Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignancies worldwide, resulting in high morbidity and
mortality, with 854,000 new cases and 810,000 cases of mortality
each year (1). HCC is also the fifth
most commonly occurring cancer, as well as the third leading cause
of cancer-related death worldwide (2). Multiple therapeutic strategies, such as
liver resection, transarterial chemoembolization, radiotherapy and
sorafenib administration, have been used for the treatment of
patients with HCC. However, the prognosis of patients with HCC
remains poor due to its complexity, high recurrence and early
vascular invasion (3,4). Increasing evidence has revealed that
HCC is associated with multi-gene mutations (5). Molecular targeting has been suggested
as a novel therapeutic approach for patients with advanced HCC, as
it significantly increases the survival time of patients and has
favorable curative effects (6).
Therefore, understanding the development of HCC and identifying
novel molecular therapeutic targets has received increasing
attention.
Lysine demethylase 6A (KDM6A; also known as
ubiquitously transcribed X chromosome tetratricopeptide repeat
protein) is a Jumonji-C domain-containing histone demethylase that
catalyzes the removal of histone H3 lysine-27 trimethylation
(7,8). As a member of the KDM6 family, KDM6A is
also a key molecule that regulates cell fate decisions and cell
identity during normal development by controlling the expression of
pluripotency and lineage-specific genes (9,10). It
has also been reported that KDM6A is frequently targeted by somatic
inactivating mutations in various types of cancer, such as
esophageal and urethral cancer, as well as pancreatic ductal
adenocarcinoma and multiple myeloma (11). Mechanistically, KDM6A mutations
suppress cell proliferation by activating retinoblastoma
protein-related genes, which suggests that KDM6A serves as a
putative tumor suppressor (12).
Furthermore, the biological function of KDM6A in a number of
malignancies, including bladder and lung cancer, as well as
pancreatic ductal adenocarcinoma, has been reported (13-16).
However, the cellular and molecular mechanisms underlying KDM6A in
HCC are not completely understood.
The present study investigated the importance of
KDM6A during HCC progression. The results indicated that KDM6A
expression was downregulated in human HCC tissues compared with
corresponding normal control tissues. In addition, KDM6A
overexpression inhibited HCC cell proliferation in vitro and
in vivo. Therefore, the present study identified a potential
molecular mechanism underlying the role of KDM6A during HCC
development.
Materials and methods
HCC tissue samples
A total of 30 paired primary HCC tissues and
adjacent normal tissues(distance, <2 cm) were obtained from
patients (age, 32-68 years; mean age, 46±3.2 years; 22 male
patients and 8 female patients) who had undergone a hepatectomy
between February and July 2019 at Gansu Provincial Hospital
(Lanzhou, China). The inclusion criteria were as follows: i)
Patients clinically diagnosed with liver cancer following surgery;
ii) R0 resection based on histological examinations; and iii)
paired normal tissue was adjacent to tumor tissue (distance, <2
cm). The exclusion criteria were as follows: i) Patients with
distant metastasis; and ii) patients who had received radiotherapy
or chemotherapy before surgery. Paired samples were subjected to
RNA extraction for reverse transcription-quantitative PCR (RT-qPCR)
and immunohistochemistry (IHC) analyses. The present study was
approved by the Institutional Review Board of The Institute for
Gansu Provincial Hospital (Lanzhou, China). All patients provided
written informed consent.
IHC
Tumor tissues and paired normal tissues were fixed
in 4% formalin at room temperature for 24 h, embedded in paraffin
and cut into 5-µm thick consecutive sections. Paraffin-embedded
tissue sections were deparaffinized in xylene, rehydrated using a
descending ethanol gradient before being subjected to antigen
retrieval in a sodium citrate solution (pH 6.0) for 20 min at 98˚C
and endogenous peroxidase activity blocking with 3% hydrogen
peroxide. The sections were washed three times with 0.01 M PBS for
5 min each and blocked in 0.01 M PBS containing 0.3% Triton X-100
and 5% BSA (Gibco; Thermo Fisher Scientific, Inc.) at room
temperature for 1 h. Subsequently, sections were incubated with an
anti-KDM6A antibody (cat. no. orb333886; 1:100; Biorbyt Ltd.) at
4˚C overnight. After washing with 0.01 M PBS, sections were
incubated with an anti-rabbit horseradish peroxidase-conjugated
secondary antibody (cat. no. 7074; 1:5,000; Cell Signaling
Technology, Inc.) at room temperature for 1 h. Subsequently, DAB
substrate was used to visualize KDM6A protein expression levels.
Stained sections were observed in five randomly selected,
independent high-power microscopic field of view using an inverted
light microscope (magnification, x400).
RNA extraction and RT-qPCR
Tissues were snap-frozen in liquid nitrogen and
stored at -80˚C until further analysis. Total RNA was extracted
from tumor tissues and adjacent noncancerous tissues using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). RNA (2 µg) was reverse transcribed into cDNA using the
PrimeScript™ RT Reagent kit (Takara Bio, Inc.), according to the
manufacturer's instructions, with the following thermocycling
conditions: 37˚C for 15 min and 85˚C for 5 sec. Subsequently, qPCR
was performed using SYBR® Premix Ex Taq (Takara Bio,
Inc.). The sequences of the primers used for qPCR are listed in
Table I. The following thermocycling
conditions were used for qPCR: Initial denaturation for 1 min at
95˚C; 40 cycles of denaturation for 20 sec at 95˚C, annealing at
56˚C for 60 sec and extension at 72˚C for 2 min; followed by final
extension at 72˚C for 6 min. mRNA expression levels were quantified
using the 2-∆∆Cq method (17) and normalized to the internal
reference gene GAPDH.
 | Table IPrimer and shRNA sequences. |
Table I
Primer and shRNA sequences.
| Gene | Sequence (5'→3') |
|---|
| KDM6A | F:
TTCCTCGGAAGGTGCTATTCA |
| | R:
GAGGCTGGTTGCAGGATTCA |
| GAPDH | F:
ATGACCCCTTCATTGACCTCA |
| | R:
GAGATGATCACCCTTTTGGCT |
| KDM6A-SCR | F:
TTCTCCGAACGTGTCACGT |
| | R:
ACGTGACACGTTCGGAGAA |
| KDM6A-sh1 | F:
CCGGGCACATAGACTAAGGAATAAACTCGAGTTTATTCCTTAGTCTATGTGCTTTTTG |
| | R:
AATTCAAAAAGCACATAGACTAAGGAATAAACTCGAGTTTATTCCTTAGTCTATGTGC |
| KDM6A-sh2 | F:
CCGGGCAGCACGAATTAAGTATTTACTCGAGTAAATACTTAATTCGTGCTGCTTTTTG |
| | R:
AATTCAAAAAGCAGCACGAATTAAGTATTTACTCGAGTAAATACTTAATTCGTGCTGC |
Cell culture
Human HCC cell lines (YY-8103, SNU-398, MHCC97-L,
Hep3B, LM3 and Huh7) and 293T cells were purchased from The Cell
Bank of Type Culture Collection of the Chinese Academy of Sciences.
Cells were cultured in DMEM (Invitrogen; Thermo Fisher Scientific,
Inc.) supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin
and 10% FBS (Gibco; Thermo Fisher Scientific, Inc.) at 37˚C with 5%
CO2. The Huh7, LM3 and Hep3B cell lines have high
invasive characteristics, whereas the YY-8103, SNU-398 and MHCC-97L
cell lines have less invasive characteristics (18).
Cell transfection
A KDM6A coding sequence was constructed and inserted
into the p23-3xflag-GFP vector (Invitrogen; Thermo Fisher
Scientific, Inc.) to generate a KDM6A overexpression vector. The
empty p23 vector was used as the negative control. Lentiviral short
hairpin (sh)RNAs targeting KDM6A were designed and constructed into
pLKO.1-TRC vectors (Shanghai GenePharma Co., Ltd.). A non-targeting
scrambled (SCR) oligonucleotide (Shanghai GenePharma Co., Ltd.)
constructed into pLKO.1-TRC vectors served as the negative control.
The sequences of the shRNAs are provided in Table I.
To produce lentiviral particles for KDM6A
overexpression and knockdown, the core plasmid (1.5 µg) was
co-transfected with the packaging plasmids 3.0 µg pMD2.G and 3.0 µg
psPAX2 (Shanghai GenePharma Co., Ltd) into 293 T cells
(5x105 cells per well) using a calcium phosphate
co-precipitation method in six-well plates (19). At 12 h post-transfection, the medium
was refreshed. At 24 and 48 h post-transfection, the cell culture
supernatants containing the virus were collected and filtered
through a 0.45 µm membrane. Subsequently, the virus was
concentrated by centrifugation at 50,000 x g for 140 min at 4˚C.
The pellet was resuspended in DMEM solution, aliquoted and stored
at -80˚C until further use. For the transduction process, Huh7 and
LM3 cells (1x106 cells per well) were grown to 60%
confluence in 6-well plates and transduced with 40 µl viral
supernatant (1x107 units per well) and 5 µg/ml polybrene
(Hanheng Biological Technology Co., Ltd.). After 24 h at 37˚C, the
medium was refreshed and the cells were transferred to 10-cm
dishes. After 2 days at 37˚C, KDM6A-overexpression Huh7 and LM3
cells were screened by green fluorescence via flow cytometry using
an IX-71 flow cytometer (Olympus Corporation). KDM6A-knockdown
YY-8103 and SNU-398 cells were cultured and screened in medium
containing 3 µg/ml puromycin (Hanheng Biological Technology Co.,
Ltd.) for 4 days at 37˚C. Subsequently, individual
puromycin-resistant colonies were isolated and used for subsequent
experiments. Transfection efficiency was verified by western
blotting.
Western blotting
Total protein was extracted from cells using RIPA
Lysis Buffer (Thermo Fisher Scientific, Inc.) supplemented with
phenylmethylsulfonyl fluoride (Thermo Fisher Scientific, Inc.).
Total cell lysates were centrifuged at 10,000 x g for 15 min at
4˚C, following which the supernatant was collected. Protein
concentration of the supernatant was determined using Bradford
reagent (Sigma-Aldrich; Merck KGaA). Protein (20 µg per lane) was
separated via 10% SDS-PAGE and transferred onto nitrocellulose
membranes (EMD Millipore). The membranes were blocked with 5%
fat-free milk for 1 h at room temperature. Subsequently, the
membranes were incubated at 4˚C overnight with the following
primary antibodies: Anti-KDM6A (1:1,000; cat. no. orb333886;
Biorbyt Ltd.), anti-transforming growth factor (TGF)-β (1:1,000;
cat. no. 3711; Cell Signaling Technology, Inc.),
anti-phosphorylated (p)-smad2 (1:1,000; cat. no. 18338; Cell
Signaling Technology, Inc.), anti-Smad2 (1:1,000; cat. no.
12570-1-AP; ProteinTech Group, Inc.), anti-p-smad4 (1:1,000; cat.
no. 10231-8-AP; ProteinTech Group, Inc.), anti-smad4 (1:1,000; cat.
no. 10231-1-AP; ProteinTech Group, Inc.), anti-proliferating cell
nuclear antigen (PCNA; 1:1,000; cat. no. 10205-2-AP; ProteinTech
Group, Inc.), anti-Ki67 (1:1,000; cat. no. 27309-1-AP; ProteinTech
Group, Inc.), anti-GAPDH (1:1,000; cat. no. 10494-1-AP; ProteinTech
Group, Inc.) and anti-Flag (1:4,000; cat. no. F7425; Sigma-Aldrich;
Merck KGaA). Following primary incubation, the membranes were
incubated with an anti-rabbit (1:5,000; cat. no. 7074; Cell
Signaling Technology, Inc.) or anti-mouse (1:3,000; cat. no. 7076;
Cell Signaling Technology, Inc.) horseradish peroxidase-conjugated
secondary antibody for 1 h at room temperature. The immunoreactive
protein bands were visualized using an enhanced chemiluminescence
kit (Pierce; Thermo Fisher Scientific, Inc.) and a Gel Dox XR
system (Bio-Rad Laboratories, Inc.). GAPDH was used as the loading
control. ImageJ software (version 1.8.0, National Institutes of
Health) was used for quantification of western blotting.
Crystal violet assay
Wild-type and transfected cells were seeded
(1x103 cells/well) into 6-well plates. Cells were
cultured in DMEM (Invitrogen; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS and the medium was changed every 3 days.
After 2 weeks, the medium was removed and cells were fixed with 20%
methanol at room temperature for 10 min. Subsequently, cells were
stained with 0.5% crystal violet at room temperature for 10 min.
Stained cells were washed to PBS and observed using a light
microscope (magnification, x4). Subsequently, 1 ml glacial acetic
acid was added to each well and the optical density of each well
was measured at a wavelength of 600 nm using a microplate
reader.
MTT assay
Cells were seeded (1x103 cells/well) into
96-well plates in triplicate. After incubation for 1-7 days at
37˚C, 20 µl MTT solution (5 mg/ml) was added to each well and
incubated at 37˚C for 4 h. Subsequently, the culture medium was
removed, 200 µl DMSO was added and the plates were gently agitated
to dissolve the formazan crystals. The absorbance of each well was
measured at a wavelength of 570 nm using an automatic microplate
reader.
In vivo tumorigenesis analysis
A total of 8 male BALB/c nude mice (age, ~5 weeks;
median weight, 20 g; weight range, 18-21 g) were obtained from
Beijing Huafukang Bioscience Co., Ltd. Mice were housed with free
access to regular chow diet and water under specific pathogen-free
conditions in laboratory cages at 23±3˚C with 35±5% humidity and
12-h light/dark cycles. The mice divided into were divided into two
groups (n=4 per group): i) Huh7 p23 negative control Vector and ii)
Huh7 KDM6A. KDM6A overexpression vector- or p23 negative control
vector-transfected Huh7 cells (1x106 cells in a total
volume of 100 µl PBS) were then subcutaneously injected using a 1
ml injection syringe into the right flank of each mouse in their
corresponding groups. Tumor size was measured on a weekly basis.
Tumor volume was calculated according to the following formula:
Tumor volume (mm3)=0.5xlengthxwidth2. At the
end of the experiment (week 4), mice were sacrificed by cervical
dislocation. The tumors were excised, photographed and weighed. The
maximum tumor volume observed was 609 mm3. All animal
experiments were performed according to guidelines of the
Institutional Animal Care and Use Committee of Gansu Provincial
Hospital (20) and were approved by
Ethics Committee of the Gansu Provincial Hospital.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism (version 5; GraphPad Software, Inc.). Data are presented as
the mean ± SD. Comparisons between two groups were determined using
the unpaired Student's t-test. Comparisons between multiple groups
were determined using one-way ANOVA followed Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference. All experiments were performed in triplicate.
Results
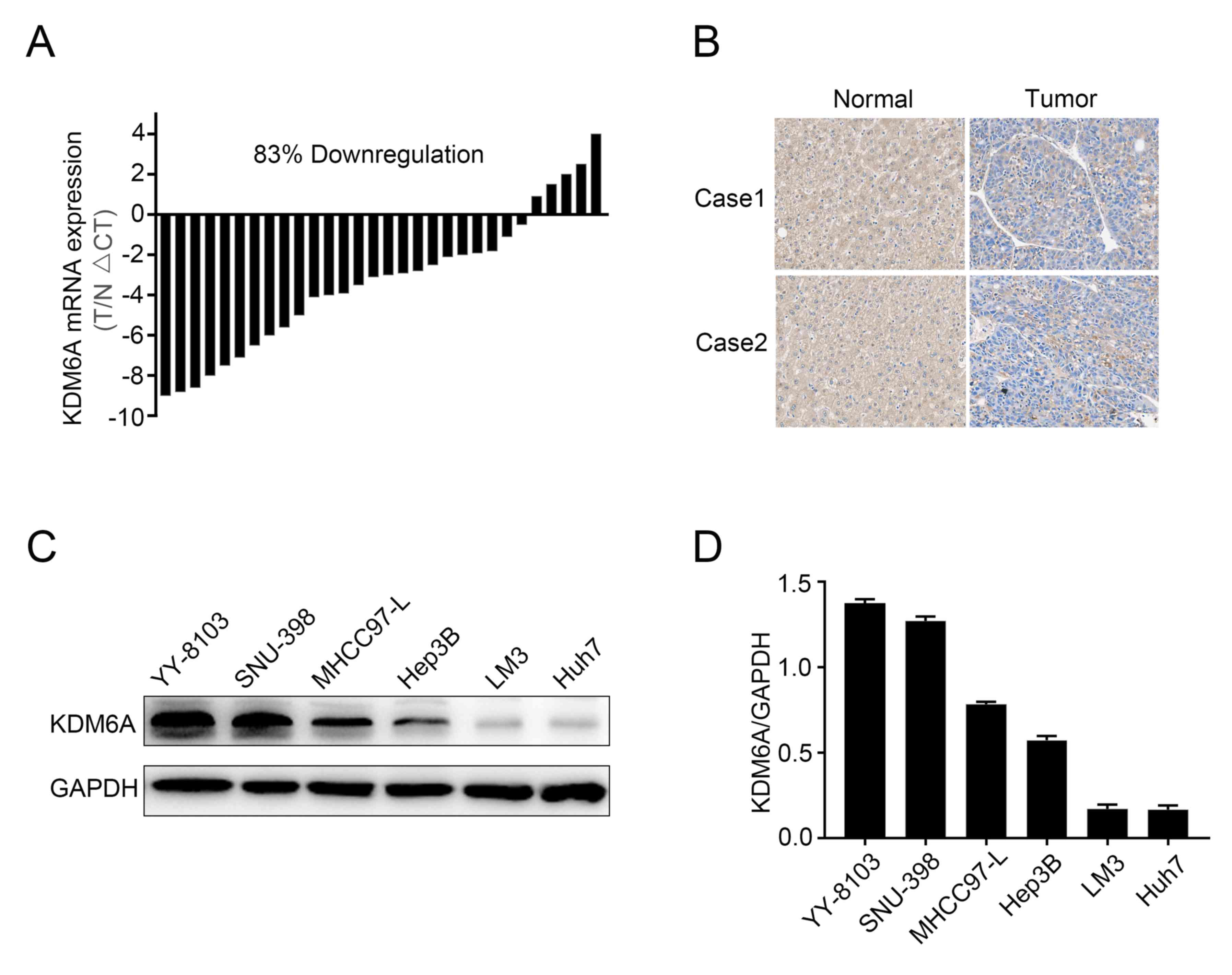
KDM6A is downregulated in HCC
tissues
To explore the potential roles of KDM6A in HCC, the
mRNA expression levels of KDM6A in 30 paired HCC and corresponding
normal tissues were examined. KDM6A mRNA levels were significantly
decreased in 25/30 (83%) HCC samples compared with the
corresponding normal samples (Fig.
1A). IHC analysis indicated that KDM6A was primarily expressed
in the cytoplasm of both normal and tumor tissues. Consistent with
the RT-qPCR results, HCC tissues displayed a decreased staining
intensity compared with the corresponding normal tissues in two
patients (Fig. 1B), which indicated
that KDM6A expression was decreased in HCC tissues compared with
corresponding normal tissues. In addition, the expression of KDM6A
in several HCC cell lines was assessed. KDM6A expression was
notably higher in YY-8103 and SNU-398 cells compared with Huh7 and
LM3 cells (Fig. 1C and D).
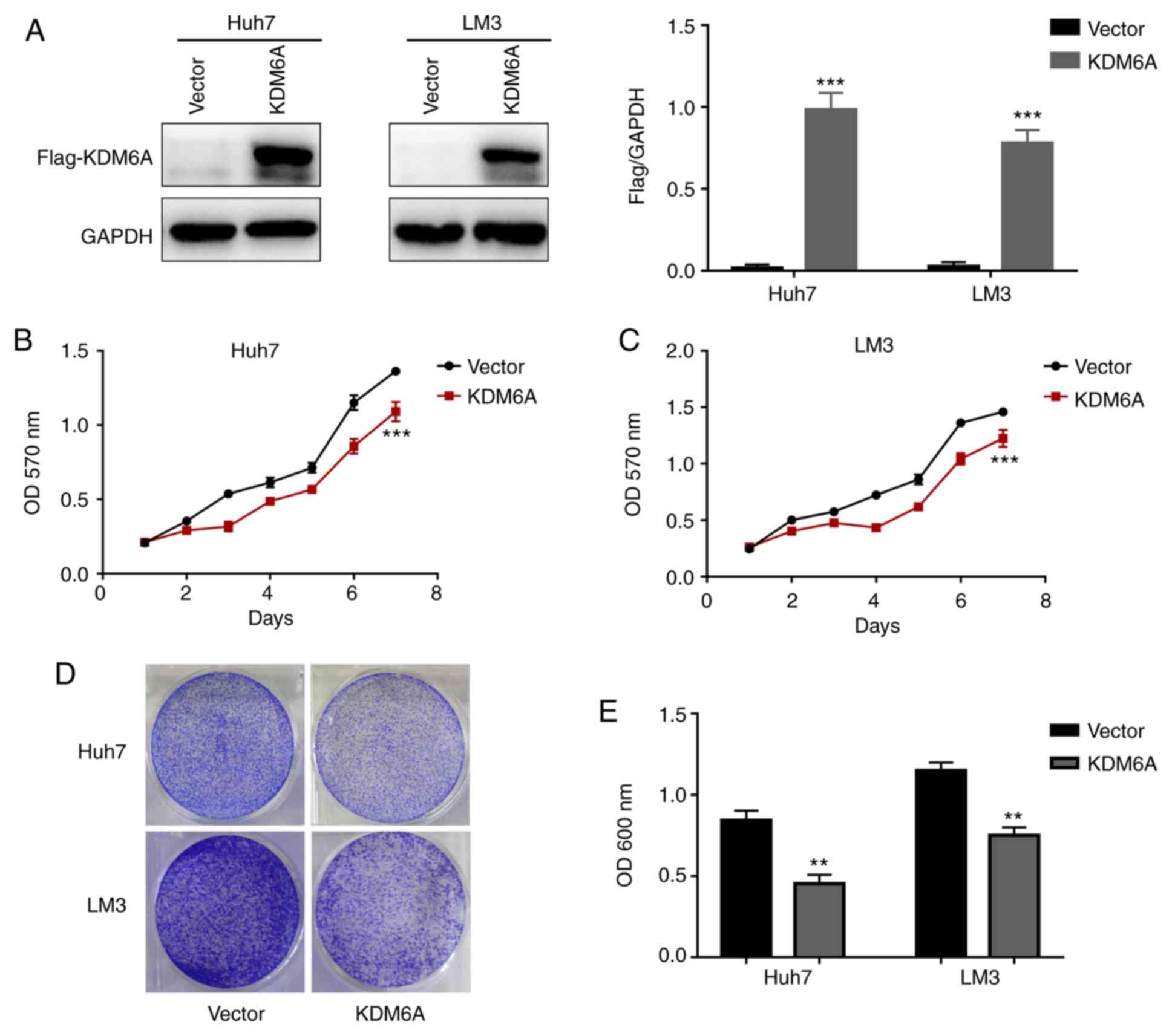
KDM6A overexpression inhibits HCC cell
proliferation in vitro
Based on the aforementioned clinical data, it was
hypothesized that KDM6A may influence HCC cell proliferation in
vitro. To evaluate the biological function of KDM6A in HCC
cells, Huh7 and LM3 cell lines were transfected with empty
(control) or KDM6A overexpression vectors. Western blotting was
performed to confirm the transfection efficiency of KDM6A
overexpression in HCC cells (Fig.
2A).
The MTT assay results indicated that the absorbance
values of KDM6A-overexpression Huh7 and LM3 cells were
significantly lower compared with empty vector-transfected cells on
day 7 (Fig. 2B and C). In addition, the results of the crystal
violet assay indicated that KDM6A overexpression markedly inhibited
Huh7 and LM3 cell proliferation compared with empty
vector-transfected cells (Fig. 2D).
Similarly, the absorbance values of KDM6A-overexpression Huh7 and
LM3 cells were significantly lower compared with empty
vector-transfected cells (Fig.
2E).
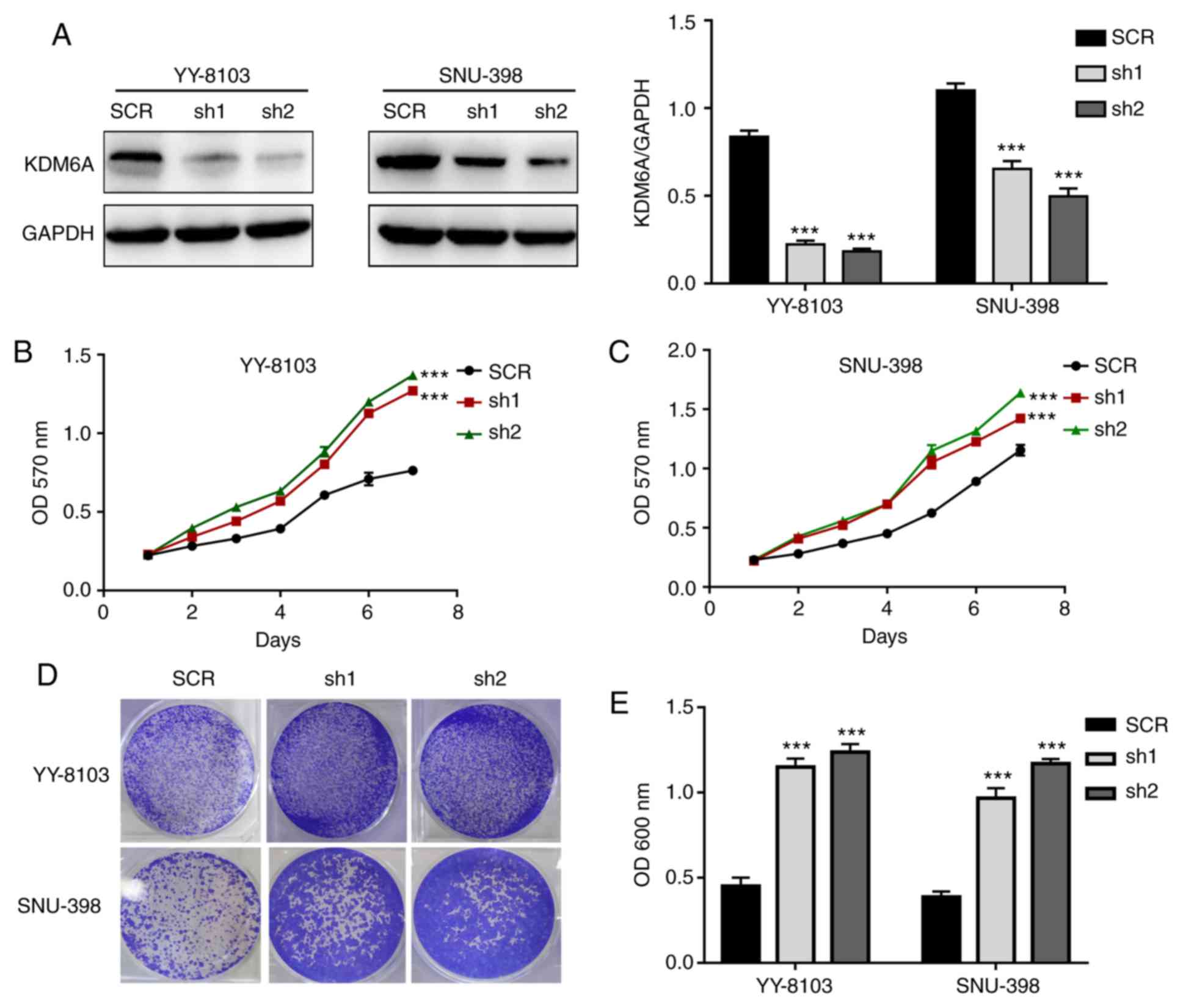
KDM6A knockdown promotes HCC cell
proliferation in vitro
Using gene-specific shRNAs, KDM6A expression was
successfully knocked down in YY-8103 and SNU-398 cells (Fig. 3A). Similar to the overexpression
experiments, the effect of KDM6A knockdown on HCC cell
proliferation was assessed. The results of the MTT assay indicated
that the absorbance values of KDM6A-knockdown YY-8103 and SNU-398
cells were significantly higher compared with SCR-transfected cells
on day 7 (Fig. 3B and C). Consistently, the crystal violet assay
results indicated that KDM6A knockdown markedly promoted YY-8103
and SNU-398 cell proliferation compared with SCR-transfected cells
(Fig. 3D). Furthermore, the
absorbance values of crystal violet in KDM6A-knockdown YY-8103 and
SNU-398 cells were significantly higher compared with
SCR-transfected cells (Fig. 3E).
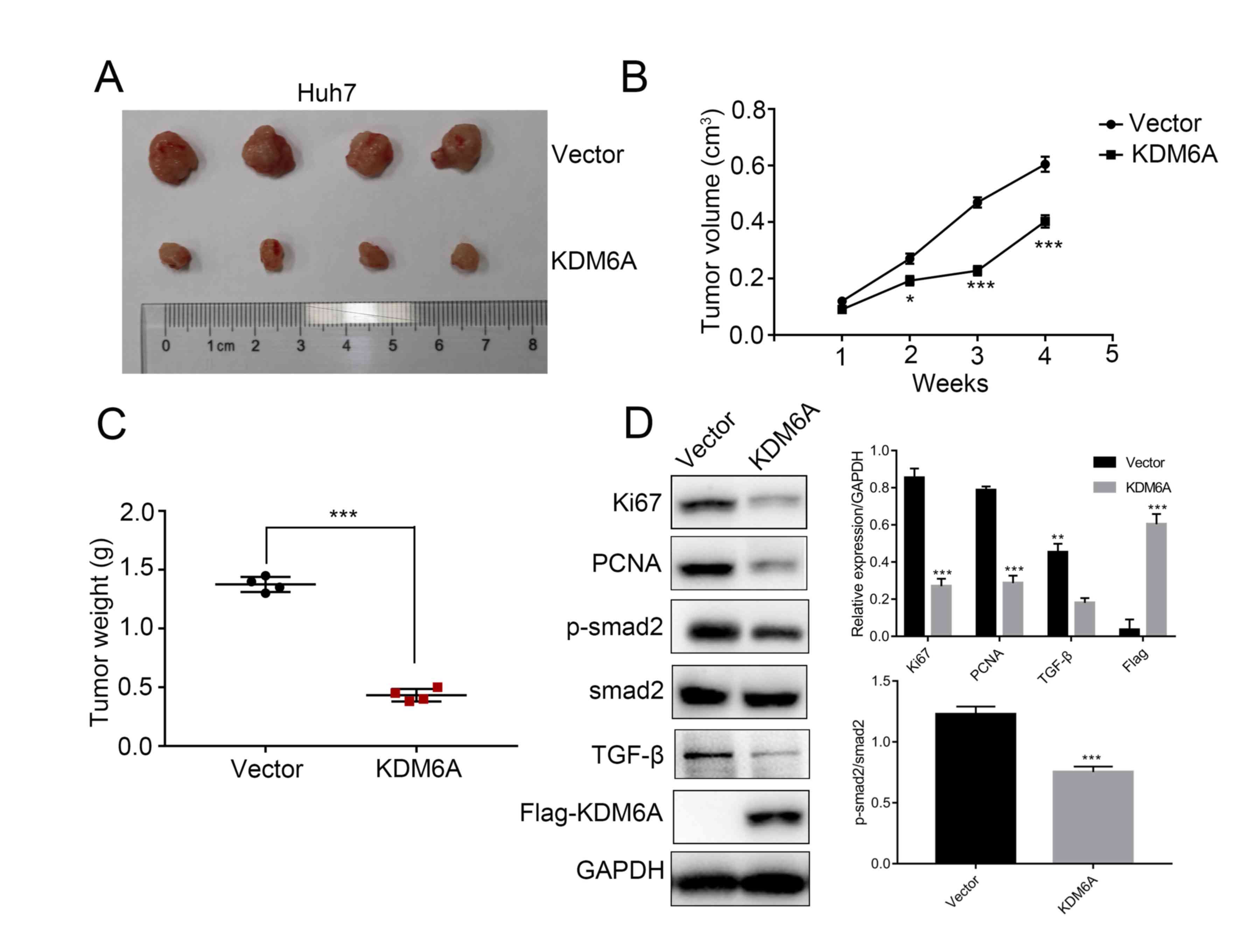
KDM6A overexpression suppresses HCC
cell tumorigenesis in vivo
Based on the in vitro results, empty vector-
and KDM6A overexpression vector-transfected Huh7 cells were
subcutaneously injected into the right flank of nude mice.
Subsequently, the tumors were excised and photographed. Consistent
with the in vitro results, KDM6A overexpression notably
suppressed tumor growth compared with the empty vector control
group in the xenograft mouse model (Fig.
4A). In particular, KDM6A overexpression tumors were lighter
and grew at a reduced rate compared with empty vector control
tumors (P<0.001; Fig. 4B and
C). The results suggested that KDM6A
may serve as a tumor suppressor during HCC cell tumorigenesis in
vivo. In addition, the western blotting results indicated that
the protein expression levels of TGF-β, p-smad2, Ki67 and PCNA were
significantly decreased in KDM6A overexpression tumors compared
with empty vector control tumors (Fig.
4D).
KDM6A inhibits TGF-β/SMAD signaling in
HCC cells
To explore the molecular mechanism underlying the
effects of KDM6A on HCC, p-smad2, total smad2, p-smad4, total smad4
and TGF-β protein expression levels were measured in
KDM6A-overexpression and KDM6A-knockdown cells. The results
indicated that the expression levels of TGF-β and p-Smad2 were
significantly decreased in KDM6A-overexpression cells compared with
empty vector control cells (Fig. 5A
and B). By contrast, KDM6A-knockdown
cells displayed significantly increased TGF-β and p-smad2
expression levels compared with SCR-transfected cells (Fig. 5C and D). However, there was no significant
difference in the expression levels of p-smad4 in
KDM6A-overexpression or KDM6A-knockdown cells compared with the
corresponding control cells. In addition, KDM6A overexpression
significantly decreased the expression levels of proliferation
markers, such as Ki67 and PCNA, compared with empty vector control
cells. By contrast, KDM6A knockdown significantly increased the
expression levels of Ki67 and PCNA compared with SCR-transfected
cells (Fig. 5).
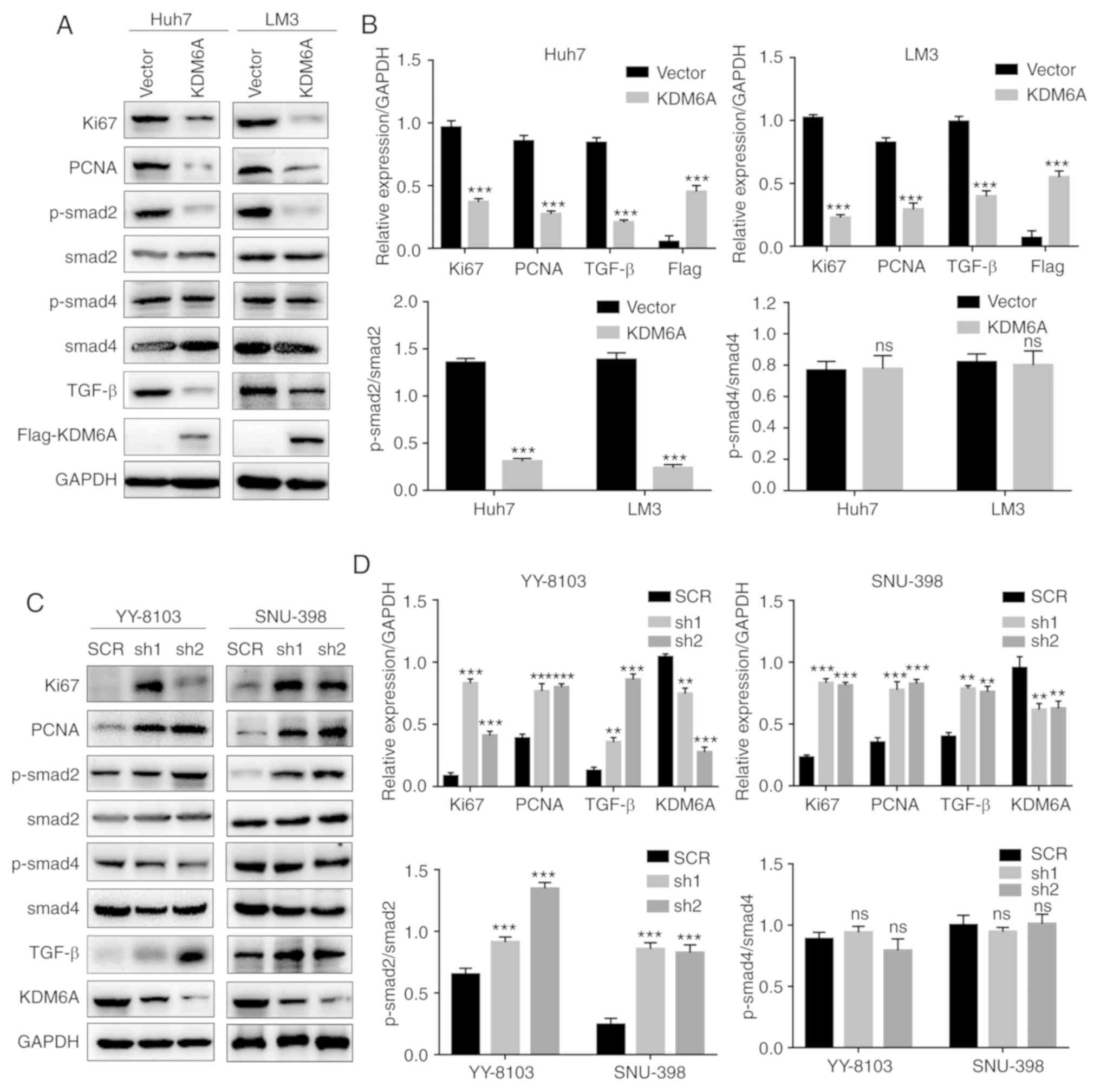 | Figure 5KDM6A negatively regulates the
TGF-β/SMAD signaling pathway. (A) The effect of KDM6A
overexpression on the expression of TGF-β, p-smad2, p-smad4, Ki67
and PCNA in Huh7 and LM3 cells was (A) determined by western
blotting and (B) semi-quantified. The effect of KDM6A knockdown on
the expression of TGF-β, p-smad2, p-smad4, Ki67 and PCNA in YY-8103
and SNU-398 was (C) determined by western blotting and (D)
semi-quantified. **P<0.01 and
***P<0.001 vs. vector or SCR. KDM6A, lysine
demethylase 6A; TGF-β, transforming growth factor-β; p,
phosphorylated; PCNA, proliferating cell nuclear antigen; SCR,
scrambled; sh, short hairpin RNA; ns, not significant. |
Discussion
A number of inactivating somatic mutations and copy
number losses related to KDM6A have been identified in numerous
malignancies, including pancreatic ductal adenocarcinoma, multiple
myeloma, and esophageal and urethral cancer (11). KDM6A gene loss induces squamous-like
metastatic pancreatic cancer, specifically in women, by
deregulating the COMPASS-like complex (15). Furthermore, loss of KDM6A has been
correlated with a poor prognostic subtype of human pancreatic
cancer, where it serves a functional role as a tumor suppressor
(13). KDM6A can also inhibit the
invasion of HCT-116 colon cancer cells by upregulating E-cadherin
expression, whereas KDM6A knockdown enhances epithelial-mesenchymal
transition (EMT)-mediated stem cell properties in breast cancer
cells (21,22). Moreover, Terashima et al
(16) reported that KDM6A served an
antagonistic role in TGF-β-induced lung cancer cell EMT and
migration.
The present study indicated that KDM6A mRNA levels
were significantly downregulated in HCC tissues compared with
corresponding control tissues. Western blotting was performed to
verify the transfection efficiency of KDM6A overexpression and
knockdown in HCC cell lines. HCC cell proliferation was
significantly inhibited by KDM6A overexpression compared with empty
vector-transfected cells. Conversely, KDM6A knockdown promoted HCC
cell proliferation compared with SCR-transfected cells.
Furthermore, KDM6A overexpression also suppressed HCC cell
tumorigenesis in vivo. Further mechanistic studies indicated
that the expression levels of TGF-β and p-smad2 were significantly
decreased by KDM6A overexpression compared with empty
vector-transfected cells. By contrast, KDM6A knockdown increased
TGF-β and p-smad2 expression levels, which are related to tumor
progression (23), compared with
SCR-transfected cells. Therefore, the present study indicated that
KDM6A may inhibit HCC cell proliferation by negatively regulating
TGF-β/SMAD signaling. TGF-β can influence gene expression by
interacting with SMAD protein transcription factors, such as smad2,
smad3 and smad4, to induce heterodimer formation (24,25).
TGF-β signaling serves a complex role during the development and
progression of various diseases (26). Specifically, activation of the TGF-β
signaling pathway has been associated with increased growth and
invasion of malignant cells during the late stages of tumor
progression (27-29).
To the best of our knowledge, the present study was the first study
to indicate that KDM6A may be involved in HCC cell
proliferation.
In summary, the present study demonstrated that
KDM6A may serve a critical role in HCC cell proliferation. The
results provided a mechanistic understanding of the role of KDM6A
during tumor development, suggesting that KDM6A may serve as a
potential target for the diagnosis or treatment of HCC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL and JY conducted the experiments. XZ and HL
provided the clinical samples. XZ, HL and JG analyzed the data. YL,
XZ, HL and JG drafted and revised the manuscript. JG designed and
supervised the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Gansu Provincial Hospital (approval no.
KY-20190089; Lanzhou, China). Written informed consent was obtained
from all subjects and the study was conducted in accordance with
the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
European Association for the Study of the
Liver. Electronic address: easloffice@easloffice.eu; European
Association for the Study of the Liver. EASL Clinical Practice
Guidelines: Management of hepatocellular carcinoma. J Hepatol.
69:182–236. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lu XJ, Shi Y, Chen JL and Ma S:
Krüppel-like factors in hepatocellular carcinoma. Tumour Biol.
36:533–541. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yu WB, Rao A, Vu V, Xu L, Rao JY and Wu
JX: Management of centrally located hepatocellular carcinoma:
Update 2016. World J Hepatol. 9:627–634. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Portolani N, Coniglio A, Ghidoni S,
Giovanelli M, Benetti A, Tiberio GA and Giulini SM: Early and late
recurrence after liver resection for hepatocellular carcinoma:
Prognostic and therapeutic implications. Ann Surgery. 243:229–235.
2006.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hu J and Gao DZ: Distinction immune genes
of hepatitis-induced heptatocellular carcinoma. Bioinformatics.
28:3191–3194. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Marquardt JU, Galle PR and Teufel A:
Molecular diagnosis and therapy of hepatocellular carcinoma (HCC):
An emerging field for advanced technologies. J Hepatol. 56:267–275.
2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hong S, Cho YW, Yu LR, Yu H, Veenstra TD
and Ge K: Identification of JmjC domain-containing UTX and JMJD3 as
histone H3 lysine 27 demethylases. Proc Natl Acad Sci USA.
104:18439–18444. 2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lee MG, Villa R, Trojer P, Norman J, Yan
KP, Reinberg D, Di Croce L and Shiekhattar R: Demethylation of
H3K27 regulates polycomb recruitment and H2A ubiquitination.
Science. 318:447–450. 2007.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Jiang W, Wang J and Zhang Y: Histone
H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm
differentiation from human ESCs by regulating WNT signaling
pathway. Cell Res. 23:122–130. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Schuettengruber B, Bourbon HM, Di Croce L
and Cavalli G: Genome Regulation by Polycomb and Trithorax: 70
Years and Counting. Cell. 171:34–57. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
van Haaften G, Dalgliesh GL, Davies H,
Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O'Meara S, Teague
J, et al: Somatic mutations of the histone H3K27 demethylase gene
UTX in human cancer. Nat Genet. 41:521–523. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Terashima M, Ishimura A, Yoshida M, Suzuki
Y, Sugano S and Suzuki T: The tumor suppressor Rb and its related
Rbl2 genes are regulated by Utx histone demethylase. Biochem
Biophys Res Commun. 399:238–244. 2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Watanabe S, Shimada S, Akiyama Y, Ishikawa
Y, Ogura T, Ogawa K, Ono H, Mitsunori Y, Ban D, Kudo A, et al: Loss
of KDM6A characterizes a poor prognostic subtype of human
pancreatic cancer and potentiates HDAC inhibitor lethality. Int J
Cancer. 145:192–205. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ler LD, Ghosh S, Chai X, Thike AA, Heng
HL, Siew EY, Dey S, Koh LK, Lim JQ, Lim WK, et al: Loss of tumor
suppressor KDM6A amplifies PRC2-regulated transcriptional
repression in bladder cancer and can be targeted through inhibition
of EZH2. Sci Transl Med. 9(eaai8312)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Andricovich J, Perkail S, Kai Y, Casasanta
N, Peng W and Tzatsos A: Loss of KDM6A activates super-enhancers to
induce gender-specific squamous-like pancreatic cancer and confers
sensitivity to BET inhibitors. Cancer Cell. 33:512–526.e8.
2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Terashima M, Ishimura A, Wanna-Udom S and
Suzuki T: Epigenetic regulation of epithelial-mesenchymal
transition by KDM6A histone demethylase in lung cancer cells.
Biochem Biophys Res Commun. 490:1407–1413. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wang Y, Liu DP, Chen PP, Koeffler HP, Tong
XJ and Xie D: Involvement of IFN regulatory factor (IRF)-1 and
IRF-2 in the formation and progression of human esophageal cancers.
Cancer Res. 67:2535–2543. 2007.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Qiu Z, Zou K, Zhuang L, Qin J, Li H, Li C,
Zhang Z, Chen X, Cen J, Meng Z, et al: Hepatocellular carcinoma
cell lines retain the genomic and transcriptomic landscapes of
primary human cancers. Sci Rep. 6(27411)2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Vatandoost J and Kafi Sani K: A study of
recombinant factor IX in Drosophila insect S2 cell lines through
transient gene expression technology. Avicenna J Med Biotechnol.
10:265–268. 2018.PubMed/NCBI
|
|
20
|
Sikes RS: 2016 Guidelines of the American
Society of Mammalogists for the use of wild mammals in research and
education. J Mammal. 97:663–688. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zha L, Cao Q, Cui X, Li F, Liang H, Xue B
and Shi H: Epigenetic regulation of E-cadherin expression by the
histone demethylase UTX in colon cancer cells. Med Oncol.
33(21)2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Choi HJ, Park JH, Park M, Won HY, Joo HS,
Lee CH, Lee JY and Kong G: UTX inhibits EMT-induced breast CSC
properties by epigenetic repression of EMT genes in cooperation
with LSD1 and HDAC1. EMBO Rep. 16:1288–1298. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Dai J, Xu M, Zhang X, Niu Q, Hu Y, Li Y
and Li S: Bi-directional regulation of TGF-β/Smad pathway by
arsenic: A systemic review and meta-analysis of in vivo and in
vitro studies. Life Sci. 220:92–105. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wrana JL, Attisano L, Wieser R, Ventura F
and Massagué J: Mechanism of activation of the TGF-beta receptor.
Nature. 370:341–347. 1994.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Moses HL, Roberts AB and Derynck R: The
discovery and early days of TGF-β: A historical perspective. Cold
Spring Harb Perspect Biol. 8(a021865)2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Derynck R, Akhurst RJ and Balmain A:
TGF-beta signaling in tumor suppression and cancer progression. Nat
Genet. 29:117–129. 2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Pasche B: Role of transforming growth
factor beta in cancer. J Cell Physiol. 186:153–168. 2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Elliott RL and Blobe GC: Role of
transforming growth factor Beta in human cancer. J Clin Oncol.
23:2078–2093. 2005.PubMed/NCBI View Article : Google Scholar
|