. |
Table II
GO enrichment analysis of
differentially expressed genes in the most significant module.
| A, Cellular
component |
|---|
| Term | Description | Count in gene
set | P-value |
|---|
| GO:0070062 | Extracellular
exosome | 7 |
8.15x10-5 |
| GO:0005576 | Extracellular
region | 4 | 0.0183 |
| GO:0071953 | Elastic fiber | 3 |
7.59x10-7 |
| B, Molecular
function |
| Term | Description | Count in gene
set | P-value |
| GO:0005516 | Calmodulin
binding | 3 | 0.0025 |
| GO:0005509 | Calcium ion
binding | 3 | 0.0328 |
| GO:0005201 | Extracellular
matrix structural constituent | 3 | 0.0274 |
| C, Biological
process |
| Term | Description | Count in gene
set | P-value |
| GO:0048251 | Elastic fiber
assembly | 3 |
2.23x10-6 |
| GO:0006939 | Smooth muscle
contraction | 2 | 0.0064 |
| GO:0006936 | Muscle
contraction | 2 | 0.0376 |
Hub gene selection and analysis
The DEGs were selected as hub genes if their cluster
degrees were ≥3.0 in the MCODE analysis. A total of eight hub genes
were identified, all of which were contained in the most
significant module. PubMed Gene was used to obtain the
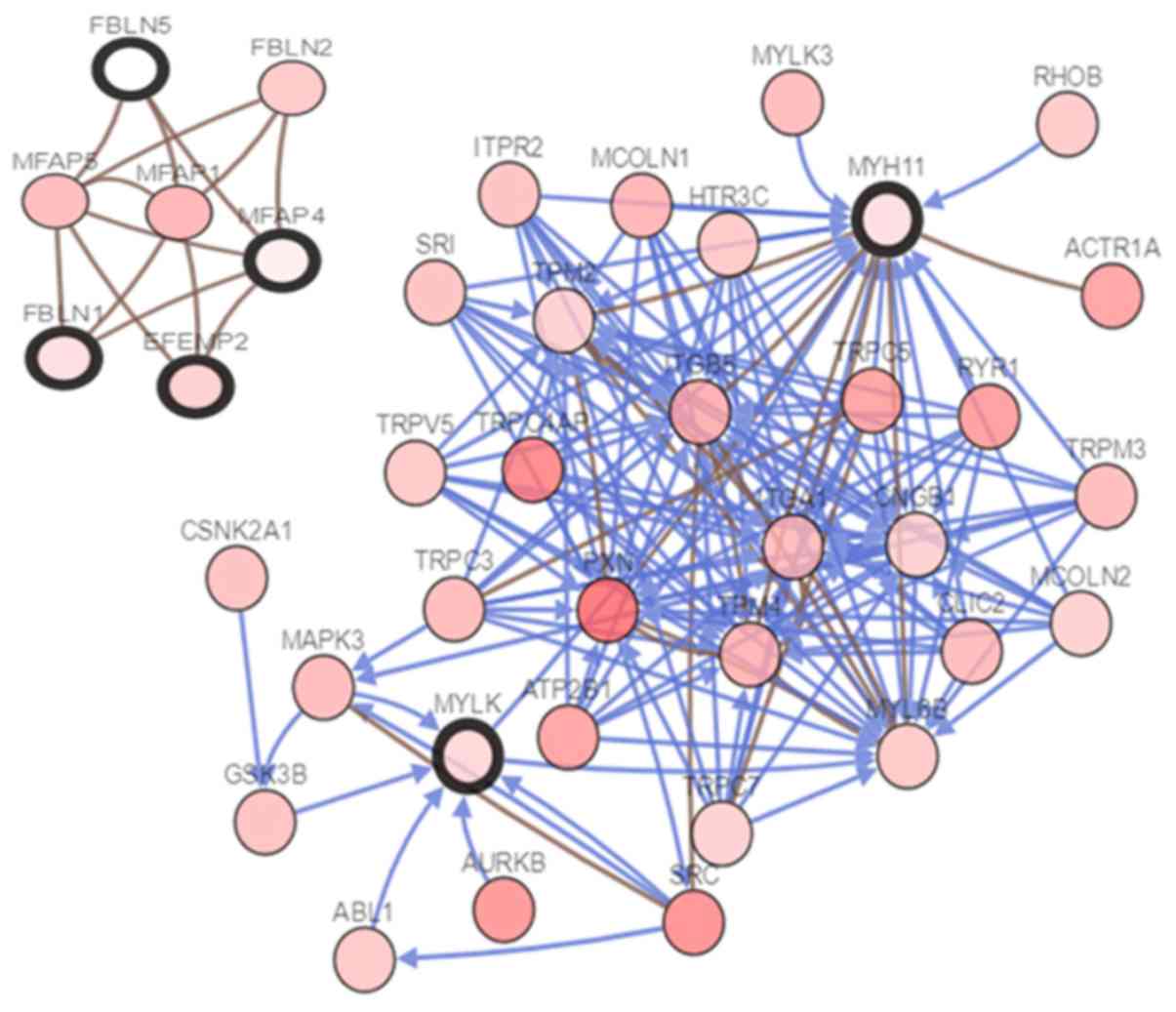
corresponding gene names, abbreviations and functions (Table III). The cBioportal online tool was
used to construct a co-expressed gene network of the hub genes
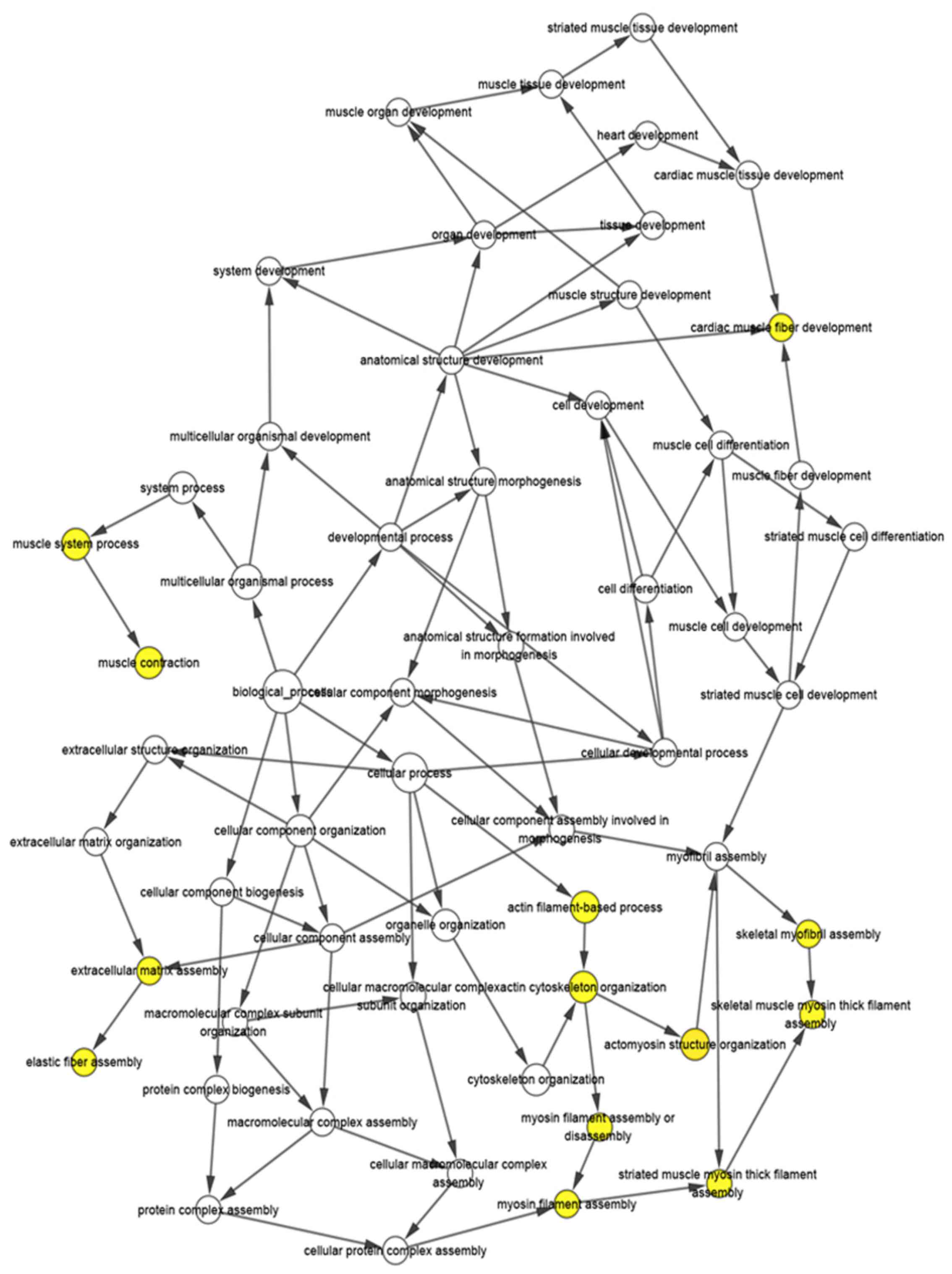
(Fig. 3) and the BP visualization
network of the hub genes was completed via BiNGO (Fig. 4). Using UCSC for hierarchical
clustering analysis, the hub genes displayed low expression in
tumor tissues, compared with normal tissues (Fig. 5).
| Table IIIFunctional roles of hub genes. |
Table III
Functional roles of hub genes.
| Gene | Full name | Function |
|---|
| CNN1 | Calponin 1 | Cell proliferation,
anchorage-independent colony formation, cell motility and
invasion. |
| CSRP1 | Cysteine and
glycine rich protein 1 | A growth factor,
cell proliferation, somatic differentiation. |
| MYLK | Myosin light chain
kinase | Catalyze the
phosphorylation of myosin light chains (MLC), cell invasion and
metastasis. |
| MYH11 | Myosin heavy chain
11 | Hydrolysis of ATP,
cell migration and adhesion, intracellular transport, signal
transduction. |
| EFEMP2 | EGF containing
fibulin extracellular matrix protein 2 | Blood coagulation,
activation of complement, determination of cell fate during
development. |
| FBLN1 | Fibulin 1 | Cell adhesion,
migration, differentiation. |
| MFAP4 | Microfibril
associated protein 4 | Cell adhesion,
intercellular interactions. |
| FBLN5 | Fibulin 5 | Angiogenesis,
epithelial cell motility, the activity of matrix metalloprotease 9
(MMP-9). |
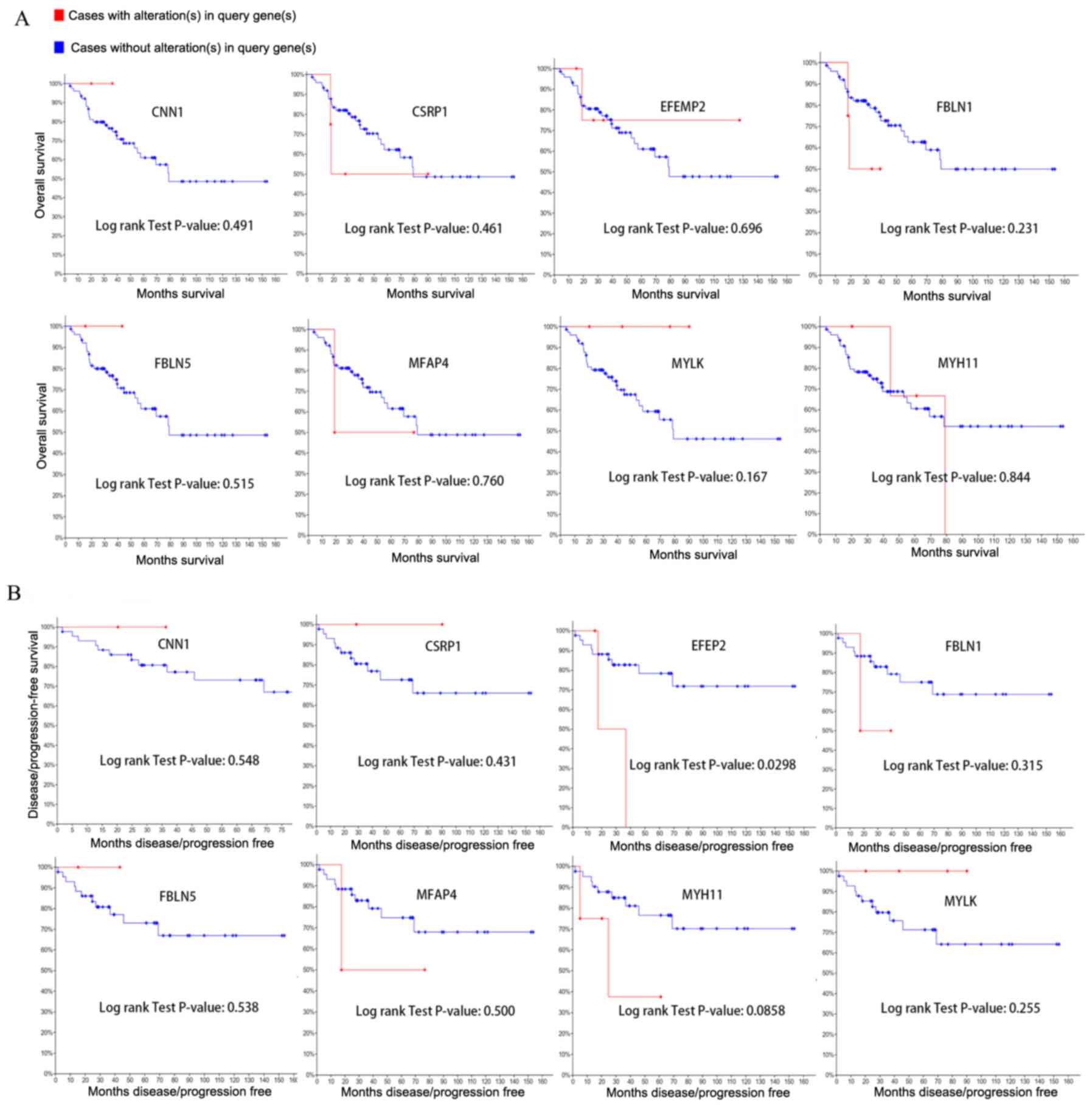
Changes in the expression of all hub genes did not
affect overall survival rate (Fig.
6). However, alteration of EGF containing fibulin extracellular
matrix protein 2 (EFEMP2) led to a decline in disease-free survival
rate.
The hub genes, cysteine and glycine rich protein 1
(CSRP1) and microfibril associated protein 4 (MFAP4), showed the
highest node degree of 5, suggesting that these genes may have
important functions in ACC carcinogenesis and progression.
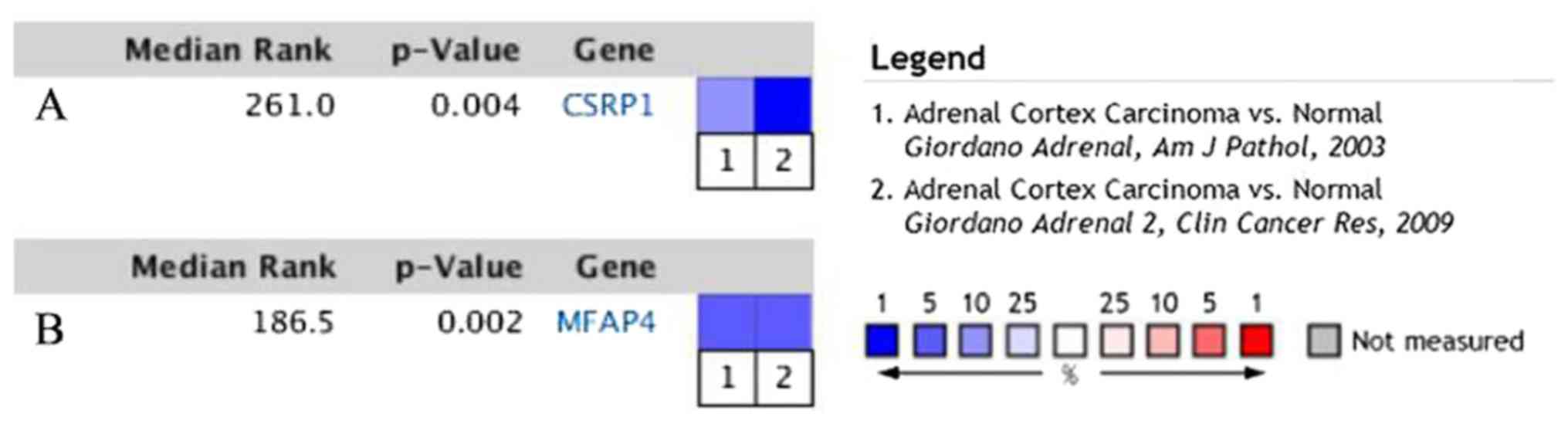
Subsequently, further verification was carried out via the Oncomine
database. CSRP1 and MFAP4 were significantly downregulated in
different studies (14,15) (Fig.
7). Among these studies (14,15),
only Giordano et al's study (14) was provided with sufficient
information of clinicopathological features (including capsular
invasion, histological grade and vascular invasion), hence, which
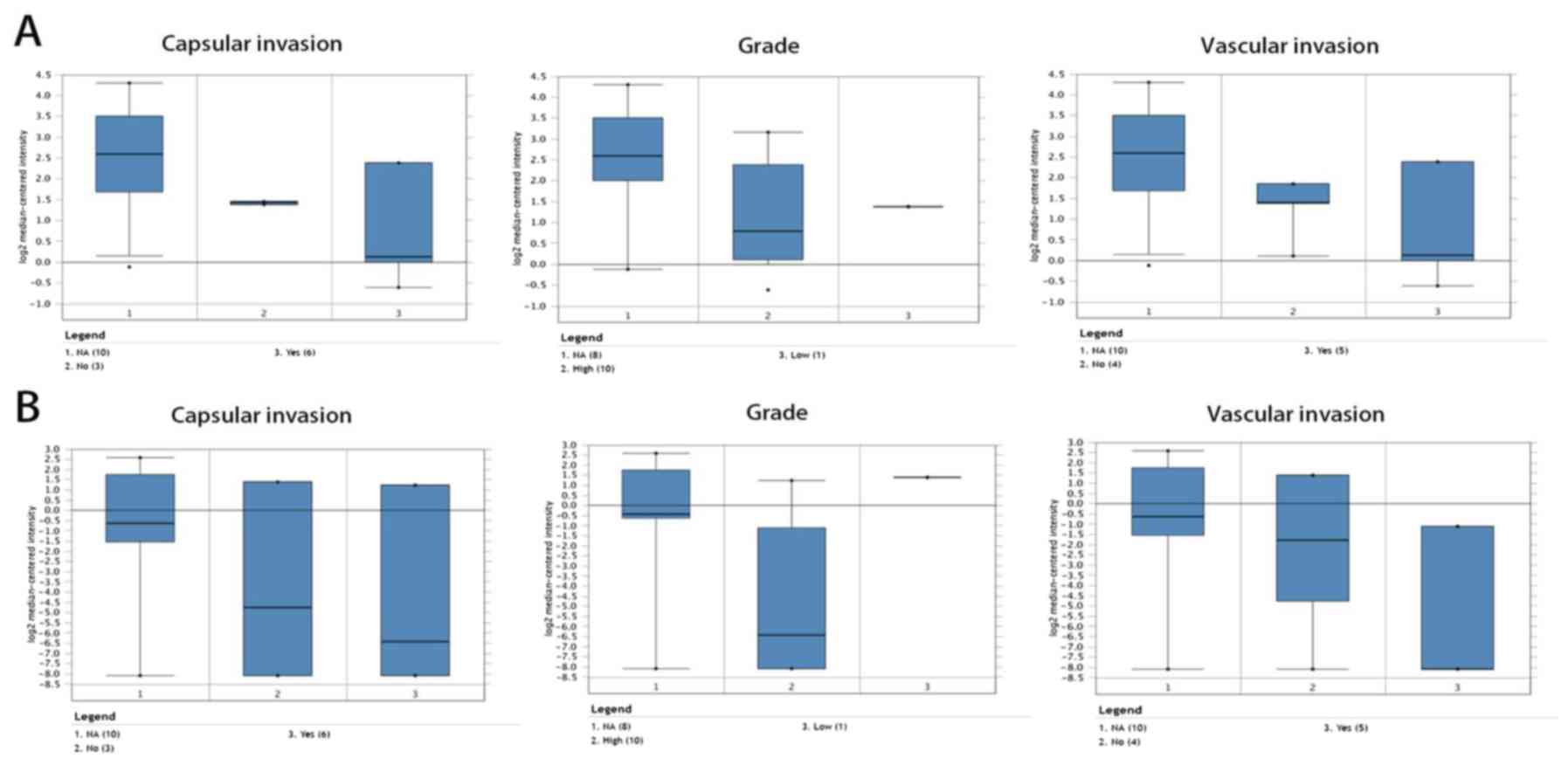
was used to perform clinical correlation analysis. The results
revealed that lower mRNA levels of CSRP1 and MFAP4 were associated
with adverse capsular invasion, grade and vascular invasion
(Fig. 8).
Discussion
The incidence of ACC is low; however, due to its
high potential for malignancy and metastasis, the 5-year survival
rate of patients is only 16-47% (16). In addition, ACC is difficult to
diagnose, even with imaging, hormone tests and postoperative
diagnostic methods. Therefore, understanding the mechanisms of
carcinogenesis and progression in ACC is of great importance to
search for potential diagnostic markers and therapeutic
targets.
Microarray technology has enabled assessment of
genetic expression changes in ACC, which has provided insight into
the molecular mechanism of this disease and has already been used
extensively in cancer research. In the present study, three ACC
mRNA matrix datasets from the GEO database were screened, allowing
the identification of 96 DEGs. Enrichment analysis indicated that
these DEGs were associated with some tumor-related BPs, such as
‘cell adhesion’ and ‘negative regulation of inflammatory response’
and may regulate ACC tumorigenesis and progression through the
binding to other functional proteins (calcium-binding proteins,
actin and integrin). In addition, the major functional region of
these regulatory processes appeared to be extracellular locations.
Cell adhesion is mediated by adhesion molecules, including members
of the immunoglobulin superfamily and the integrin family.
Integrins have crucial activities in regulating immune cell
function, including transport of immune cells into tissues,
activation of effector cells and formation of immune synapses
between immune cells and tumor cells (14). Therefore, research on integrins is an
active field in basic oncology. The present study identified that
DEGs were significantly enriched in integrin regulation, suggesting
that the selected DEGs play a crucial role in ACC carcinogenesis
and progression.
The inflammatory response is also closely related to
tumor progression. Although the immune system can recognize and
kill tumor cells, the inflammatory response induced by immunization
can also promote the proliferation of tumor cells and inhibit the
anticancer response (17). Thus,
inflammatory processes, as well as major metabolites involved in
inflammation, including adiponectin and high-density lipoprotein,
are strongly associated with the risk and invasiveness of solid
tumors (18-20).
The GO enrichment analysis also demonstrated that DEGs are involved
in ‘negative regulation of inflammatory response’ (Table I). However, the concrete inflammatory
regulatory mechanisms of DEGs relied on further research. In
summary, the enrichment analysis results of the current study are
consistent with previous oncology research (12,21).
Using MCODE, the most significant module in the PPI
network was obtained and eight hub genes with degree ≥3 were
identified. The hub genes identified in the present study were all
downregulated in ACC, compared with normal tissue. This result was
not in agreement with a previous study by Xiao et al
(22). Several reasons might account
for this difference. A possibility is the use of different study
groups. From six datasets in the GEO database, Xiao et al
(22) considered DNA topoisomerase
II α (TOP2A), NDC80 kinetochore complex component, centrosomal
protein 55, cyclin-dependent kinase inhibitor 3 and
cyclin-dependent kinase 1, as five key genes that affect the
progression and prognosis of ACC. In their analysis, the specimens
of GSE33371 were from breast cancer and the specimens of GSE75415
were from adrenal cortical tumors of children. The dataset
selection in the current study, however, involved a different study
group. Alternatively, differences in preliminary screening of the
mRNA expression datasets could also explain the discrepancies
between the two studies. Unlike the previous study by Xiao et
al (22), a preliminary
screening of the mRNA datasets was conducted before obtaining the
DEGs. The criteria were LogFC ≥0.5 and P<0.01, to ensure that
the genes entering the analysis reached the pre-set threshold for
statistical significance. Thus, differences in screening conditions
and analysis likely explain the different results between previous
research and the present study.
Compared with upregulated hub genes identified in
previous studies, the present results suggested that downregulated
hub genes are also important in carcinogenesis. The following
descriptions of the ACC-associated downregulated genes speculated
how they may contribute to ACC onset.
In ovarian cancer, calponin 1 (CNN1) is an important
tumor suppressor gene (23). Low
expression of CNN1 in peritumoral vessels is negatively related to
the expression of vascular endothelial growth factor, which is
involved in the generation of tumor blood vessels (24). In addition, CNN1 was associated with
the progression and prognosis of bladder cancer in a previous study
(25).
CSRP1 and MFAP4 are expressed at low levels in some
tumors, yet this was shown to have different consequences in
different tumor types or stages. CSRP1 was hypothesized to be a
tumor suppressor gene in colorectal cancer (26). In addition, CSRP1 may be inactivated
due to abnormal methylation and may be an important diagnostic
marker for liver cancer (27).
However, celecoxib may exhibit anti-gastric cancer effects by
suppressing expression of CSRP1(28). In the present study, CSRP1 was
downregulated in ACC, which indicated that CSRP1 may be a tumor
suppressor gene. MFAP4 is a tumor suppressor gene in prostate
cancer and it displays low expression in breast cancer (29,30). By
contrast, downregulated MFAP4 may lead to adverse clinical
incidents in ovarian cancer (31).
This contradiction may be explained by the fact that, in early
stage cancer, MFAP4 facilitates inflammatory cell recruitment and
assists immunological cancer surveillance to restrain cancer cell
survival (32). However, in advance
stage, alteration of the tumor microenvironment results in
decreased immune function of lymphocytes and MFAP4 predominantly
promoted cancer cell proliferation and migration (33). Similarly, it's putative that low
expression of MFAP4 ineffectively activate immune and inflammatory
cells to suppress malignant progression of ACC.
Fibulin (FBLN) 1 and -5 belong to the FBLN protein
family, which is involved in maintaining the stability of the basal
membrane, elastic fibers and loose connective tissue. Schluterman
et al (34) demonstrated that
loss of fibulin 5 (FBLN5) expression promoted tumor progression by
increasing the level of reactive oxygen species. In most human
carcinomas, especially in kidney, breast, ovarian, colon and
malignant metastatic carcinoma, FBLN5 was downregulated compared
with normal tissues (35). In
addition, FBLN5 is also a target for transforming growth factor-β
in endothelial cells, suggesting that FBLN5 may be a therapeutic
target (36).
The myosin heavy chain 11 (MYH11) gene encodes the
smooth muscle myosin heavy chain and mutations in MYH11 were mainly
associated with aortic aneurysm and acute myeloid leukemia
(37,38). Carcinoma metastasis and invasion are
driven by cell movement, a process involving myosin/actin
contraction and cell contact point degradation (39). Mutation and downregulation of MYH11
were associated with colon cancer and mucosal polyp syndrome
(40). MYH11 was also downregulated
in breast and bladder carcinoma (41).
EFEMP2 is an extracellular matrix protein necessary
for elastic fiber formation and connective tissue development,
processes that are highly associated with tumor invasion and
metastasis (42). The expression of
EFEMP2 in bladder cancer tissues was significantly lower than that
in normal tissues in previous study (43). Zhou et al (43) confirmed that low expression of EFEMP2
could reduce the expression of epithelial marker E-cadherin, as
well as increase the expressive levels of mesenchymal markers
N-cadherin, vimentin, Snail and Slug and key factors of the
Wnt/β-catenin signaling pathway (β-catenin, c-Myc and cyclin D1).
Their observations demonstrated that EFEMP2 inhibited tumor
progression and metastasis in bladder cancer (43). However, to the best of our knowledge,
studies on EFEMP2 in ACC have not yet been performed; thus, the
findings of the present study may provide insight for
adrenocortical tumorigenesis.
Myosin light chain kinase (MYLK) regulates myosin
activity through phosphorylation and dephosphorylation of the
myosin light chain. Therefore, it is involved in many physiological
processes, such as cell adhesion, cell proliferation, cell
migration and infiltration (44).
MYLK can increase the expression of epidermal growth factor
receptor and activate the ERK/JNK signal pathway, which can ablate
the adhesion between cells and increase the aggressiveness of
breast cancer cells (45). In
addition, MYLK expression is low in prostate cancer, bladder
cancer, non-small cell carcinoma and gastric cancer, which
indicates this gene may greatly impact on carcinogenesis and
malignant progression (46,47).
ACC is difficult to diagnose, even with
postoperative pathological analysis. Previous studies and published
guidelines (48-50)
indicated that histopathological features alone cannot predict
malignant or metastatic occurrence and that regular and long-term
follow-up is necessary for most cases. Thus, there is a clear need
to find rapid and accurate tools for diagnosis of adrenocortical
cancer.
Certain previous studies have revealed that Ki-67
and minichromosome maintenance protein are reliable indicators of
benign and malignant adrenal tumors (51,52).
Additionally, various studies have used pituitary-tumor
transforming gene 1(53), telomerase
activity (54) and vascular
endothelial growth factor (55) as
diagnostic markers of ACC. However, studies of these ACC diagnostic
markers have not reached a uniform and reliable conclusion.
Nowadays, gene expression analysis has been used to screen
molecular markers for cancer diagnosis and prognosis. Microarray
technology may become the method of choice for the detection of
malignant adrenal tissue. In the present study, bioinformatics
analysis was used to identify eight key downregulated genes. It was
verified that these genes were associated with ACC in terms of
molecular function, biological processes and cytology. Moreover,
using cluster analysis in the USCS Cancer Genomics Browser, it was
demonstrated that the selected hub genes can distinguish normal
adrenal tissues from ACC tissues. However, there were many samples
that did not display expression of the hub genes, which suggested
that these hub genes are not differentially expressed in all ACC
tissues. This condition will impose some limitations on
diagnosis.
Analysis of the relationships between gene
expression levels and clinical phenotypes is another important
issue in oncology. Using Kaplan-Meier analysis, the association
between overall survival rate, disease-free survival rate and the
downregulated hub genes was assessed. This analysis showed that
alterations of all hub genes did not affect overall survival rate.
However, downregulated EFEMP2 resulted in a decrease on
disease-free survival rate. The lack of effects on survival may be
due to several reasons. Firstly, survival analyses in cBioPortal
database were performed on the basis of the relationship between
gene mutation and prognosis, whereas genetic low expression may
result from promoter methylation, histone modification or protein
acetylation, not just mutation. Thus, low expression of the eight
hub genes in ACC may possessed low frequency of mutation, which led
the prognostic difference insignificant. Other previous studies
demonstrated a similar lack of conformity. For example, in
bioinformatics analysis conducted by Li et al (56), the TOP2A oncogene did not affect
overall and disease-free survival rates. However, some previous
clinical studies demonstrated that TOP2A was significantly related
to the survival rate of patients with hepatocellular carcinoma
(57,58). Secondly, carcinogenesis and
progression of tumors are the result of multi-gene dysregulation
and different genes can have various effects on tumor prognosis.
Although the low-expression genes identified in ACC in the present
study are involved in multiple key steps of tumorigenesis and
progression, their effects on prognosis may be less than the
effects of high-expression genes identified in previous studies
(22,59). Compared with other urologic
neoplasms, ACC has a low incidence (0.7-2/million), which may lead
the insufficiency of datasets and samples. Small sample size may
skew the results of prognostic analysis (60).
Capsular invasion, histological grade and vascular
invasion are common yet informative clinicopathological parameters.
These indicators can reflect the tendency of tumor progression and
reveal the differentiated degree of neoplasms. Thus, several
previous studies have analyzed the relationships between gene
expression and these clinical parameters in different cancer, such
as hepatocellular carcinoma, thyroid carcinoma and pheochromocytoma
(61-63).
Therefore, these clinicopathological features are commonly used in
cancer research. In the present study, the expression levels of
CSRP1 and MFAP4 were associated with capsular invasion, grade and
vascular invasion, which suggested that CSRP1 and MFAP4 may promote
progression of ACC.
The present study also has some limitations. First,
the clinical data of ACC are insufficient. Due to the low incidence
of ACC, there were few qualified datasets for bioinformatics
analysis. Moreover, subtypes of ACC were not considered in the
bioinformatics analysis. The main subtype of ACC is adrenal
epithelial cell carcinoma, which accounts for >95% of all
subtypes. Other rare subtypes include oncocytic adrenal neoplasms,
myxoid adrenal cortical carcinoma and adrenal carcinosarcoma.
Different subtypes may have different mechanisms of carcinogenesis
and progression, but there is a lack of data and relevant research
to verify this possibility.
In conclusion, the hub genes screened in the present
study were downregulated and these genes were associated with ACC
carcinogenesis and progression. Identification of these hub genes
improves the gene expression profile of ACC and provides important
molecular biological insight for the diagnosis, treatment and
prognosis of ACC. Nevertheless, further studies are needed to
elucidate how the biological functions of these genes contribute to
ACC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TC designed the study and revised the manuscript. FX
performed the Gene Expression Omnibus database analysis, analyzed
the data. FX and PZ performed bioinformatics analyses and assisted
with analysis of other data. FX, MY and XY wrote the manuscript,
collected data, performed revision of the manuscript and created
the figures. All authors have read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Erickson LA, Rivera M and Zhang J:
Adrenocortical carcinoma: Review and update. Adv Anat Pathol.
21:151–159. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Stigliano A, Cerquetti L, Lardo P,
Petrangeli E and Toscano V: New insights and future perspectives in
the therapeutic strategy of adrenocortical carcinoma (Review).
Oncol Rep. 37:1301–1311. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang HW, Wu YH, Hsieh JY, Liang ML, Chao
ME, Liu DJ, Hsu MT and Wong TT: Pediatric primary central nervous
system germ cell tumors of different prognosis groups show
characteristic miRNome traits and chromosome copy number
variations. BMC Genomics. 11(132)2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Soon PS, Gill AJ, Benn DE, Clarkson A,
Robinson BG, McDonald KL and Sidhu SB: Microarray gene expression
and immunohistochemistry analyses of adrenocortical tumors identify
IGF2 and Ki-67 as useful in differentiating carcinomas from
adenomas. Endocr Relat Cancer. 16:573–583. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Szabó PM, Wiener Z, Tömböl Z, Kovács A,
Pócza P, Horányi J, Kulka J, Riesz P, Tóth M, Patócs A, et al:
Differences in the expression of histamine-related genes and
proteins in normal human adrenal cortex and adrenocortical tumors.
Virchows Arch. 455:133–142. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gene Ontology Consortium. Gene ontology
consortium: Going forward. Nucleic Acids Res. 43 (Database
Issue):D1049–D1056. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kanehisa M, Goto S, Furumichi M, Tanabe M
and Hirakawa M: KEGG for representation and analysis of molecular
networks involving diseases and drugs. Nucleic Acids Res. 38
(Database Issue):D355–D360. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8(R183)2007.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C
and Jensen LJ: STRING v9.1: Protein-protein interaction networks,
with increased coverage and integration. Nucleic Acids Res. 41
(Database Issue):D808–D815. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Rivera CG, Vakil R and Bader JS: NeMo:
Network module identification in cytoscape. BMC Bioinformatics.
11(S61)2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of gene ontology
categories in biological networks. Bioinformatics. 21:3448–3449.
2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Goldman M, Craft B, Swatloski T, Ellrott
K, Cline M, Diekhans M, Ma S, Wilks C, Stuart J, Haussler D and Zhu
J: The UCSC cancer genomics browser: Update 2013. Nucleic Acids
Res. 41 (Database Issue):D949–D954. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Giordano TJ, Thomas DG, Kuick R, Lizyness
M, Misek DE, Smith AL, Sanders D, Aljundi RT, Gauger PG, Thompson
NW, et al: Distinct transcriptional profiles of adrenocortical
tumors uncovered by DNA microarray analysis. Am J Pathol.
162:521–531. 2003.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Giordano TJ, Kuick R, Else T, Gauger PG,
Vinco M, Bauersfeld J, Sanders D, Thomas DG, Doherty G and Hammer
G: Molecular classification and prognostication of adrenocortical
tumors by transcriptome profiling. Clin Cancer Res. 15:668–676.
2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Else T, Kim AC, Sabolch A, Raymond VM,
Kandathil A, Caoili EM, Jolly S, Miller BS, Giordano TJ and Hammer
GD: Adrenocortical carcinoma. Endocr Rev. 35:282–326.
2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Harjunpää H, Llort Asens M, Guenther C and
Fagerholm SC: Cell adhesion molecules and their roles and
regulation in the immune and tumor microenvironment. Front Immunol.
10(1078)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Revilla G, Corcoy R, Moral A, Escolà-Gil
JC and Mato E: Cross-talk between inflammatory mediators and the
epithelial mesenchymal transition process in the development of
thyroid carcinoma. Int J Mol Sci. 20: pii(E2466)2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Antunes DM, Rodrigues MFSD, Guimarães DM,
Duarte CME, Miguita L, Corrêa L, DE Oliveira APL, Fernandes KPS and
Nunes FD: Nonsteroidal anti-inflammatory drugs modulate gene
expression of inflammatory mediators in oral squamous cell
carcinoma. Anticancer Res. 39:2385–2394. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Qu X, Tang Y and Hua S: Immunological
approaches towards cancer and inflammation: A cross talk. Front
Immunol. 9(563)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Macciò A and Madeddu C: Blocking
inflammation to improve immunotherapy of advanced cancer.
Immunology. 159:357–364. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Xiao H, Xu D, Chen P, Zeng G, Wang X and
Zhang X: Identification of five genes as a potential biomarker for
predicting progress and prognosis in adrenocortical carcinoma. J
Cancer. 9:4484–4495. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yamane T, Asanoma K, Kobayashi H, Liu G,
Yagi H, Ohgami T, Ichinoe A, Sonoda K, Wake N and Kato K:
Identification of the critical site of calponin 1 for suppression
of ovarian cancer properties. Anticancer Res. 35:5993–5999.
2015.PubMed/NCBI
|
|
24
|
Yanagisawa Y, Takeoka M, Ehara T, Itano N,
Miyagawa S and Taniguchi S: Reduction of Calponin h1 expression in
human colon cancer blood vessels. Eur J Surg Oncol. 34:531–537.
2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liu Y, Wu X, Wang G, Hu S, Zhang Y and
Zhao S: CALD1, CNN1, and TAGLN identified as potential prognostic
molecular markers of bladder cancer by bioinformatics analysis.
Medicine (Baltimore). 98(e13847)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhou CZ, Qiu GQ, Wang XL, Fan JW, Tang HM,
Sun YH, Wang Q, Huang F, Yan DW, Li DW and Peng ZH: Screening of
tumor suppressor genes on 1q31.1-32.1 in Chinese patients with
sporadic colorectal cancer. Chin Med J (Engl). 121:2479–2486.
2008.PubMed/NCBI
|
|
27
|
Hirasawa Y, Arai M, Imazeki F, Tada M,
Mikata R, Fukai K, Miyazaki M, Ochiai T, Saisho H and Yokosuka O:
Methylation status of genes upregulated by demethylating agent
5-aza-2'-deoxycytidine in hepatocellular carcinoma. Oncology.
71:77–85. 2006.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jin GH, Xu W, Shi Y and Wang LB: Celecoxib
exhibits an anti-gastric cancer effect by targeting focal adhesion
and leukocyte transendothelial migration-associated genes. Oncol
Lett. 12:2345–2350. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Davalieva K, Kostovska IM, Kiprijanovska
S, Markoska K, Kubelka-Sabit K, Filipovski V, Stavridis S, Stankov
O, Komina S, Petrusevska G and Polenakovic M: Proteomics analysis
of malignant and benign prostate tissue by 2D DIGE/MS reveals new
insights into proteins involved in prostate cancer. Prostate.
75:1586–1600. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yang J, Song H, Chen L, Cao K, Zhang Y, Li
Y and Hao X: Integrated analysis of microfibrillar-associated
proteins reveals MFAP4 as a novel biomarker in human cancers.
Epigenomics. 11:1635–1651. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhao H, Sun Q, Li L, Zhou J, Zhang C, Hu
T, Zhou X, Zhang L, Wang B, Li B, et al: High expression levels of
AGGF1 and MFAP4 predict primary platinum-based chemoresistance and
are associated with adverse prognosis in patients with serous
ovarian cancer. J Cancer. 10:397–407. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Niu D, Peatman E, Liu H, Lu J, Kucuktas H,
Liu S, Sun F, Zhang H, Feng T, Zhou Z, et al:
Microfibrillar-associated protein 4 (MFAP4) genes in catfish play a
novel role in innate immune responses. Dev Comp Immunol.
35:568–579. 2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Schlosser A, Pilecki B, Hemstra LE,
Kejling K, Kristmannsdottir GB, Wulf-Johansson H, Moeller JB,
Füchtbauer EM, Nielsen O, Kirketerp-Møller K, et al: MFAP4 promotes
vascular smooth muscle migration, proliferation and accelerates
neointima formation. Arterioscler Thromb Vasc Biol. 36:122–133.
2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Schluterman MK, Chapman SL, Korpanty G,
Ozumi K, Fukai T, Yanagisawa H and Brekken RA: Loss of fibulin-5
binding to beta1 integrins inhibits tumor growth by increasing the
level of ROS. Dis Model Mech. 3:333–342. 2010.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Dou CY, Cao CJ, Wang Z, Zhang RH, Huang
LL, Lian JY, Xie WL and Wang LT: EFEMP1 inhibits migration of
hepatocellular carcinoma by regulating MMP2 and MMP9 via ERK1/2
activity. Oncol Rep. 35:3489–3495. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Topalovski M, Hagopian M, Wang M and
Brekken RA: Hypoxia and transforming growth factor β cooperate to
induce fibulin-5 expression in pancreatic cancer. J Biol Chem.
291:22244–22252. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Pannu H, Tran-Fadulu V, Papke CL, Scherer
S, Liu Y, Presley C, Guo D, Estrera AL, Safi HJ, Brasier AR, et al:
MYH11 mutations result in a distinct vascular pathology driven by
insulin-like growth factor 1 and angiotensin II. Hum Mol Genet.
16:2453–2462. 2007.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Assaf N, El-Cheikh J, Bazarbachi A, Salem
Z, Farra C, Chakhachiro Z, Nassif S, Zaatari G and Mahfouz R:
Molecular profiling of adult acute myeloid and lymphoid leukemia in
a major referral center in Lebanon: A 10-year experience report and
review of the literature. Mol Biol Rep. 46:2003–2011.
2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Friedl P and Wolf K: Tumour-cell invasion
and migration: Diversity and escape mechanisms. Nat Rev Cancer.
3:362–374. 2003.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lee WS, Seo G, Shin HJ, Yun SH, Yun H,
Choi N, Lee J, Son D, Cho J, Kim J, et al: Identification of
differentially expressed genes in microsatellite stable HNPCC and
sporadic colon cancer. J Surg Res. 144:29–35. 2008.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Alhopuro P, Karhu A, Winqvist R, Waltering
K, Visakorpi T and Aaltonen LA: Somatic mutation analysis of MYH11
in breast and prostate cancer. BMC Cancer. 8(263)2008.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Song L, Li XX, Liu XY, Wang Z, Yu Y, Shi
M, Jiang B and He XP: EFEMP2 suppresses the invasion of lung cancer
cells by inhibiting epithelial-mesenchymal transition (EMT) and
down-regulating MMPs. Onco Targets Ther. 13:1375–1396.
2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Zhou Q, Chen S, Lu M, Luo Y, Wang G, Xiao
Y, Ju L and Wang X: EFEMP2 suppresses epithelial-mesenchymal
transition via Wnt/β-catenin signaling pathway in human bladder
cancer. Int J Biol Sci. 15:2139–2155. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Khapchaev AY and Shirinsky VP: Myosin
light Chain kinase MYLK1: Anatomy, interactions, functions, and
regulation. Biochemistry (Mosc). 81:1676–1697. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Kim DY and Helfman DM: Loss of MLCK leads
to disruption of cell-cell adhesion and invasive behavior of breast
epithelial cells via increased expression of EGFR and ERK/JNK
signaling. Oncogene. 35:4495–4508. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Dai Y, Li D, Chen X, Tan X, Gu J, Chen M
and Zhang X: Circular RNA myosin light chain kinase (MYLK) promotes
prostate cancer progression through modulating Mir-29a expression.
Med Sci Monit. 24:3462–3471. 2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Zhong Z, Huang M, Lv M, He Y, Duan C,
Zhang L and Chen J: Circular RNA MYLK as a competing endogenous RNA
promotes bladder cancer progression through modulating VEGFA/VEGFR2
signaling pathway. Cancer Lett. 403:305–317. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Nakamura Y, Yamazaki Y, Felizola SJ, Ise
K, Morimoto R, Satoh F, Arai Y and Sasano H: Adrenocortical
carcinoma: Review of the pathologic features, production of adrenal
steroids, and molecular pathogenesis. Endocrinol Metab Clin North
Am. 44:399–410. 2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Lowery AJ, Walsh S, McDermott EW and
Prichard RS: Molecular and therapeutic advances in the diagnosis
and management of malignant pheochromocytomas and paragangliomas.
Oncologist. 18:391–407. 2013.PubMed/NCBI View Article : Google Scholar
|
|
50
|
de Wailly P, Oragano L, Radé F, Beaulieu
A, Arnault V, Levillain P and Kraimps JL: Malignant
pheochromocytoma: New malignancy criteria. Langenbecks Arch Surg.
397:239–246. 2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Aporowicz M, Czopnik P, Kubicka E,
Piotrowska A, Dziegiel P, Bolanowski M and Domoslawski P:
Minichromosome maintenance proteins MCM-3, MCM-5, MCM-7, and Ki-67
as proliferative markers in adrenocortical tumors. Anticancer Res.
39:1151–1159. 2019.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Saiegh L, Sheikh-Ahmad M, Shechner C, Reut
M, Darawsha Y, Zolotov S, Shefer H, Bejar I and Bejar J:
Metallothionein protein and minichromosome maintenance protein-2
expression in adrenocortical tumors. Ann Endocrinol (Paris).
80:324–328. 2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Romero Arenas MA, Whitsett TG, Aronova A,
Henderson SA, LoBello J, Habra MA, Grubbs EG, Lee JE, Sircar K,
Zarnegar R, et al: Protein expression of PTTG1 as a diagnostic
biomarker in adrenocortical carcinoma. Ann Surg Oncol. 25:801–807.
2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Hirano Y, Nobata S, Takahashi H, Kageyama
S, Sudoko H, Ushiyama T, Suzuki K and Fujita K: Histologically
benign but telomerase positive adrenal pheochromocytoma. Int J
Urol. 9:697–699. 2002.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Kołomecki K, Stepień H and Narebski JM:
Vascular endothelial growth factor and basic fibroblast growth
factor evaluation in blood serum of patients with hormonally active
and inactive adrenal gland tumours. Cytobios. 101:55–64.
2000.PubMed/NCBI
|
|
56
|
Li L, Lei Q, Zhang S, Kong L and Qin B:
Screening and identification of key biomarkers in hepatocellular
carcinoma: Evidence from bioinformatic analysis. Oncol Rep.
38:2607–2618. 2017.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Panvichian R, Tantiwetrueangdet A,
Angkathunyakul N and Leelaudomlipi S: TOP2A amplification and
overexpression in hepatocellular carcinoma tissues. Biomed Res Int.
2015(381602)2015.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Wong N, Yeo W, Wong WL, Wong NL, Chan KY,
Mo FK, Koh J, Chan SL, Chan AT, Lai PB, et al: TOP2A overexpression
in hepatocellular carcinoma correlates with early age onset,
shorter patients survival and chemoresistance. Int J Cancer.
124:644–652. 2009.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Guo J, Gu Y, Ma X, Zhang L, Li H, Yan Z,
Han Y, Xie L and Guo X: Identification of hub genes and pathways in
adrenocortical carcinoma by integrated bioinformatic analysis. J
Cell Mol Med. 24:4428–4438. 2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Button KS, Ioannidis JP, Mokrysz C, Nosek
BA, Flint J, Robinson ES and Munafò MR: Power failure: Why small
sample size undermines the reliability of neuroscience. Nat Rev
Neurosci. 14:365–376. 2013.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Celestino R, Nome T, Pestana A, Hoff AM,
Gonçalves AP, Pereira L, Cavadas B, Eloy C, Bjøro T,
Sobrinho-Simões M, et al: CRABP1, C1QL1 and LCN2 are biomarkers of
differentiated thyroid carcinoma, and predict extrathyroidal
extension. BMC Cancer. 18(68)2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Pinato DJ, Black JR, Trousil S, Dina RE,
Trivedi P, Mauri FA and Sharma R: Programmed cell death ligands
expression in phaeochromocytomas and paragangliomas: Relationship
with the hypoxic response, immune evasion and malignant behavior.
Oncoimmunology. 6(e1358332)2017.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Zhang L, Luo B, Dang YW, He RQ, Peng ZG,
Chen G and Feng ZB: Clinical significance of microRNA-196b-5p in
hepatocellular carcinoma and its potential molecular mechanism. J
Cancer. 10:5355–5370. 2019.PubMed/NCBI View Article : Google Scholar
|