Introduction
Ischemic heart disease is a leading cause of
mortality worldwide (1).
Furthermore, acute myocardial infarction (AMI) is the most severe
type of ischemic heart disease and has a high mortality rate
(2). Timely reperfusion remains
critical for the treatment of AMI; however, reperfusion may
exacerbate metabolic dysfunction and structural damage to the
myocardium, which is known as myocardial ischemia/reperfusion (I/R)
injury (3). Myocardial I/R is
characterized by endothelial dysfunction, cellular calcium
overload, oxidative stress, inflammatory response and myocyte
death, which all amplify tissue injury (4). Therefore, alleviating I/R injury during
myocardial reperfusion represents a crucial clinical challenge.
The ubiquitously expressed Rho-kinase, which is a
serine/threonine kinase, has been reported to serve an important
role in a number of major cardiovascular diseases, such as
hypertension, heart failure, pulmonary hypertension,
atherosclerosis, myocardial infarction and myocardial I/R (5-11).
Previous studies have demonstrated that Rho-kinase activation is
involved in the pathogenesis of I/R in vivo (12-16).
It has also been shown that fasudil, a Rho-kinase inhibitor,
inhibits the activation of Rho-kinase during I/R and reduces
infarct size and myocyte apoptosis in rats (17,18).
Moreover, there has been increasing research into Rho-kinase as a
potential therapeutic target in myocardial I/R.
Statins, which are competitive inhibitors of
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, are used
for treatment of dyslipidemias and prevention of cardiovascular
diseases (19), as they decrease
cholesterol biosynthesis, reduce low-density lipoprotein (LDL)
cholesterol and triglyceride levels, and increase levels of
high-density lipoprotein cholesterol (20). Previous studies have also reported
that statins may exert cardiovascular protective effects and may
inhibit Rho-kinase activity independent of LDL reduction, which are
referred to as pleiotropic effects (21-25).
However, the effect of statins on Rho-kinase activity in heart I/R
injury in vivo requires further investigation. Therefore, it
was hypothesized that cardioprotection of statins may be associated
with inhibition of Rho-kinase in myocardial I/R injury.
Materials and methods
Animals
Female Wistar rats (age, 130 days; weight, 250-300
g) were obtained from Shandong University and acclimatized for 1
week prior to any experimentation. Rats were allowed free access to
water and standard chow diet. All animals were housed under the
following conditions: Standard lighting at 12:12 h light-dark
cycle, temperature, 22±0.5˚C and humidity, 60±10%. The present
study was approved by the Ethics Committees of the Second Hospital
of Shandong University, and all procedures were performed in
accordance with the Institutional Animal Care and Use Committee and
National Institutes of Health Guidelines.
Randomized assignment and treatment
groups
A total of 60 rats were randomly and equally
allocated into 5 groups (n=12/group): i) Sham group; ii) I/R group;
iii) I/R + atorvastatin group; iv) I/R + fasudil group; and v) I/R
+ atorvastatin + fasudil group. The sham group was used as a
control and received a standard diet and water prior to the sham
operation. The I/R group was used as a control and received a
standard diet and water before I/R. The I/R + atorvastatin group
was orally administered atorvastatin (10 mg/kg body weight daily)
for 2 weeks prior to I/R. The I/R + fasudil group was administered
intravenously fasudil (10 mg/kg body weight daily) for 2 weeks
prior to I/R. The I/R + atorvastatin + fasudil group was
administered the same doses of atorvastatin and fasudil prior to
I/R.
In vivo heart I/R injury model
Rats were anesthetized with sodium pentobarbital (50
mg/kg, intraperitoneally) and subjected to 30 min left anterior
descending coronary artery (LAD) ligation using a 4-0 silk suture,
followed by a 180 min reperfusion according to our previous
published protocol (13). The sham
control animals were subjected to the entire surgical procedure and
silk suture was passed beneath the coronary artery, but the LAD
coronary artery was not ligated. At the end of reperfusion, rats
were exposed to overdoses of isoflurane for ~10 min and
euthanatized by exsanguination.
Biochemical testing
Following reperfusion, arterial blood samples were
collected and placed in heparinized centrifuge tubes, and were
centrifuged at 1,500 x g for 20 min at 4˚C. The supernatant was
collected and stored at -80˚C. Serum IL-6 and TNF-α activities were
measured using Rat IL-6 and TNF-α ELISA kits (Bio-Swamp, Inc.; cat.
no. RA20607; cat. no. RA20035, respectively), according to the
manufacturer's protocols. Total nitric oxide (NO) production was
determined by measuring the concentration of nitrate and nitrite, a
stable metabolite of NO, by the modified Griess reaction method;
the procedure involved the use of the Total NO kit (Beyotime
Institute of Biotechnology), according to the manufacturer's
protocols. Each sample supernatant (100 µl) was reacted with
Nitrate Reductase for 30 min and Griess reagent for 10 min at room
temperature in the dark. Nitrite levels were determined by
measuring absorbance at 540 nm using an OPTImax multiplate reader.
The levels of nitrite were normalized to standard values.
Determination of myocardial infarct
size
At the end of reperfusion, the coronary artery was
re-occluded and Evans blue dye solution (3 ml, 2% wt/vol) was
injected into the left ventricle to determine between ischemic
[area at risk (AAR), unstained] and non-ischemic myocardium (area
not at risk, stained blue). The hearts were subsequently harvested,
rinsed in normal saline and the atria, right ventricle and great
vessels were removed. The heart was excised and cut into transverse
slices (thickness, 1 mm). The AAR was separated from the area not
at risk and subsequently incubated with nitro-blue tetrazolium
(NBT, 1% wt/vol, 15 min at 37˚C) to distinguish between ischemic
(stained blue) and infarcted tissue (unstained). Then, the AAR and
infarct size were calculated after weighing the respective tissue
samples, and infarct size was expressed as the percentage of the
AAR.
Evaluation of apoptosis in heart
tissue sections
TUNEL was used to evaluate apoptotic activity
following I/R injury. Heart sections were fixed in 4%
paraformaldehyde for 24 h at room temperature and embedded in
paraffin. Each section (thickness, 6 µm) was deparaffinized with
xylene and rehydrated with serial changes ethanol (100, 95, 90, 80,
70, 60 and 50%). TUNEL staining was performed using an in
situ Cell Death Detection kit (Roche Diagnostics) according to
the manufacturer's protocol. The TdT reaction was carried out for 1
h at 37˚C in a humidified chamber, and then DAB
chromogen (OriGene Technologies, Inc; cat. no. ZLI-9019) was
applied. Hematoxylin was used as a counterstain. The results were
viewed using a confocal FV 1000 SPD Laser Scanning microscope
(Olympus Corporation; magnification, x400). TUNEL-positive cells
were determined by randomly selecting 10 fields of view and were
expressed as a percentage of normal nuclei.
Determination of superoxide dismutase
(SOD) and malondialdehyde (MDA)
A total of 20 mg myocardial tissue was minced and
homogenized in ice-cold RIPA buffer (Sigma-Aldrich; Merck KGaA).
Homogenates were subject to centrifugation at 13,000 x g for 15 min
at 4˚C to obtain the supernatant as sample tissue for total protein
preparation. The protein concentration was determined by
bicinchoninic acid (BCA) assay kit (Beyotime Institute of
Biotechnology). Myocardial tissue SOD activity and MDA content were
assayed, according to the manufacturer's protocols (Jiancheng
Biotech Ltd.; cat.no. A001-3-2; cat. no. A003-1-2). The results of
SOD and MDA assays were expressed as units per mg of protein.
Western blot analysis for measurement
of Rho-kinase activity and cleaved Caspase-3
Rho-kinase activity was assessed by examining the
phosphorylation state of myosin phosphatase targeting subunit 1
(MYPT-1), a well-established Rho-kinase specific substrate
(26). Western blotting was
performed on heart tissue obtained from the AAR. Tissue proteins
were extracted using the Membrane and Cytosol Protein Extraction
kit (Beyotime Institute of Biotechnology; cat. no. P0033) and
protein concentration was determined using the BCA assay kit
(Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. After protein quantification, equal
amounts of protein (50 µg) were separated on 10% Tris-glycine SDS
gel by electrophoresis and subsequently transferred to PVDF
membranes. After blocking with 5% BSA (Beyotime Institute of
Biotechnology) in Tris-buffer for 1 h at room temperature,
membranes were incubated overnight at 4˚C with the
primary antibodies: β-actin (1:1,000; Santa Cruz Biotechnology,
Inc.; cat. no. sc-69879), rabbit polyclonal MYPT-1 antibody (1:500;
Bioworld Technology, Inc.; cat. no. BS8367), rabbit polyclonal
phosphorylated (p)-MYPT-1 antibody (1:500; Bioworld Technology,
Inc.; cat. no. BS64148) and rabbit monoclonal cleaved caspase-3
antibody (1:1,000; Cell Signaling Technology, Inc.; cat. no. 9664).
Then, membranes were incubated at room temperature for 2 h with
horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (1:10,000; Santa Cruz Biotechnology, Inc.; cat. no.
sc-2004). Immunoreactive bands were visualized with the SuperSignal
West Pico enhanced chemoluminescence kit (Piece; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. Band
intensities were quantified using a densitometer analysis system
Quantity One 4.6.6 (Bio-Rad Laboratories, Inc.).
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical analysis of the results was carried out via one-way
ANOVA followed by the Tukey's post-hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Statistical analyses were performed using SPSS 19.0 (SPSS,
Inc.).
Results
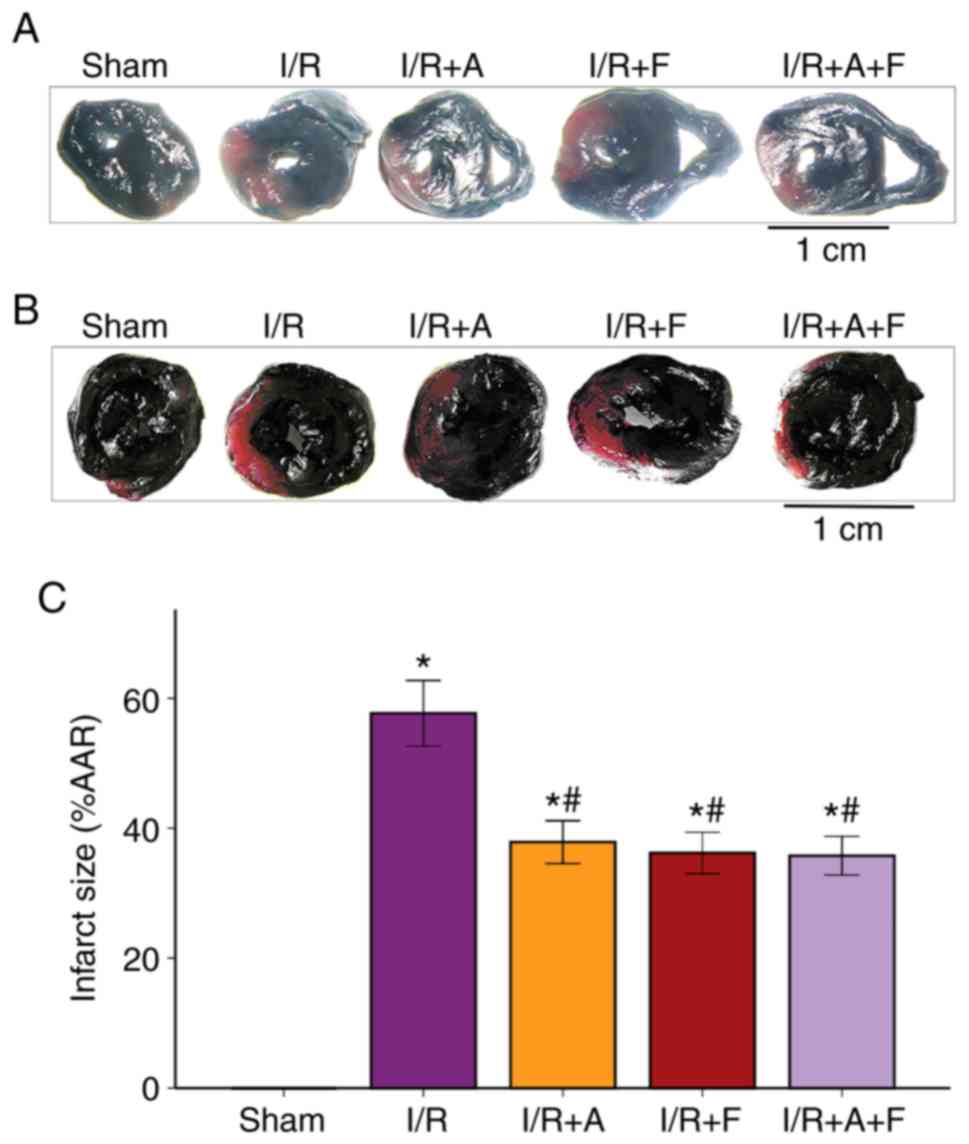
Atorvastatin and fasudil reduce
myocardial infarct size in heart I/R
It was identified that the average infarct size of
the heart was 57.68±3.95% in the I/R group. Furthermore,
administration of atorvastatin or fasudil caused a significant
reduction in infarct size; the infarct size of the heart was
37.87±5.19 and 36.21±5.01% in the I/R + atorvastatin group and I/R
+ fasudil group, respectively. Moreover, it was found that there
were no significant differences between the I/R + atorvastatin
group, I/R + fasudil group and I/R + atorvastatin + fasudil group
(Fig. 1). Therefore, the present
results suggested that atorvastatin and fasudil reduced myocardial
infarct size in rat heart I/R injury.
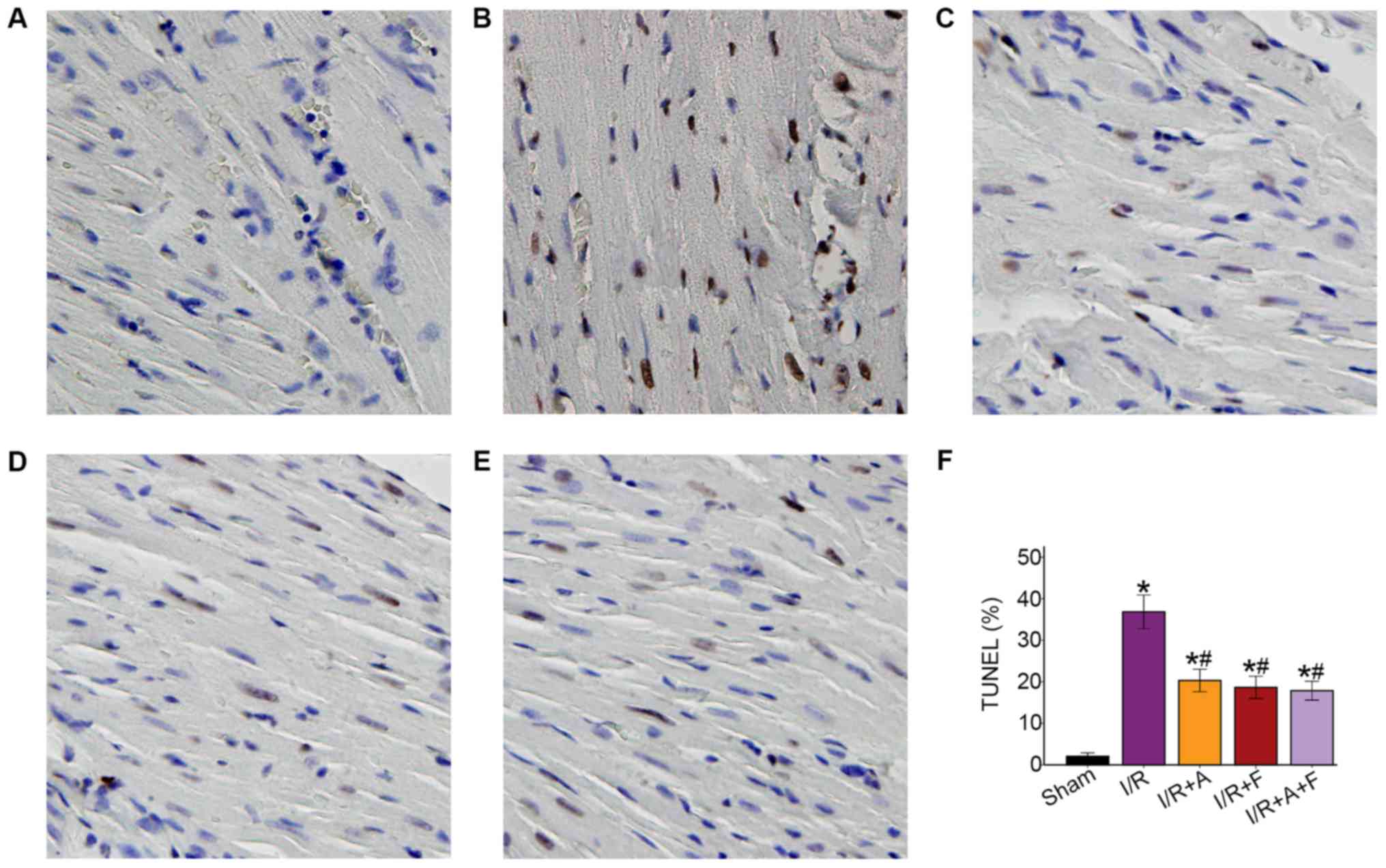
Atorvastatin and fasudil reduce cell
apoptosis of the heart in I/R
Following examination of microscopy images, it was
indicated that apoptotic cell nuclei were stained dark brown, while
healthy myocardial cell nuclei were stained blue. The apoptotic
rate was 2.04±1.34% in the sham group, while the number of
TUNEL-positive cells was significantly increased in I/R group
(36.83±6.39%). In addition, it was demonstrated that the apoptotic
rates were significantly reduced in the I/R + atorvastatin, I/R +
fasudil and I/R + atorvastatin + fasudil groups (P<0.05 vs. I/R
group); however, there were no significant differences among these
three groups (Fig. 2).
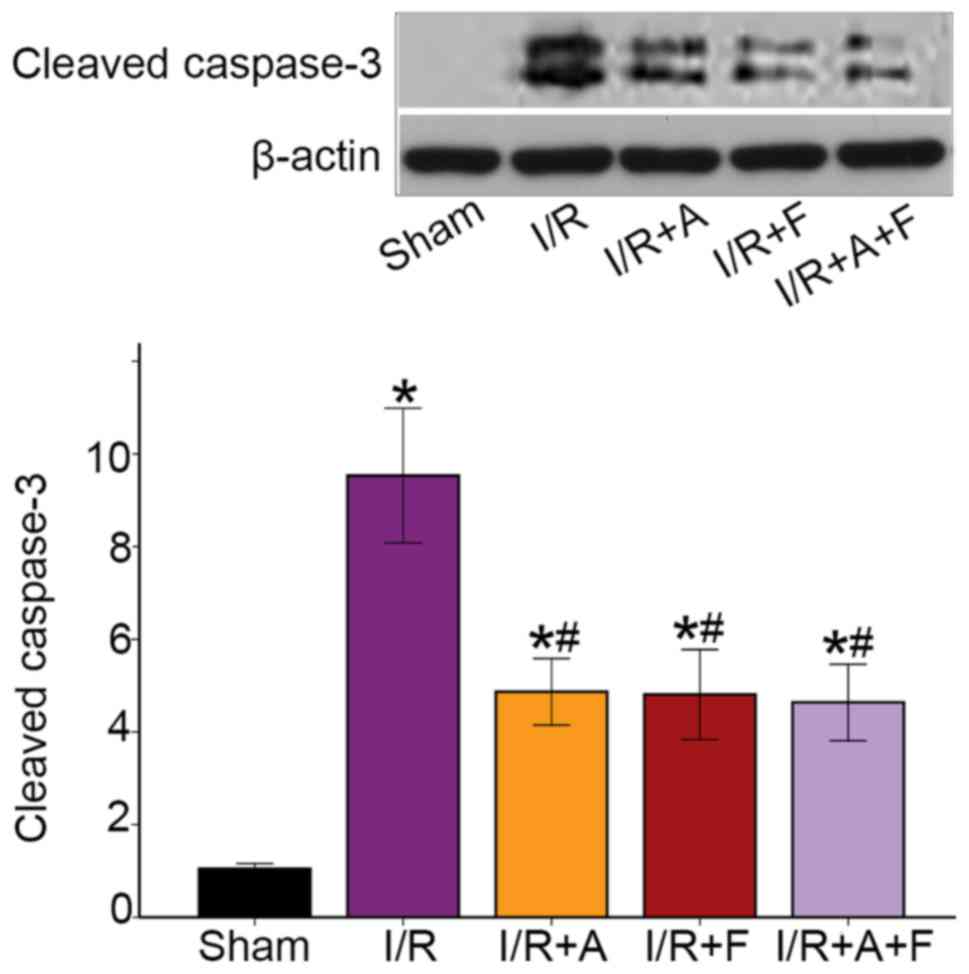
Activation of caspase-3 is a hallmark of apoptotic
cell death, and caspase-3 cleavage is indicative of its activation
(27). The western blotting results
identified that I/R caused a significant increase in the protein
expression of cleaved capsase-3. Moreover, it was found that
capsase-3 activity was attenuated in the I/R + atorvastatin, I/R +
fasudil and I/R + atorvastatin + fasudil groups (Fig. 3). Thus, the results suggested that
atorvastatin and fasudil reduced cell apoptosis in rat heart I/R
injury.
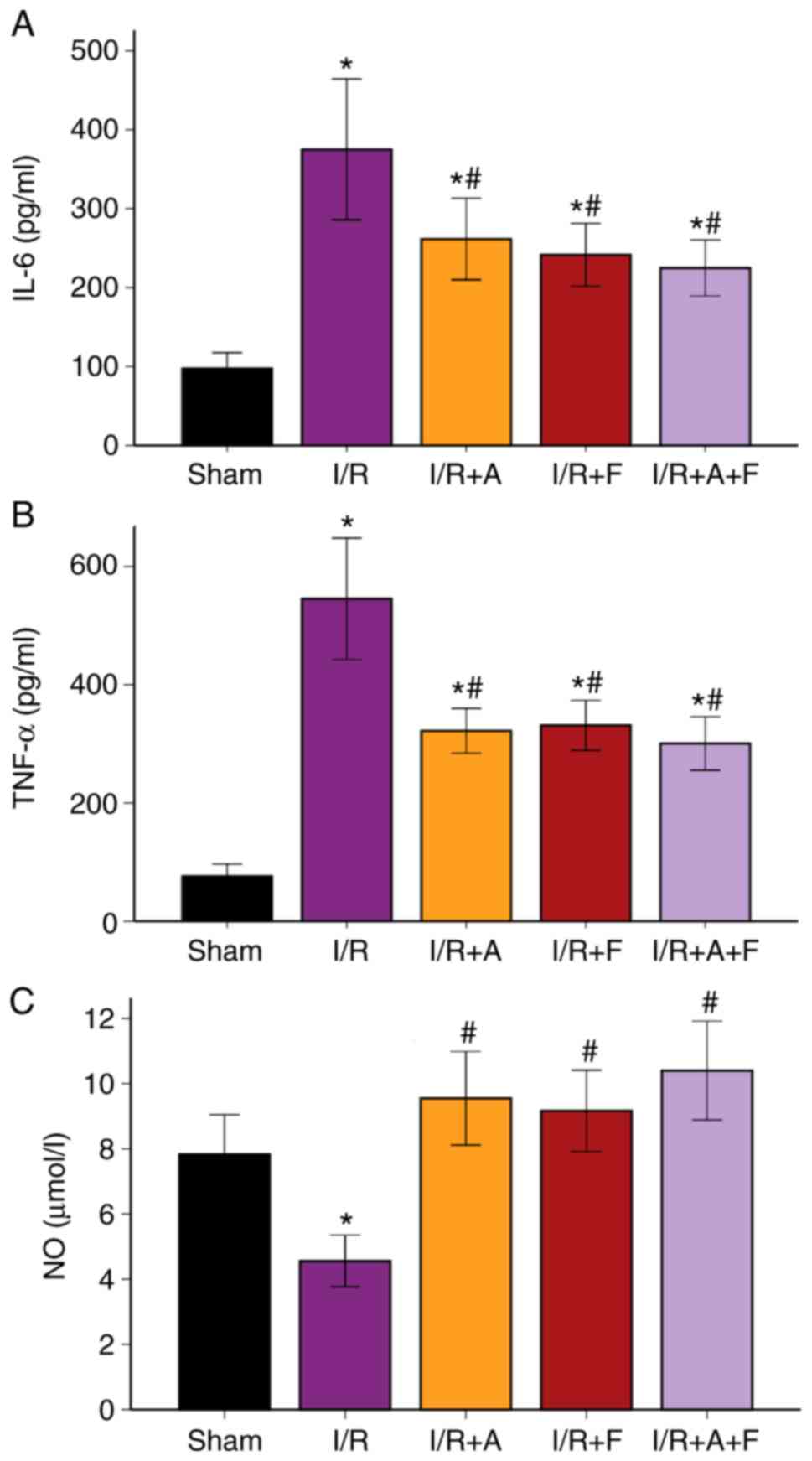
Expression levels of plasma IL-6,
TNF-α and NO in the different groups
Compared with the sham group, the serum
concentrations of IL-6 and TNF-α were significantly elevated in the
I/R group (P<0.05). Furthermore, these increases in IL-6 and
TNF-α levels were significantly suppressed in the I/R +
atorvastatin, I/R + fasudil and I/R + atorvastatin + fasudil groups
(P<0.05 vs. I/R group); however, there were no significant
differences among these three groups (Fig. 4A and B; Table I).
It was also found that NO production in the I/R group was
significantly decreased compared with the sham group (P<0.05).
Moreover, when treated with atorvastatin, fasudil or their
combination, NO production increased compared with the I/R group
(P<0.05), but there were no significant differences among these
three groups (Fig. 4C; Table I).
 | Table ISerum levels of IL-6, TNF-α and
NO. |
Table I
Serum levels of IL-6, TNF-α and
NO.
| Variables | Sham | I/R | I/R + A | I/R + F | I/R + A + F |
|---|
| IL-6, pg/ml | 97.5±31.36 |
375.1±140.39a |
261.5±81.28a,b |
241.5±62.61a,b |
224.9±55.77a,b |
| TNF-α, pg/ml | 76.2±32.55 |
545.5±161.27a |
322.1±59.68a,b |
331.3±66.54a,b |
300.875±71.42a,b |
| NO, µmol/l | 7.83±1.91 |
4.56±1.25a |
9.55±2.26b |
9.17±1.96b |
10.40±2.39b |
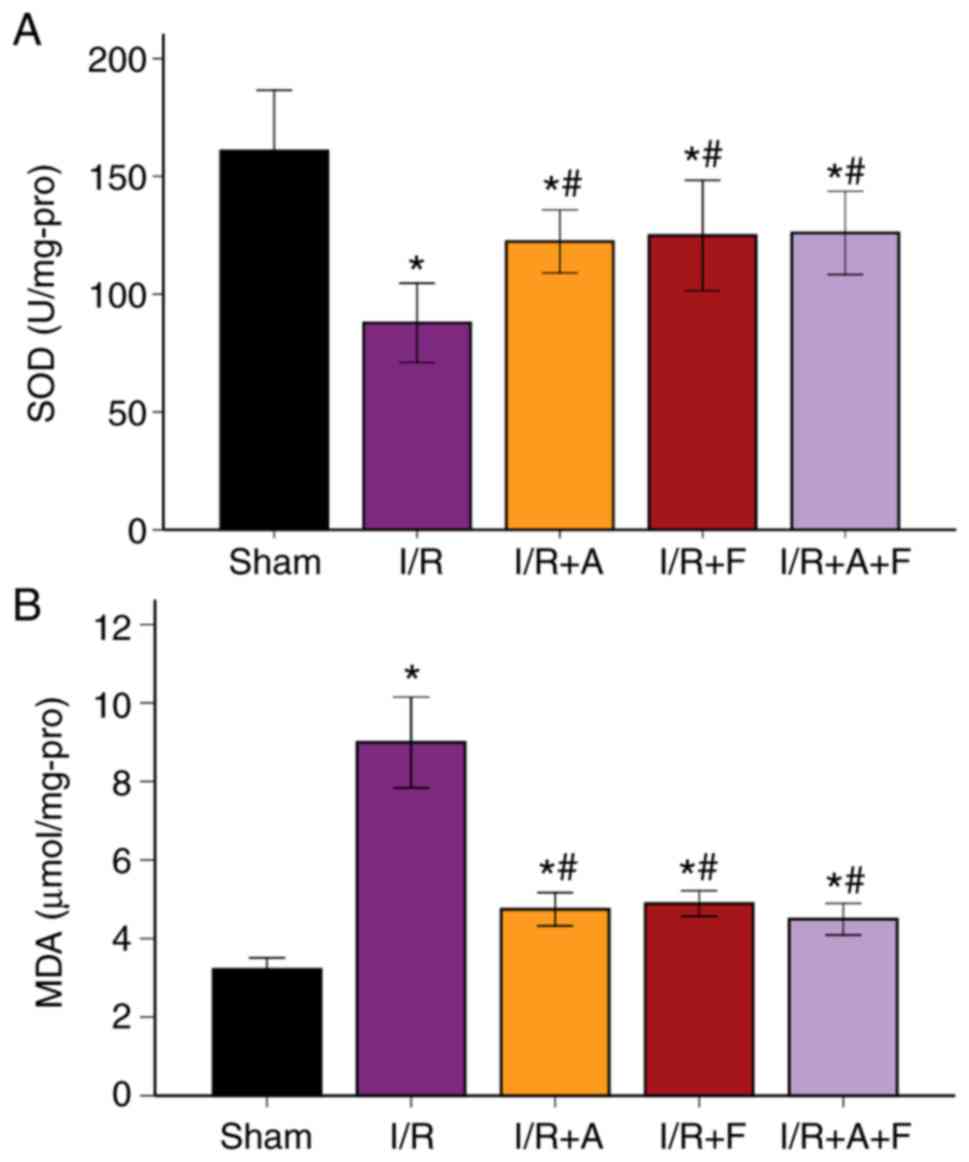
SOD activity and MDA levels in the
different groups
It was indicated that MDA level was increased
following I/R injury, while SOD decreased significantly (P<0.05
vs. sham group). In addition, when treated with atorvastatin,
fasudil or their combination, the level of MDA was significantly
suppressed, while SOD activity significantly increased (P<0.05
vs. I/R group; Fig. 5A and B;
Table II). Therefore, the results
suggested that atorvastatin and fasudil antagonized oxidative
stress induced by I/R injury.
| Table IISOD activity and MDA levels. |
Table II
SOD activity and MDA levels.
| Variables | Sham | I/R | I/R + A | I/R + F | I/R + A + F |
|---|
| SOD, U/mg
protein | 160.8±40.63 |
87.8±26.44a |
122.3±21.08a,b |
124.9±36.96a,b |
126.0±27.81a,b |
| MDA, µmol/mg
protein | 3.21±0.46 |
8.99±1.82a |
4.74±0.67a,b |
4.89±0.51a,b |
4.49±0.64a,b |
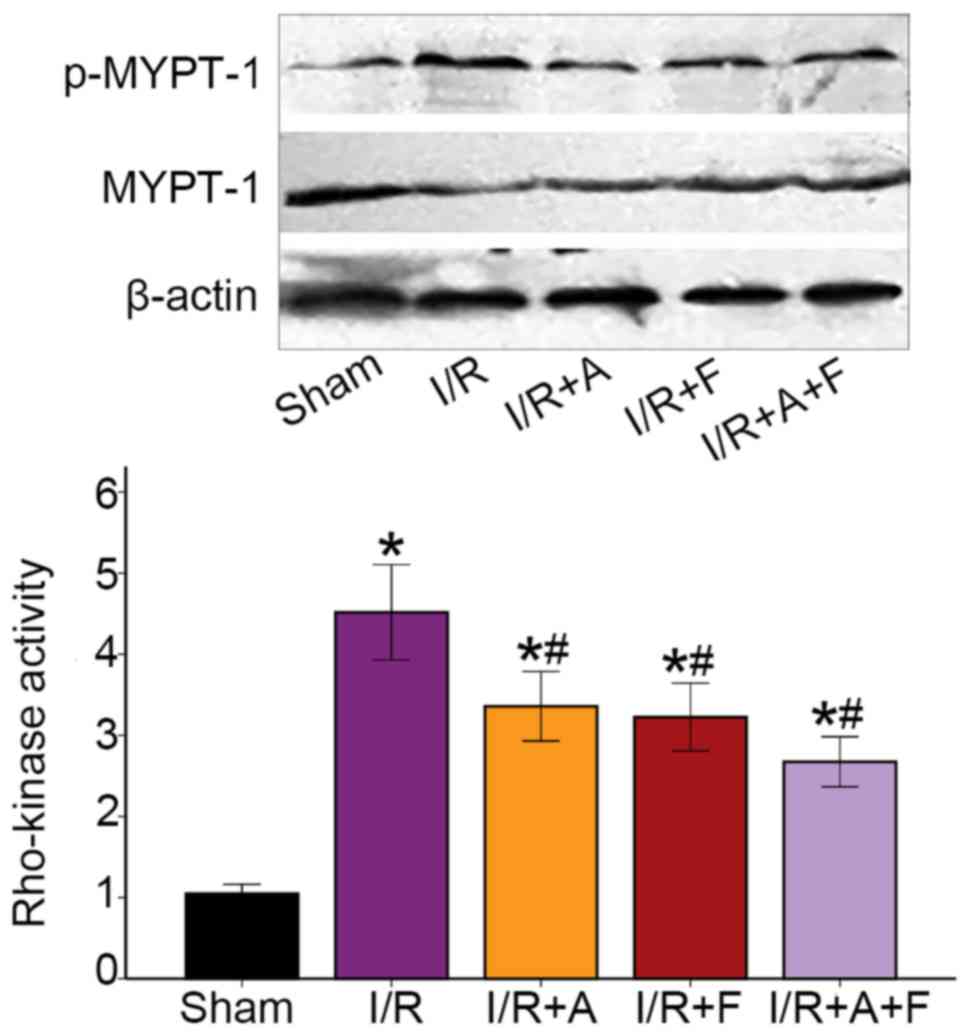
Rho-kinase activity in the different
groups
Rho-kinase activity was assessed by examining the
phosphorylation of MYPT-1 using western blot analysis and it was
found that the phosphorylation of MYPT-1 was significantly
increased during I/R protocol (P<0.05 vs. the sham group).
Furthermore, atorvastatin, fasudil or their combination therapy
resulted in significant reduction in p-MYPT-1/MYPT-1 ratio
(P<0.05 vs. I/R group; Fig. 6).
Collectively, the results indicated that atorvastatin, similar to
fasudil, inhibited Rho-kinase activity in heart I/R injury.
Discussion
In the present study, the effect of atorvastatin and
fasudil was examined in heart I/R injury in the rat models. The
major findings of the present in vivo study were as follows:
i) Both fasudil and atorvastatin significantly attenuated
myocardial I/R injury in rat models, including myocardial infarct
size, cardiomyocyte apoptosis, oxidative stress and inflammatory
response; and ii) Rho-kinase inhibition was involved in the
cardiovascular protective effects of atorvastatin in myocardial
I/R.
Statins have been used to prevent coronary artery
disease and stroke by lowering serum LDL cholesterol and inhibiting
hepatic cholesterol biosynthesis (19). Previous studies have shown that
statins have antiproliferative, antithrombotic, anti-inflammatory
and anti-atherogenic effects, as well as their LDL
cholesterol-lowering effects (24,28-30).
Moreover, it has been revealed that atorvastatin provides
cardioprotective effects against heart I/R injury via reducing
myocardial infarct size and cardiomyocyte apoptosis (31-33).
The average plasma half-life of atorvastatin is ~14 h, but the
actual half-life of inhibition of HMG-COA reductase is 20-30 h due
to the influence of its active metabolites (34). In the present study, the drugs were
administered 2 weeks prior to surgery and atorvastatin was selected
as the statin test agent. The present results indicated that
pretreatment with atorvastatin attenuated myocardial infarction and
myocardial apoptosis. Furthermore, it was found that atorvastatin
resulted in a 44.8% reduction in apoptotic cardiomyocytes and a
34.3% reduction in myocardial infarct size, thus suggesting the
cardiac protection provided by atorvastatin against heart I/R
injury.
Oxidative stress, endothelial dysfunction and
inflammation are among the most common mechanisms of heart I/R
injury (35,36). It has been shown that statins
increase endothelial NO production, which is impaired by I/R
(33). Furthermore, previous studies
have reported that low-dose atorvastatin increases
anti-inflammatory activity and increases NO concentration against
I/R injury in isolated hearts of rats (37). The present results indicated that I/R
increased levels of IL-6, TNF-α and MDA, while it decreased SOD and
NO production. It was also observed that preventively administered
atorvastatin attenuated the levels of IL-6, TNF-α and MDA, and
upregulated SOD activity and NO production. Therefore, these
results indicate that atorvastatin may attenuate I/R heart injury,
which may be mediated by reducing oxidative and inflammatory
responses, and activating the NO pathway.
The Rho-kinase pathway, which serves an important
role in a number of cardiovascular diseases, has been shown to be
involved in heart I/R injury (5).
In vitro studies reported that increased Rho-kinase activity
downregulates NO production and that Rho-kinase inhibitors increase
NO production (38,39). It has also been revealed that
atorvastatin prevents pulmonary vascular remodeling by inhibiting
RhoA/Rho-kinase (40). Moreover,
atorvastatin may upregulate NO levels via Rho-kinase signaling
(41). Similar to our previous
studies showing that the activation of Rho-kinase can be
significantly upregulated by heart I/R injury (13,42), the
present results suggested that myocardial I/R caused a significant
increase in Rho-kinase activity. Furthermore, Rho-kinase activity
was assessed by examining the phosphorylation of MYPT-1 in the
present study. It was found that I/R heart injury resulted in a
4.5-fold increase in p-MYPT1 expression, thus indicating the
activation of Rho-kinase following I/R injury. As fasudil is a
specific and potent antagonist for Rho-kinase, it was identified
that Rho-kinase activity was significantly decreased when treated
with fasudil prior to I/R. The present study also examined whether
Rho-kinase inhibition was involved in the cardiac protection of
atorvastatin. A significant decrease in Rho-kinase activity was
observed as a result of administration of atorvastatin and the
combination of fasudil + atorvastatin. Therefore, it was speculated
that inhibition of Rho-kinase may be involved in the
cardioprotective effect of atorvastatin in heart I/R injury.
However, there are several limitations that require
mentioning for the present study. Firstly, hemodynamic parameters
were not determined and thus will be considered in future studies.
In addition, it was found that atorvastatin lowered plasma cytokine
levels and inflammation, but the results lack inflammatory
infiltrate in myocardial tissues. Moreover, the underlying
mechanism of Rho-kinase inhibition via atorvastatin in I/R injury
remains to be fully elucidated. Future studies are required to
assess whether atorvastatin inhibits Rho-kinase activity in a dose-
and time-dependent manner. Furthermore, the present study only
assessed the effect of atorvastatin on Rho-kinase inhibition by
evaluating MYPT-1 phosphorylation, and therefore additional
effectors of Rho-kinase are to be considered in future experiments.
The aim of this study was also to identify the possible protective
effects of atorvastatin, as continuous oral administration of
atorvastatin could be beneficial for patients with acute myocardial
infarction/percutaneous coronary intervention. Therefore, clinical
trials registration will be considered in future research.
In conclusion, the present results indicated that
atorvastatin may have a cardiovascular protective effect against
I/R-induced injury, including inhibition of Rho-kinase activity.
Thus, these findings may provide a novel approach for the process
of statin administration and may provide new therapeutic strategies
for myocardial I/R injury.
Acknowledgements
Not applicable.
Funding
The authors gratefully acknowledge research support
provided by Youth Foundation of the National Natural Science
Foundation of China (grant no. 81600284) and Shandong Key Research
and Development Project (grant no. 2016GSF201196).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ designed the experiments and drafted the
manuscript. CC and XL mainly performed the experiments and analyzed
the data. SB and QL performed some of the experiments. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committees of the Second Hospital of Shandong University (Jinan,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Laflamme MA and Murry CE: Heart
regeneration. Nature. 473:326–335. 2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Szummer K, Jernberg T and Wallentin L:
From early pharmacology to recent pharmacology interventions in
acute coronary syndromes: JACC State-of-the-Art review. J Am Coll
Cardiol. 74:1618–1636. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Fan ZX and Yang J: The role of microRNAs
in regulating myocardial ischemia reperfusion injury. Saudi Med J.
36:787–793. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Shimokawa H, Sunamura S and Satoh K:
RhoA/Rho-kinase in the cardiovascular system. Circ Res.
118:352–366. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhou Q and Liao JK: Rho kinase: An
important mediator of atherosclerosis and vascular disease. Curr
Pharm Des. 15:3108–3115. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang J, Xu DL, Liu XB, Bi SJ, Zhao T, Sui
SJ, Ji XP and Lu QH: Darapladib, a lipoprotein-associated
phospholipase A2 inhibitor, reduces rho kinase activity in
atherosclerosis. Yonsei Med J. 57:321–327. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Patel P, Parikh M, Shah H and Gandhi T:
Inhibition of RhoA/Rho kinase by ibuprofen exerts cardioprotective
effect on isoproterenol induced myocardial infarction in rats. Eur
J Pharmacol. 791:91–98. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tanna AP and Johnson M: Rho Kinase
inhibitors as a novel treatment for glaucoma and ocular
hypertension. Ophthalmology. 125:1741–1756. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ocaranza MP, Moya J, Jalil JE, Lavandero
S, Kalergis AM, Molina C, Gabrielli L, Godoy I, Córdova S, Castro
P, et al: Rho-kinase pathway activation and apoptosis in
circulating leucocytes in patients with heart failure with reduced
ejection fraction. J Cell Mol Med. 24:1413–1427. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Fazakas C, Nagaraj C, Zabini D, Végh AG,
Marsh LM, Wilhelm I, Krizbai IA, Olschewski H, Olschewski A and
Bálint Z: Rho-kinase inhibition ameliorates Dasatinib-induced
endothelial dysfunction and pulmonary hypertension. Front Physiol.
9(537)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bao W, Hu E, Tao L, Boyce R, Mirabile R,
Thudium DT, Ma XL, Willette RN and Yue TL: Inhibition of Rho-kinase
protects the heart against ischemia/reperfusion injury. Cardiovasc
Res. 61:548–558. 2004.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhang J, Li XX, Bian HJ, Liu XB, Ji XP and
Zhang Y: Inhibition of the activity of Rho-kinase reduces
cardiomyocyte apoptosis in heart ischemia/reperfusion via
suppressing JNK-mediated AIF translocation. Clin Chim Acta.
401:76–80. 2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang J, Bian HJ, Li XX, Liu XB, Sun JP,
Li N, Zhang Y and Ji XP: ERK-MAPK signaling opposes rho-kinase to
reduce cardiomyocyte apoptosis in heart ischemic preconditioning.
Mol Med. 16:307–315. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Li Y, Zhu W, Tao J, Xin P, Liu M, Li J and
Wei M: Fasudil protects the heart against ischemia-reperfusion
injury by attenuating endoplasmic reticulum stress and modulating
SERCA activity: The differential role for PI3K/Akt and JAK2/STAT3
signaling pathways. PLoS One. 7(e48115)2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Jiang ZH, Zhang TT and Zhang JF:
Protective effects of fasudil hydrochloride post-conditioning on
acute myocardial ischemia/reperfusion injury in rats. Cardiol J.
20:197–202. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang YS, Tang LJ, Tu H, Wang SJ, Liu B,
Zhang XJ, Li NS, Luo XJ and Peng J: Fasudil ameliorates the
ischemia/reperfusion oxidative injury in rat hearts through
suppression of myosin regulatory light chain/NADPH oxidase 2
pathway. Eur J Pharmacol. 822:1–2. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Huang YY, Wu JM, Su T, Zhang SY and Lin
XJ: Fasudil, a rho-kinase inhibitor, exerts cardioprotective
function in animal models of myocardial ischemia/reperfusion
injury: A meta-analysis and review of preclinical evidence and
Possible Mechanisms. Front Pharmacol. 9(1083)2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sirtori CR: The pharmacology of statins.
Pharmacol Res. 88:1–3. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Istvan ES and Deisenhofer J: Structural
mechanism for statin inhibition of HMG-CoA reductase. Science.
292:1160–1164. 2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Oesterle A, Laufs U and Liao JK:
Pleiotropic effects of statins on the cardiovascular system. Circ
Res. 120:229–243. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tramontano AF, O'Leary J, Black AD,
Muniyappa R, Cutaia MV and El-Sherif N: Statin decreases
endothelial microparticle release from human coronary artery
endothelial cells: Implication for the Rho-kinase pathway. Biochem
Biophys Res Commun. 320:34–38. 2004.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yamanouchi D, Banno H, Nakayama M,
Sugimoto M, Fujita H, Kobayashi M, Kuwano H and Komori K:
Hydrophilic statin suppresses vein graft intimal hyperplasia via
endothelial cell-tropic Rho-kinase inhibition. J Vasc Surg.
42:757–764. 2005.PubMed/NCBI View Article : Google Scholar
|
|
24
|
McNeish AJ, Jimenez-Altayo F, Cottrell GS
and Garland CJ: Statins and selective inhibition of Rho kinase
protect small conductance calcium-activated potassium channel
function (K(Ca)2.3) in cerebral arteries. PLoS One.
7(e46735)2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ma MM, Li SY, Wang M and Guan YY:
Simvastatin attenuated cerebrovascular cell proliferation in the
development of hypertension through Rho/Rho-kinase pathway. J
Cardiovasc Pharmacol. 59:576–582. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Narumiya S and Thumkeo D: Rho signaling
research: History, current status and future directions. FEBS Lett.
592:1763–1776. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Nagata S: Apoptosis and clearance of
apoptotic cells. Annu Rev Immunol. 36:489–517. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Li M, Liu Y, Dutt P, Fanburg BL and Toksoz
D: Inhibition of serotonin-induced mitogenesis, migration, and ERK
MAPK nuclear translocation in vascular smooth muscle cells by
atorvastatin. Am J Physiol Lung Cell Mol Physiol. 293:L463–L471.
2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ridker PM, Danielson E, Fonseca FA, Genest
J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ,
MacFadyen JG, et al: Rosuvastatin to prevent vascular events in men
and women with elevated C-reactive protein. N Engl J Med.
359:2195–2207. 2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Rawlings R, Nohria A, Liu PY, Donnelly J,
Creager MA, Ganz P, Selwyn A and Liao JK: Comparison of effects of
rosuvastatin (10 mg) versus atorvastatin (40 mg) on rho kinase
activity in caucasian men with a previous atherosclerotic event. Am
J Cardiol. 103:437–441. 2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Sun T, Zhang HJ, Krittanawong C, Wang S,
Tao Y, Li Z, Yin Q, Zhang D, Wang Q, Huang J, Zhang J, Li Z and
Cheng Y: Acute atorvastatin treatment restores the cardioprotective
effects of ischemic postconditioning in hyperlipidemic rats.
Oncotarget. 8:55187–55193. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
El Desoky ES, Hassan AKM, Salem SY, Fadil
SA and Taha AF: Cardioprotective effect of atorvastatin alone or in
combination with remote ischemic preconditioning on the biochemical
changes induced by ischemic/reperfusion injury in a mutual
prospective study with a clinical and experimental animal arm. Int
J Cardiol. 222:866–873. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Rohilla A, Ahmad A, Khan MU and Khanam R:
A comparative study on the cardioprotective potential of
atorvastatin and simvastatin in hyperhomocysteinemic rat hearts.
Eur J Pharmacol. 764:48–54. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ye YC, Zhao XL and Zhang SY: Use of
atorvastatin in lipid disorders and cardiovascular disease in
Chinese patients. Chin Med J. 128:259–266. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Sydow K and Munzel T: ADMA and oxidative
stress. Atheroscler Suppl. 4:41–51. 2003.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chatterjee A, Black SM and Catravas JD:
Endothelial nitric oxide (NO) and its pathophysiologic regulation.
Vascular Pharmacol. 49:134–140. 2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lunder M, Ziberna L, Janić M, Jerin A,
Skitek M, Sabovič M and Drevenšek G: Low-dose atorvastatin,
losartan, and particularly their combination, provide
cardiovascular protection in isolated rat heart and aorta. Heart
Vessels. 28:246–254. 2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Takemoto M, Sun J, Hiroki J, Shimokawa H
and Liao JK: Rho-kinase mediates hypoxia-induced downregulation of
endothelial nitric oxide synthase. Circulation. 106:57–62.
2002.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Rikitake Y, Kim HH, Huang Z, Seto M, Yano
K, Asano T, Moskowitz MA and Liao JK: Inhibition of Rho kinase
(ROCK) leads to increased cerebral blood flow and stroke
protection. Stroke. 36:2251–2257. 2005.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wang Q, Guo YZ, Zhang YT, Xue JJ, Chen ZC,
Cheng SY, Ou MD, Cheng KL and Zeng WJ: The Effects and mechanism of
atorvastatin on pulmonary hypertension due to left heart disease.
PLoS One. 11(e0157171)2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
An L, An S, Jia Z, Wang H, Yang Z, Xu C,
Teng X, Wang J, Liu X, Cao Q and Wang S: Atorvastatin improves left
ventricular remodeling and cardiac function in rats with congestive
heart failure by inhibiting RhoA/Rho kinase-mediated endothelial
nitric oxide synthase. Exp Ther Med. 17:960–966. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zhang J, Liu XB, Cheng C, Xu DL, Lu QH and
Ji XP: Rho-kinase inhibition is involved in the activation of
PI3-kinase/Akt during ischemic-preconditioning-induced
cardiomyocyte apoptosis. Int J Clin Exp Med. 7:4107–4114.
2014.PubMed/NCBI
|