Introduction
Internal organs of the human and other vertebrates
exhibit a highly conserved left-right (LR) asymmetry arrangement
which is the base for their function and position (1,2). The
normal organ asymmetry arrangement across the LR axis is termed as
situs solitus (3,4). LR asymmetry disorders occur as a result
of genetic defects in the motile cilia of the embryonic node and
nodal signaling pathway (5,6). The cilia-mediated fluid flow plays a
critical role in the establishment of LR asymmetry arrangement
during early embryonic development (7). Human LR asymmetry disorders with a
minimum incidence of 1 per 8,000 newborns, can be divided into two
categories, including situs inversus (complete mirror-image
reversal of the internal organs) and situs ambiguus (abnormal
arrangement of the internal organs, also named heterotaxy)
(1,4). Situs inversus is also named situs
inversus with dextrocardia for mirror-image anatomical topography
of the heart (8,9). Situs inversus occurs in approximately
half of patients with primary ciliary dyskinesia (PCD), while situs
ambiguus occurs in no less than 12% of PCD patients (8). PCD is a ciliary disorder characterized
by LR asymmetry disorder, neonatal respiratory distress, male
infertility, and chronic oto-sino-pulmonary disease (10,11).
Other rare symptoms including female infertility, hydrocephalus and
retinitis pigmentosa have also been observed (6,12). Most
of PCD cases are inherited in an autosomal recessive inheritance
pattern, and a few have autosomal dominant or X-linked inheritance
(8,13). Mutations in the genes associated with
the establishment and function of nodal cilia are one of the
genetic causes of human LR asymmetry disorders (4,12). No
less than 69 genes have been reported to be associated with human
LR asymmetry disorders (5,13-22).
Mutations in the coiled-coil domain containing 114 gene
(CCDC114), encoding a ciliary protein necessary for the
attachment of the outer dynein arms (ODAs) to the axoneme of cilia,
are reported to cause a subtype of PCD named ciliary dyskinesia,
primary, 20 (CILD20) (23).
In the present study, a combination of exome
sequencing and Sanger sequencing was used to identify the
disease-causing gene and variant for a Han-Chinese patient with
situs inversus. As a result, a homozygous variant, c.584T>C
(p.L195P), in the CCDC114 gene, was identified in the
patient with situs inversus.
Patient and methods
Participators and clinical data
A 62-year-old female patient diagnosed with
mirror-image dextrocardia using a 12-lead electrocardiogram, in
addition to a 59-year-old unrelated healthy female without any
off-target condition, were recruited from the Third Xiangya
Hospital, Central South University (Changsha, China) in December,
2018. The medical record of the patient was scrutinized and routine
physical and radiological examinations were performed on the
patient. The medical history of her deceased parents was also
scanned. Written informed consent was obtained from the patient and
the healthy individual before venous blood was sampled. The entire
study was approved by the Institutional Review Board of the Third
Xiangya Hospital, Central South University (Changsha, China) and
carried out in accordance with the Declaration of Helsinki as
revised in 2013.
Exome capture
After being extracted from peripheral blood samples
by using the standard method described in a previous study
(24), the genomic DNA (gDNA) of the
proband was quantified by Qubit dsDNA HS Assay Kit (Invitrogen;
Thermo Fisher Scientific, Inc.), and its integrity and purity were
qualified on 1% agarose gel. An exome library was constructed with
1 µg gDNA. It was sheared into fragments using Covaris E220
(Covaris, Inc.) and those with sizes of between 150 and 250 bp were
selected using Agencourt AMPure XP Kit (Beckman Coulter Inc.).
After being subjected to end-repairing, A-tailing reactions and
adaptor ligation, the ligated fragments were enriched via
amplification, purification and hybridization, using Agilent
SureSelect Human All Exon V6 (cat. no. 5190-8872; Agilent
Technologies, Inc.) and Agencourt AMPure XP Kit (Beckman Coulter
Inc.). Following circularization with T4 DNA Ligase (cat. no.
L6030-LC-L; Enzymatics Inc.; Qiagen GmbH), captured enrichment was
processed by rolling circle amplification to form DNA nanoballs,
which were then loaded onto a sequencing chip with a concentration
of 40 ng/µl to be sequenced using MGISEQ-2000RS High-throughput
Sequencing Set (FCL PE100; cat. no. 1000012554; BGI group).
Paired-end sequencing with read lengths of 100 bp was performed on
BGISEQ-500 sequencing platforms (BGI group) using combinatorial
probe-anchor synthesis. All of the aforementioned experimental
operations were performed in accordance to the manufacturers'
protocols.
Read mapping and variant analysis
The raw data was processed to clean data which was
mapped to the human reference genome (GRCh37/hg19) using Burrows
Wheeler Aligner (BWA; version 0.7.15) (25). Remove of duplicated sequence by
Picard (version 2.5.0, https://broadinstitute.github.io/picard/) and
realignment by Genome Analysis Toolkit (GATK v.3.3.0, https://gatk.broadinstitute.org/hc/en-us) were
performed for accurate variant calling (26). Insertions-deletions (indels) and
single nucleotide polymorphisms (SNPs) were called using
HaplotypeCaller of GATK. Variants were annotated by SnpEff software
(http://snpeff.sourceforge.net/SnpEff_manual.html)
(27), and filtered against public
databases including the Single Nucleotide Polymorphism database
(dbSNP build 141) (28), 1000
Genomes Project (29) and the NHLBI
exome sequencing project (ESP) 6500(30), and an in-house exome database of
BGI-Shenzhen (BGI group). The causative variants were confirmed by
Sanger sequencing with ABI 3500 sequencer (Applied Biosystems;
Thermo Fisher Scientific, Inc.) (31). The following are the sequences of
primers for potential causative variant in the patient: Forward,
5'-CAGTCCAGCCTCCAGTCATC-3' and reverse,
5'-TTTTCACGCTTCTCCAGGAC-3'.
Bioinformatic analyses
The possible impacts of variants on protein
structure or function were predicted by several bioinformatic
prediction software programs including MutationTaster (32), Protein Analysis Through Evolutionary
Relationships (PANTHER) (33) and
Protein Variation Effect Analyzer (PROVEAN) (34,35). A
further conservative assessment of the amino acid at the variant
position among different species was performed with the National
Center for Biotechnology Information-Basic Local Alignment Search
Tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (36,37).
Protean program of DNAStar's Lasergene v7.1.0 (DNASTAR, Inc.) was
used to predict whether the candidate variant affected the protein
secondary structure (38).
Quantitative PCR (qPCR)
qPCR was performed to check the possible deletion
involving candidate variant position using a
LightCycler® 480 Instrument II (Roche) with qPCR SYBR
Green Master Mix (Vazyme Biotech Co., Ltd.). The primers for the
possible deletion were as follows: 5'-CAGTCCAGCCTCCAGTCATC-3' and
5'-ACCTGCGCCTCCATCTCG-3'. The gDNA sample of the healthy individual
was used as the reference control. The glyceraldehyde-3-phosphate
dehydrogenase gene (GAPDH) served as the reference gene and
the primers were: 5'-CACTCCTCCACCTTTGACGC-3' and
5'-CCACCACCCTGTTGCTGTAG-3'. Each reaction was conducted in
triplicate. A repeated experiment was performed with the reference
ATP binding cassette subfamily A member 4 gene (ABCA4) to
ensure repeatability, and the primers were as previously published
(39): 5'-ACCCAAGTATGGCCCGTCCA-3'
and 5'-TCCCATCCATCTGTTGCAGG-3'. The relative copy numbers of the
candidate gene were evaluated using the comparative quantification
cycle (2-ΔΔCq) method (40). Statistical analysis was performed
using the Microsoft Excel 2016 software (Microsoft, Inc.) and
GraphPad Prism v8.0.2 (GraphPad Software, Inc.) using a two-tailed
unpaired Student's t-test.
Results
Clinical findings
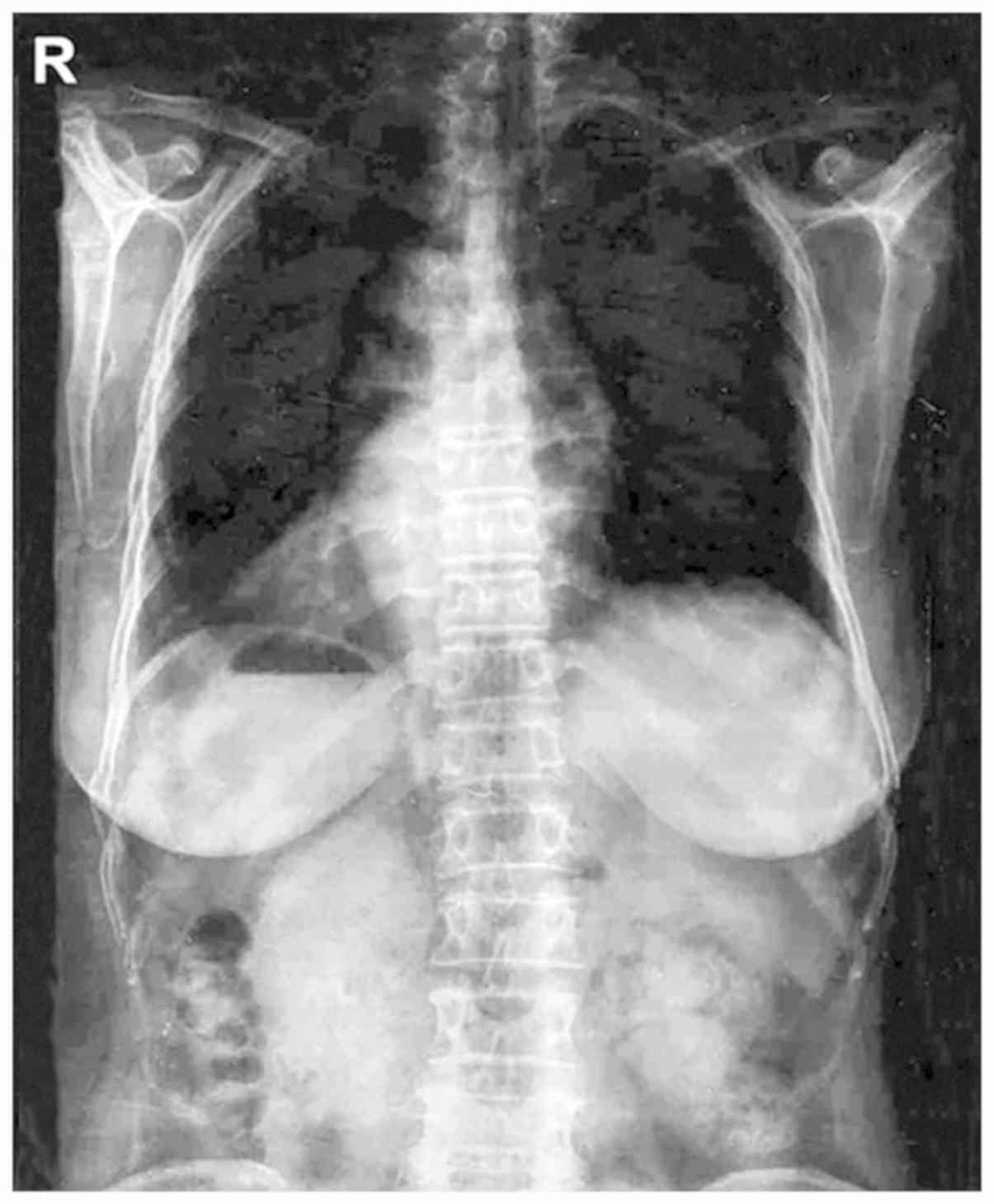
Chest radiograph revealed the mirror-image reversal
of the heart and gastric bubble in the patient (Fig. 1), and no other classical PCD symptoms
were observed, which agreed with the diagnosis of situs inversus
(Table I). The medical history of
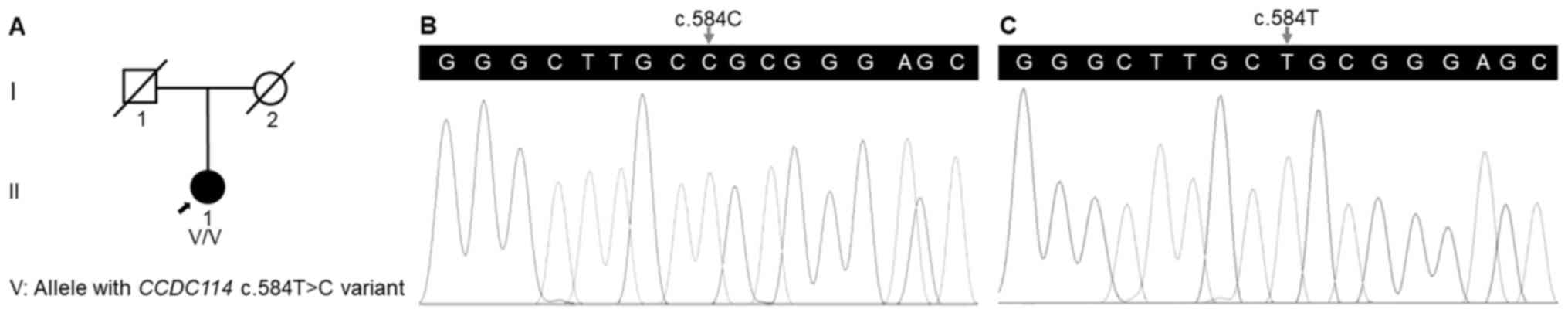
her deceased parents suggested a negative family history in her
family (Fig. 2A).
 | Table IClinical characteristics and
genotypes of patients with CCDC114 variants. |
Table I
Clinical characteristics and
genotypes of patients with CCDC114 variants.
| Study (ref.) | Patient | Sex | Age (years) | Situs | Neo RDS | Bxsis | Sinusitis | Otitis media | Other symptoms | Variant | Zygosity |
|---|
| Onoufriadis | Family 1 | II:3 | M | NA | SS | − | + | + | + | − | c.742G>A | Homozygote |
| et al,
2013 | | II:4 | M | NA | DEX | NA | + | + | − | − | | |
| (23) | | II:7 | F | NA | NA | NA | NA | NA | NA | NA | | |
| | | III:1 | M | NA | SS | + | NA | + | + | CHD | | |
| | | III:2 | M | NA | SI | − | NA | + | + | − | | |
| | | III:3 | M | NA | SI | − | NA | − | + | − | | |
| | | III:5 | F | NA | SS | − | + | − | + | − | | |
| | | III:8 | M | NA | SS | − | NA | − | + | − | | |
| | Family 2 | II:2 | F | NA | SS | − | + | + | + | − | | |
| | Family 3 | I:1 | F | NA | SS | NA | + | + | + | − | | |
| | Family 4 | II:1 | M | NA | SS | − | NA | − | + | − | | |
| | | II:2 | F | NA | Abdominal SI | − | NA | − | + | − | | |
| | Family 5 | II:1 | F | NA | SS | − | NA | NA | + | − | | |
| | Family 6 | II:1 | M | NA | SS | − | NA | + | + | − | | |
| | Family 7 | II:1 | M | NA | SI with medial
heart position | − | NA | − | + | CHD | | |
| | Family 8 | II:1 | F | NA | SI | + | NA | − | − | − | | |
| | Family 9 | II:1 | F | NA | SI | − | + | + | + | CHD | c.486+1G>A | Homozygote |
| Knowles et
al | Family 10 | III:1 | F | 39 | SS | + | + | + | + | − | c.742G>A | Homozygote |
| 2013(55) | | III:3 | F | 33 | SS | + | + | + | + | − | | |
| | Family 11 | I:3 | F | 59 | SS | + | + | + | + | − | c.742G>A; | Compound |
| | | | | | | | | | | | c.487−2A>G | heterozygote |
| | Family 12 | IV:3 | M | 3 | SS | + | − | − | + | − | c.742G>A; | Compound |
| | | | | | | | | | | | c.1391+5G>A | heterozygote |
| | Family 13 | II:1 | F | 38 | SS | − | NA | NA | + | − | c.742G>A; | Compound |
| | | | | | | | | | | | c.939delT | heterozygote |
| | | II:2 | F | 34 | SS | − | + | + | + | − | | |
| Li et al,
2019(52) | Family 14 | III:1 | F | 15 | SS | − | + | + | − | Multiple organ
dysplasia, renal fibrosis and CHD | c.596C>T
(p.A199V) | Homozygote |
| Present study | Family 15 | II:1 | F | 62 | SI | − | − | − | − | − | c.584T>C
(p.L195P) | Homozygote |
Variant screening
A total of 142.67 million effective reads were
generated from exome sequencing of the patient. Among them, 99.94%
were successful in mapping to the human reference genome. The
target sequence covered 99.48% of bases at ≥x10 with an average
sequencing depth of x177.41. A total of 97,358 SNPs and 16,875
indels were detected in the patient. A variant filtering strategy
as described in recent studies was utilized for confirming the
potential causative variant for the patient (39,41): i)
Variants documented in dbSNP141, 1000 Genomes Project and NHLBI
ESP6500 with a minor allele frequency >0.1% were excluded; ii)
among the remaining, variants absent in 1,943 additional Chinese
controls without LR asymmetry disorder from an in-house exome
database of BGI-Shenzhen were preferred for the next analysis; and
iii) variants predicted to be deleterious by in silico tools
were reserved. Using the aforementioned criteria, a variant in the
CCDC114 gene (reference sequence: NM_144577.4), c.584T>C
(p.L195P), was selected as the most possible disease-causing
variant for the patient. Sanger sequencing validated the variant in
the patient (Fig. 2B) and its
absence in the healthy individual (Fig.
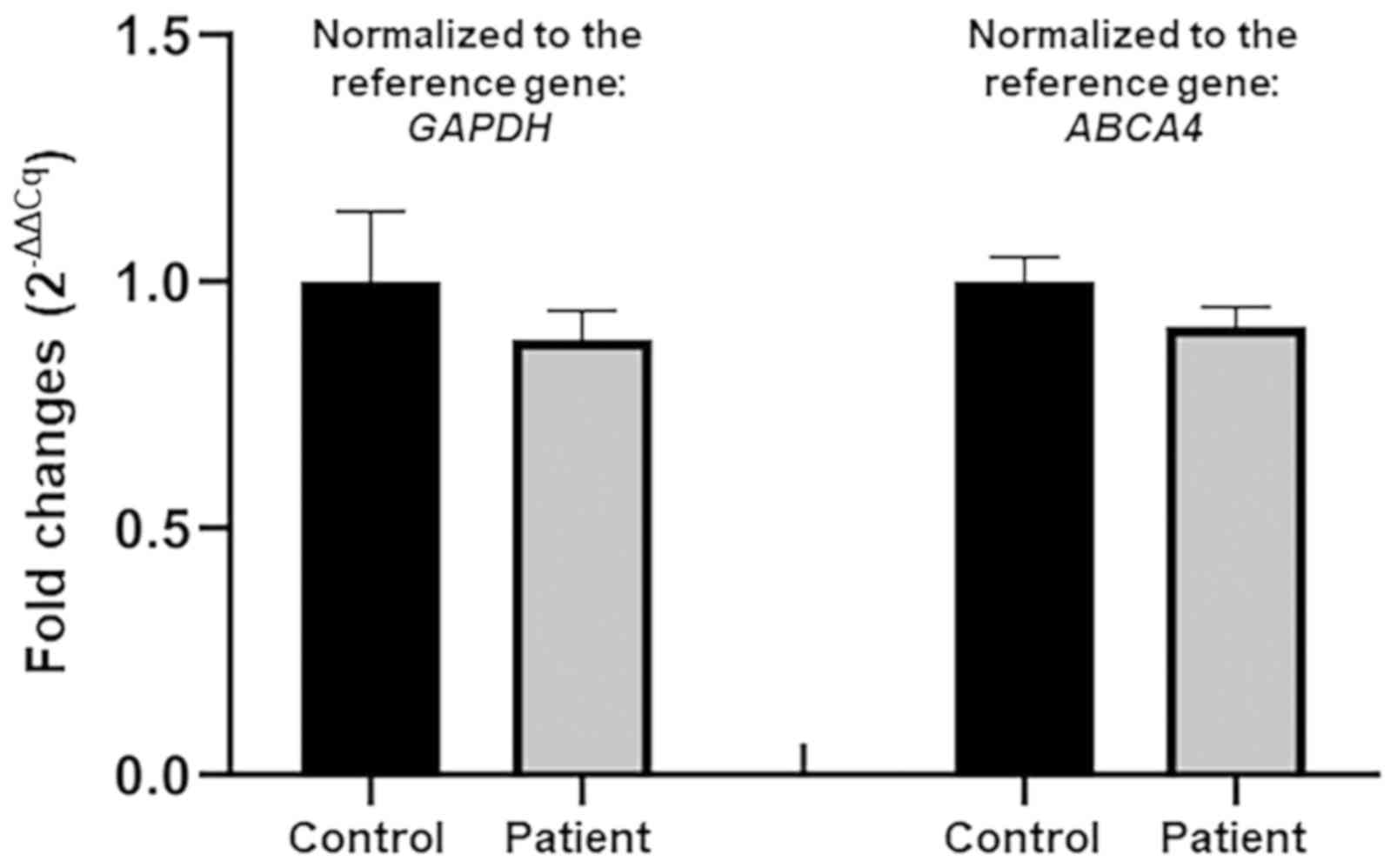
2C). The qPCR analysis confirmed no deletion of the
CCDC114 gene involving the variant position in the patient
(Fig. 3).
Bioinformatic analyses
The CCDC114 variant c.584T>C (p.L195P),
was predicted to be disease-causing by MutationTaster with a
probability score of ~1, possibly damaging by PANTHER with the
position in the protein evolutionarily conserved for >200
million years (preservation time, 220 million years), and
deleterious by PROVEAN with a score of -5.99. These indicated that
the protein structure and function were possibly affected by this
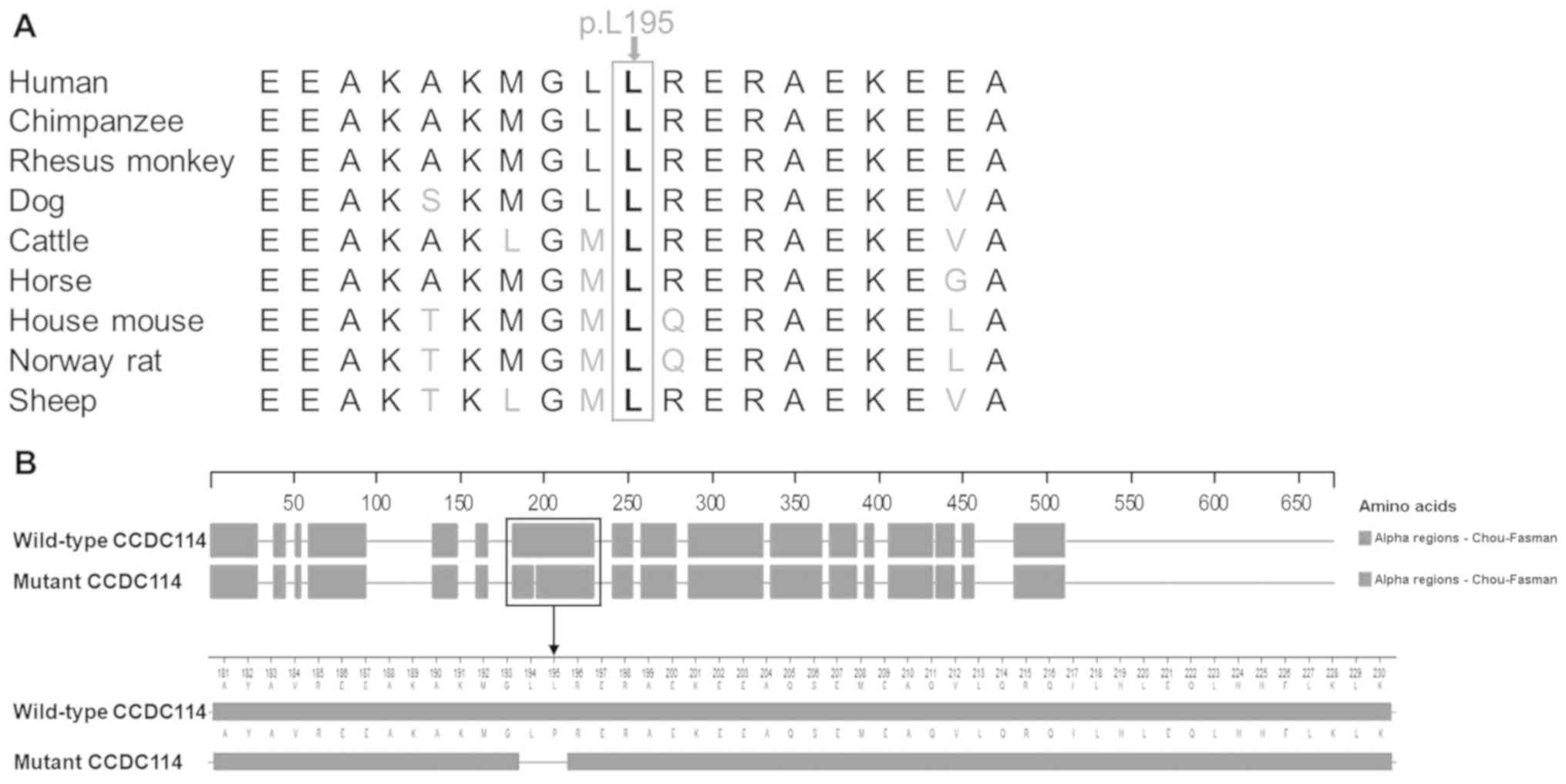
variant, which is consistent with the high conservation of the
p.L195 residue among nine vertebrates (Fig. 4A). This variant was predicted to
change the secondary structure of CCDC114 by Protean based on the
Chou-Fasman method (Fig. 4B). An
α-helix (residues 181-230) was predicted to be broken into two
α-helices (residues 181-193 and residues 196-230) by the
c.584T>C variant. Based on the above evidence, the variant
c.584T>C in the CCDC114 gene appears to be accountable
for the situs inversus in this patient.
Discussion
The normal human body displays an obvious LR
asymmetry in the placement and structure of the internal organs
(42). The perturbation of human LR
asymmetry gives rise to disorders including situs inversus and
situs ambiguous (43). The
generation of LR asymmetry depends on LR side-specific cascades of
gene expression during early embryogenesis. The process can be
divided into four distinct phases: i) the bilateral symmetry
breaking of the early embryo; ii) asymmetric signals transduced
from the node to lateral plate mesoderm (LPM); iii) cascades of LR
asymmetric gene expression in the left LPM; and iv) LR asymmetric
morphogenesis (5,44).
Abnormal embryonic nodal cilia and perturbation in
the nodal signaling pathway are two causes for human LR asymmetry
disorders (5). A group of human LR
asymmetry disorders resulting from dysmotility or absence of
embryonic nodal cilia can be regarded as a type of ciliopathy
(45). Cilia and flagella are
hair-like organelles in eukaryotic cells. A cilium is composed of a
basal body, transition zone, axoneme, ciliary membrane, and the
ciliary tip. The axoneme is the main part of the cilium, and the
basal body inside the cell plays a role in anchoring the cilium
(45-47).
The axoneme is assembled by nine peripheral microtubule doublets,
surrounding a central pair of microtubules (9+2 pattern) or absence
of the central microtubules (9+0 pattern) (48). Each peripheral doublet microtubule
possesses an outer dynein arm (ODA) and an inner dynein arm (IDA)
(6). The nodal motile cilium with a
9+0 axonemal configuration without central pair existing in the
early embryo, has dynein arms and nine peripheral doublets
(11). Gene-targeted therapy during
early embryogenesis that aimed to normalize the nodal signaling
pathway or the function of embryonic nodal cilia may be helpful for
rescuing the perturbation of human LR asymmetry.
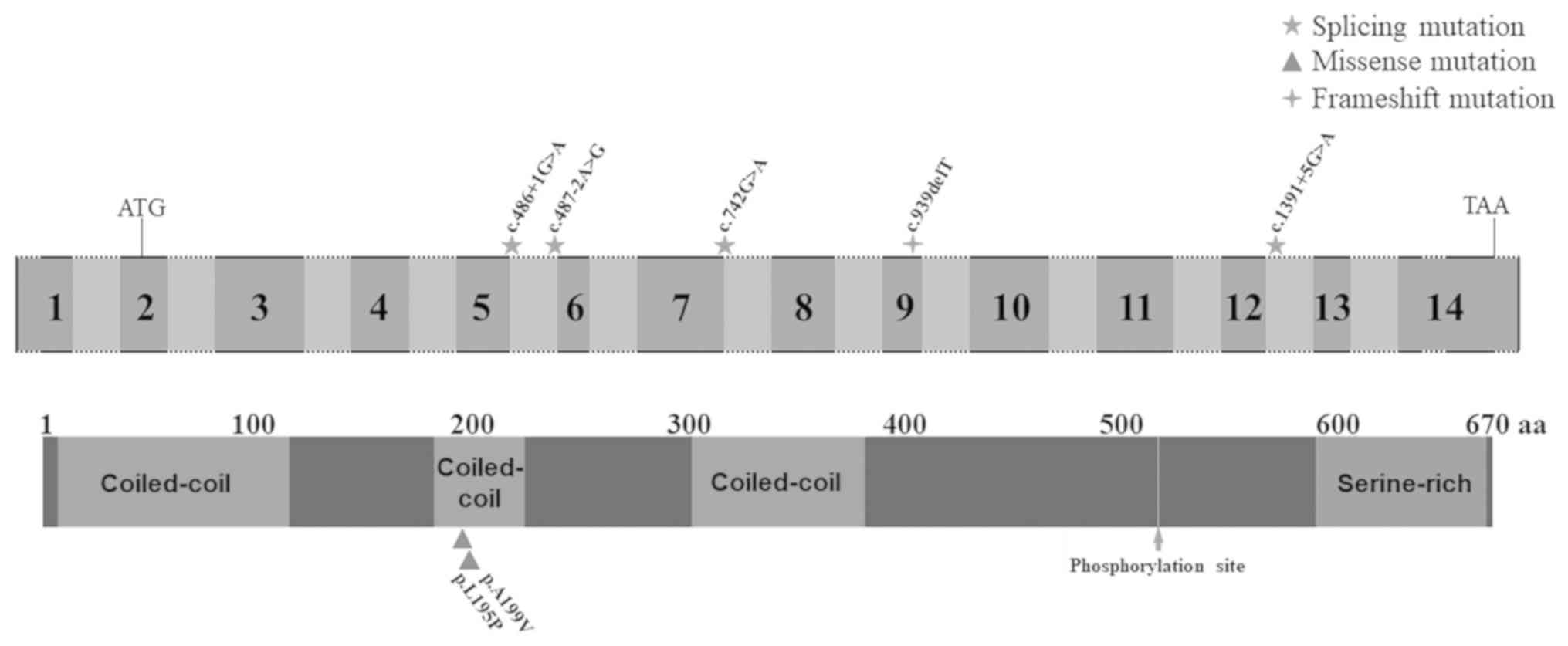
The CCDC114 gene is located at chromosome
19q13.33 and is composed of 14 exons. The ciliary protein, CCDC114,
as a component of ODA docking complex along with CCDC151, armadillo
repeat containing protein 4, and tetratricopeptide repeat domain
25, may interact with meiosis specific nuclear structural 1 and
dynein axonemal heavy chain 9 to participate in binding ODAs to
outer doublet microtubules (17,20,21,23,49). The
dynein arms containing ATPase provide a driving force (four-fifths
from ODAs) for the sliding of peripheral doublet microtubules which
generates ciliary beating (49,50). The
genetic deficiency in CCDC114 cause ODA defects, leading to
immotile cilia. Impaired motile 9+2 cilia and flagella are
responsible for PCD symptoms including neonatal respiratory
distress, chronic oto-sino-pulmonary disease, infertility, and
hydrocephalus, while human LR asymmetry disorders result from
defective embryonic nodal motile (9+0) cilia (6,10). The
nodal cilia clockwise rotary generates a leftward flow of
extraembryonic fluid, which results in the breakage of the
bilateral symmetry (51). Mutations
in the CCDC114 gene are known to cause CILD20 (OMIM 615067).
Deficiency in CCDC114 not only impairs ODA of motile cilia
but also impacts the biogenesis of primary cilia (23,52).
Various cases with mutations in ciliary protein and
displaying partial PCD symptoms but imperfect for the standard
clinical diagnostic criteria for PCD have also been reported
(14,17). In the present study, a patient
presented with situs inversus but no other PCD symptoms were
reported, and a missense variant c.584T>C (p.L195P) located in
the coiled-coil domain of the CCDC114 protein was identified in
her. The qPCR analysis confirmed no deletion of the CCDC114
gene involving the variant position, validating the homozygosity of
the c.584T>C variant. Although consanguineous marriage was
denied in this family, the parents resided within the same village
which may imply a possibility of founder effect (53). In silico tools including
MutationTaster, PANTHER and PROVEAN predicted that the c.584T>C
variant had deleterious effects on protein structure and function.
This variant was absent in dbSNP141, 1000 Genomes Project, NHLBI
ESP6500 and an in-house exome database of BGI-Shenzhen. It may
change the secondary structure of CCDC114 by breaking an α-helix
(residues 181-230) at residues 194-195 based on Protean prediction.
Further functional studies, as well as co-segregation analyses in
more families with situs inversus, are necessary for validating
accurate clinical classification of this variant. Defective motile
cilia in different tissues and organs underlie different PCD
symptoms (54). The patient carrying
homozygous CCDC114 variant c.584T>C presented with situs
inversus, suggesting that this missense variant mainly affects
embryonic nodal cilia. Published articles accessed in the PubMed
database reported 6 CCDC114 mutations associated with PCD.
Biallelic splice-site mutations in the CCDC114 gene
including homozygous mutations (c.742G>A and c.486+1G>A) and
compound heterozygous mutations (c.742G>A and c.487-2A>G,
c.742G>A and c.1391+5G>A, and c.742G>A and c.939delT) were
reported to cause classical PCD phenotype, while a homozygous
c.596C>T (p.A199V) mutation was reported to cause PCD symptoms
accompanied with other additional symptoms linked with defective
primary cilia (Table I; Fig. 5) (23,52,55). No
obvious phenotype-genotype correlation in CCDC114 is
observed. The phenotypic variation of CCDC114-associated
ciliopathies suggested that one or more types of cilia may be
involved by a single CCDC114 mutation (52), which may be a random chance
confounded by other genetic, epigenetic and environmental
factors.
In summary, using exome sequencing and a significant
filtration strategy, it was demonstrated that a CCDC114
variant, c.584T>C (p.L195P), may be responsible for situs
inversus in a Han-Chinese patient. To the best of our knowledge,
the homozygous c.584T>C variant in the CCDC114 gene is
first reported as the disease-causing variant for situs inversus in
the present study. Findings from the present study broaden the
mutational spectrum of the CCDC114 gene and assist the
genetic counseling of human LR asymmetry disorders. In the future,
more confirmation and functional studies of CCDC114
mutations, and the establishment of deficient animal models with
CCDC114-associated ciliopathies will facilitate an in-depth
comprehension of molecular and cellular mechanisms of human
ciliopathies.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81670216, 81800219 and
81873686), the Natural Science Foundation of Hunan Province (grant
no. 2018JJ2660), the Science and Technology Program of Hunan
Province (grant no. 2017SK50131), the Scientific Research Project
of Health and Family Planning Commission of Hunan Province, China
(grant nos. B20180760 and B20180834), the Lotus Scholars Program of
Hunan Province (to HD), the Hunan Provincial Innovation Foundation
For Postgraduate (grant no. CX20190252) and the Undergraduate
Innovative Training Plan Program of Central South University, China
(grant no. XCX20190629).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XC, SD and HD conceived and designed this study.
HXi, ST and HD collected the patient samples and clinical data. XC,
SD and HXu performed the experiments; XC, LY and HD analyzed the
data. XC, SD and HD wrote the manuscript. All authors read and
approved the manuscript and agreed to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work was appropriately investigated and
resolved.
Ethics approval and consent to
participate
The study was conducted in compliance with the
Declaration of Helsinki Principles and with the consent of the
Institutional Review Board of the Third Xiangya Hospital of Central
South University in Changsha, Hunan, China (approval no.
2018-S400). Written informed consent was obtained from the
participants prior to being included in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Levin M: The embryonic origins of
left-right asymmetry. Crit Rev Oral Biol Med. 15:197–206.
2004.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Nakamura T and Hamada H: Left-right
patterning: Conserved and divergent mechanisms. Development.
139:3257–3262. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Blum M and Ott T: Animal left-right
asymmetry. Curr Biol. 28:R301–R304. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Shapiro A, Davis S, Manion M and Briones
K: Primary ciliary dyskinesia (PCD). Am J Respir Crit Care Med.
198:P3–P4. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Deng H, Xia H and Deng S: Genetic basis of
human left-right asymmetry disorders. Expert Rev Mol Med.
16(e19)2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lee L: Mechanisms of mammalian ciliary
motility: Insights from primary ciliary dyskinesia genetics. Gene.
473:57–66. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Narasimhan V, Hjeij R, Vij S, Loges NT,
Wallmeier J, Koerner-Rettberg C, Werner C, Thamilselvam SK, Boey A,
Choksi SP, et al: Mutations in CCDC11, which encodes a coiled-coil
containing ciliary protein, causes situs inversus due to
dysmotility of monocilia in the left-right organizer. Hum Mutat.
36:307–318. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Shapiro AJ, Zariwala MA, Ferkol T, Davis
SD, Sagel SD, Dell SD, Rosenfeld M, Olivier KN, Milla C, Daniel SJ,
et al: Diagnosis, monitoring, and treatment of primary ciliary
dyskinesia: PCD foundation consensus recommendations based on state
of the art review. Pediatr Pulmonol. 51:115–132. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zanatta A, Zampieri F, Bonati MR, Frescura
C, Scattolin G, Stramare R and Thiene G: Situs inversus with
dextrocardia in a mummy case. Cardiovasc Pathol. 23:61–64.
2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Knowles MR, Zariwala M and Leigh M:
Primary ciliary dyskinesia. Clin Chest Med. 37:449–461.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Knowles MR, Daniels LA, Davis SD, Zariwala
MA and Leigh MW: Primary ciliary dyskinesia. Recent advances in
diagnostics, genetics, and characterization of clinical disease. Am
J Respir Crit Care Med. 188:913–922. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Boon M, Jorissen M, Proesmans M and De
Boeck K: Primary ciliary dyskinesia, an orphan disease. Eur J
Pediatr. 172:151–162. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wallmeier J, Frank D, Shoemark A,
Nöthe-Menchen T, Cindric S, Olbrich H, Loges NT, Aprea I, Dougherty
GW, Pennekamp P, et al: De novo mutations in FOXJ1 result in a
motile ciliopathy with hydrocephalus and randomization of
left/right body asymmetry. Am J Hum Genet. 105:1030–1039.
2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bonnefoy S, Watson CM, Kernohan KD, Lemos
M, Hutchinson S, Poulter JA, Crinnion LA, Berry I, Simmonds J,
Vasudevan P, et al: Biallelic mutations in LRRC56, encoding a
protein associated with intraflagellar transport, cause mucociliary
clearance and laterality defects. Am J Hum Genet. 103:727–739.
2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Vetrini F, D'Alessandro LC, Akdemir ZC,
Braxton A, Azamian MS, Eldomery MK, Miller K, Kois C, Sack V, Shur
N, et al: Bi-allelic mutations in PKD1L1 are associated with
laterality defects in humans. Am J Hum Genet. 99:886–893.
2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Fassad MR, Shoemark A, le Borgne P, Koll
F, Patel M, Dixon M, Hayward J, Richardson C, Frost E, Jenkins L,
et al: C11orf70 mutations disrupting the intraflagellar
transport-dependent assembly of multiple axonemal dyneins cause
primary ciliary dyskinesia. Am J Hum Genet. 102:956–972.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ta-Shma A, Hjeij R, Perles Z, Dougherty
GW, Abu Zahira I, Letteboer SJF, Antony D, Darwish A, Mans DA,
Spittler S, et al: Homozygous loss-of-function mutations in MNS1
cause laterality defects and likely male infertility. PLoS Genet.
14(e1007602)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Perles Z, Moon S, Ta-Shma A, Yaacov B,
Francescatto L, Edvardson S, Rein AJ, Elpeleg O and Katsanis N: A
human laterality disorder caused by a homozygous deleterious
mutation in MMP21. J Med Genet. 52:840–847. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Paff T, Loges NT, Aprea I, Wu K, Bakey Z,
Haarman EG, Daniels JMA, Sistermans EA, Bogunovic N, Dougherty GW,
et al: Mutations in PIH1D3 cause X-linked primary ciliary
dyskinesia with outer and inner dynein arm defects. Am J Hum Genet.
100:160–168. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Loges NT, Antony D, Maver A, Deardorff MA,
Güleç EY, Gezdirici A, Nöthe-Menchen T, Höben IM, Jelten L, Frank
D, et al: Recessive DNAH9 loss-of-function mutations cause
laterality defects and subtle respiratory ciliary-beating defects.
Am J Hum Genet. 103:995–1008. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wallmeier J, Shiratori H, Dougherty GW,
Edelbusch C, Hjeij R, Loges NT, Menchen T, Olbrich H, Pennekamp P,
Raidt J, et al: TTC25 deficiency results in defects of the outer
dynein arm docking machinery and primary ciliary dyskinesia with
left-right body asymmetry randomization. Am J Hum Genet.
99:460–469. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Imtiaz F, Allam R, Ramzan K and Al-Sayed
M: Variation in DNAH1 may contribute to primary ciliary dyskinesia.
BMC Med Genet. 16(14)2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Onoufriadis A, Paff T, Antony D, Shoemark
A, Micha D, Kuyt B, Schmidts M, Petridi S, Dankert-Roelse JE,
Haarman EG, et al: Splice-site mutations in the axonemal outer
dynein arm docking complex gene CCDC114 cause primary ciliary
dyskinesia. Am J Hum Genet. 92:88–98. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chen X, Deng S, Xu H, Hou D, Hu P, Yang Y,
Wen J, Deng H and Yuan L: Novel and recurring NOTCH3 mutations in
two Chinese patients with CADASIL. Neurodegener Dis. 19:35–42.
2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li H: Aligning sequence reads, clone
sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2
(q-bio.GN). Oxford University Press, 2013.
|
|
26
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The Genome Analysis Toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–11.10.33. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Cingolani P, Platts A, Wang le L, Coon M,
Nguyen T, Wang L, Land SJ, Lu X and Ruden DM: A program for
annotating and predicting the effects of single nucleotide
polymorphisms, SnpEff: SNPs in the genome of Drosophila
melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6:80–92.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
1000 Genomes Project Consortium. Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA, et al: A global reference for human genetic
variation. Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
NHLBI Exome Sequencing Project (ESP):
Exome Variant Server. http://evs.gs.washington.edu/EVS/.
Accessed 31 January, 2018.
|
|
31
|
Chen X, Yuan L, Xu H, Hu P, Yang Y, Guo Y,
Guo Z and Deng H: Novel GLI3 mutations in Chinese patients with
non-syndromic post-axial polydactyly. Curr Mol Med. 19:228–235.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Mi H, Muruganujan A, Ebert D, Huang X and
Thomas PD: PANTHER version 14: More genomes, a new PANTHER GO-slim
and improvements in enrichment analysis tools. Nucleic Acids Res.
47:D419–D426. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Choi Y and Chan AP: PROVEAN web server: A
tool to predict the functional effect of amino acid substitutions
and indels. Bioinformatics. 31:2745–2747. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xiao H, Yuan L, Xu H, Yang Z, Huang F,
Song Z, Yang Y, Zeng C and Deng H: Novel and recurring
disease-causing NF1 variants in two Chinese families with
neurofibromatosis type 1. J Mol Neurosci. 65:557–563.
2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Sayers EW, Beck J, Brister JR, Bolton EE,
Canese K, Comeau DC, Funk K, Ketter A, Kim S, Kimchi A, et al:
Database resources of the National Center for Biotechnology
Information. Nucleic Acids Res. 48:D9–D16. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Chen Q, Yuan L, Deng X, Yang Z, Zhang S,
Deng S, Lu H and Deng H: A missense variant p.Ala117Ser in the
transthyretin gene of a Han Chinese family with familial amyloid
polyneuropathy. Mol Neurobiol. 55:4911–4917. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Burland TG: DNASTAR's Lasergene sequence
analysis software. Methods Mol Biol. 132:71–91. 2000.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Xiang Q, Cao Y, Xu H, Guo Y, Yang Z, Xu L,
Yuan L and Deng H: Identification of novel pathogenic ABCA4
variants in a Han Chinese family with Stargardt disease. Biosci
Rep. 39(BSR20180872)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Xiao H, Guo Y, Yi J, Xia H, Xu H, Yuan L,
Hu P, Yang Z, He Z, Lu H and Deng H: Identification of a novel
keratin 9 missense mutation in a Chinese family with epidermolytic
palmoplantar keratoderma. Cell Physiol Biochem. 46:1919–1929.
2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Norris DP: Cilia, calcium and the basis of
left-right asymmetry. BMC Biol. 10(102)2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Huang S, Xu W, Su B and Luo L: Distinct
mechanisms determine organ left-right asymmetry patterning in an
uncoupled way. Bioessays. 36:293–304. 2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Mercola M: Left-right asymmetry: Nodal
points. J Cell Sci. 116:3251–3257. 2003.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Reiter JF and Leroux MR: Genes and
molecular pathways underpinning ciliopathies. Nat Rev Mol Cell
Biol. 18:533–547. 2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ishikawa T: Axoneme structure from motile
cilia. Cold Spring Harb Perspect Biol. 9(a028076)2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Fliegauf M, Benzing T and Omran H: When
cilia go bad: Cilia defects and ciliopathies. Nat Rev Mol Cell
Biol. 8:880–893. 2007.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Mitchison HM and Valente EM: Motile and
non-motile cilia in human pathology: From function to phenotypes. J
Pathol. 241:294–309. 2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Hjeij R, Onoufriadis A, Watson CM, Slagle
CE, Klena NT, Dougherty GW, Kurkowiak M, Loges NT, Diggle CP,
Morante NF, et al: CCDC151 mutations cause primary ciliary
dyskinesia by disruption of the outer dynein arm docking complex
formation. Am J Hum Genet. 95:257–274. 2014.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ibañez-Tallon I, Heintz N and Omran H: To
beat or not to beat: Roles of cilia in development and disease. Hum
Mol Genet. 12:R27–R35. 2003.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Eley L, Yates LM and Goodship JA: Cilia
and disease. Curr Opin Genet Dev. 15:308–314. 2005.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Li P, He Y, Cai G, Xiao F, Yang J, Li Q
and Chen X: CCDC114 is mutated in patient with a complex phenotype
combining primary ciliary dyskinesia, sensorineural deafness, and
renal disease. J Hum Genet. 64:39–48. 2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Chen H, Huang X, Yuan L, Xia H, Xu H, Yang
Y, Zheng W and Deng H: A homozygous parkin p.G284R mutation in a
Chinese family with autosomal recessive juvenile parkinsonism.
Neurosci Lett. 624:100–104. 2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Wu DH and Singaraja RR: Loss-of-function
mutations in CCDC114 cause primary ciliary dyskinesia. Clin Genet.
83:526–527. 2013.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Knowles MR, Leigh MW, Ostrowski LE, Huang
L, Carson JL, Hazucha MJ, Yin W, Berg JS, Davis SD, Dell SD, et al:
Exome sequencing identifies mutations in CCDC114 as a cause of
primary ciliary dyskinesia. Am J Hum Genet. 92:99–106.
2013.PubMed/NCBI View Article : Google Scholar
|