Introduction
Sinusoidal obstruction syndrome (SOS) is a
progressive and potentially fatal complication of radiotherapy in
patients preparing for hematopoietic stem cell transplantation and
chemotherapy for liver metastasis of colorectal cancer (1-3).
Severe SOS has a high mortality rate. SOS is considered to be
related to radiation- or chemotherapy-induced damage to the liver
microvasculature (1,4). Toxic doses of monocrotaline (MCT), a
pyrrolizidine alkaloid present in plants of genus
Crotalaria, have been used to induce SOS in rats for use as
experimental models to study liver injury in vivo.
MCT-induced liver injury is characterized by the formation of a gap
at the surface of liver sinusoidal endothelial cells (LSECs), which
leads to sinusoidal hemorrhage and centrilobular hepatocellular
necrosis. Sinusoidal destruction is accompanied by infiltration of
the centrilobular regions by inflammatory cells (4,5). Once
the liver is injured, it must be repaired and regenerated. Indeed,
rats that survive have shown resolution of MCT-induced liver
inflammation (4,5). Furthermore, hepatic tissue repair
plays a critical role in determining the final outcome of
chemical-induced hepatotoxicity (6). However, the process of liver repair
during MCT hepatotoxicity and its underlying mechanism is largely
unknown.
Macrophages play a critical role in liver repair
following acute injury induced by chemicals and
ischemia/reperfusion (I/R) (7-11).
Recently, we showed that MCT-induced liver injury is associated
with accumulation of macrophages (12). Typically, accumulation of
macrophages at the site of injury promotes the recovery of damaged
tissues. However, it is unclear whether accumulated macrophages
play a role in liver repair after MCT-induced liver injury.
C-X-C chemokine receptor type 4 (CXCR4) is a
receptor for stromal cell-derived factor 1 (SDF-1) (13). CXCR4 signaling plays a crucial role
in the mobilization and recruitment of progenitor cells from the
bone marrow (BM), which stimulates angiogenesis and recovery of
ischemic tissue (13,14). In addition, SDF-1-CXCR4 signaling
contributes to tissue repair after acute limb ischemia (15) and acute gastric ulcers (16) in mice by recruiting pro-angiogenic
macrophages from the BM. Accumulating evidence suggests that
SDF-1-CXCR4 is involved in liver repair after acute injury
(13,17). CXCR4 blockade inhibits hepatocyte
proliferation in mice treated with acetaminophen, indicating that
CXCR4 signaling promotes liver regeneration (18). Recruitment of CXCR4-expressing
hematopoietic progenitors in response to SDF-1 is an important
mechanism underlying the repair of liver tissue (19); of note, these findings indicate that
the SDF-1-CXCR4 signaling pathway promotes tissue recovery from
ischemia- or chemical-induced injury via the accumulation of
pro-angiogenic macrophages. In addition, CXCR4 also plays a role in
the development of MCT-induced pulmonary arterial hypertension and
vascular remodeling (20).
Altogether, these above findings led us to investigate whether the
SDF-1-CXCR4 axis contributes to liver repair in the context of
MCT-induced liver injury through the recruitment of
macrophages.
Here, we investigated the role of macrophages in
liver repair after MCT-induced liver injury. Further, we examined
whether SDF-1-CXCR4 axis contributes to macrophage accumulation and
tissue repair in mice after MCT-induced hepatotoxicity.
Materials and methods
Animals
Male C57BL/6 WT mice (8-10-weeks-old) were purchased
from CLEA Japan. Transgenic mice expressing green fluorescent
protein (GFP) against a C57BL/6 background were kindly provided by
Dr Okabe (Genome Information Research Center, Osaka University,
Osaka, Japan). Mice were maintained on a 12 h light/dark cycle in a
facility with constant humidity (50%±5%) and temperature (25±1˚C),
and were provided with food and water ad libitum. All experimental
procedures were approved by the Animal Experimentation and Ethics
Committee of the Kitasato University School of Medicine (2019-036,
2020-103), and were performed in following the guidelines for
animal experiments set down by the Kitasato University School of
Medicine, which are in accordance with the ‘Guidelines for Proper
Conduct of Animal Experiments’ published by the Science Council of
Japan.
Animal procedures
Animals were fasted overnight and then injected
intraperitoneally (i.p.) with 600 mg/kg MCT (Merck KGaA) dissolved
in warm pyrogen-free saline (final concentration, 2.0 mg/ml) to
induce SOS (12). A total of 70
mice were used in this study. Mice were anesthetized with
pentobarbital sodium (60 mg/kg i.p.) at 0 (n=8), 24 (n=8), 48
(n=8), 72 (n=7), 96 (n=8), and 120 h (n=8) after MCT
administration; approximately 500 µl of blood were collected from
the heart of each mice. The levels of alanine transaminase (ALT)
were measured using a Dri-Chem 7000 Chemistry Analyzer System
(FujiFilm). Immediately after blood collection, the livers were
excised and rinsed in saline. A small section of each liver was
placed in 10% formaldehyde, and the remaining liver was frozen in
liquid nitrogen and stored at -80˚C for further analysis.
Afterwards, the animals were euthanized by cervical dislocation,
and the death was verified by the lack of heartbeat, respiration
and corneal reflex.
Mice received a daily i.p. injection of a CXCR4
antagonist (AMD3100; 10 mg/kg; Sigma-Aldrich; Merck KGaA) (n=4) in
100 µl phosphate-buffered saline (PBS) (21) or vehicle (n=5). A group of mice
received a daily i.p. injection of recombinant murine SDF-1α (20
µg/kg, R&D Systems Inc.) (n=5) (22) or vehicle (n=4). An identical volume
of sterile PBS was used as the vehicle control. At 72 h, mice were
anesthetized with pentobarbital sodium (60 mg/kg i.p.), and the
blood and liver samples were collected. These mice were euthanized
by cervical dislocation.
Histology and
immunohistochemistry
Excised liver tissues were fixed immediately with
10% formaldehyde prepared in 0.1 M sodium phosphate buffer (pH
7.4). Sections (4 µm) were prepared from paraffin-embedded tissues
and stained with hematoxylin and eosin (H&E). Images of
H&E-stained sections were captured under a microscope (Biozero
BZ-9000 Series; Keyence Corporation).
Immunofluorescence analysis
Fixed liver samples were embedded in Tissue-Tek
O.C.T. Compound (Sakura Finetek USA, Inc.), frozen at -80˚C, and
cut into 8 µm sections using a cryostat. The sections were
incubated overnight at 4˚C with a rat anti-mouse CD68 monoclonal
antibody (Bio-Rad Laboratories, Inc.), Cy3-labeled mouse anti-α
smooth muscle actin (αSMA) (Sigma-Aldrich; Merck KGaA), a rat
anti-mouse CXCR4 monoclonal antibody (Invitrogen; Thermo Fisher
Scientific, Inc.), or rabbit anti-mouse SDF-1 polyclonal antibody
(Abcam). After washing with PBS, the sections were incubated for 1
h at room temperature with Alexa Fluor 488-conjugated donkey
anti-rabbit IgG and Alexa Fluor 594-conjugated donkey anti-rat IgG
(Molecular Probes; Thermo Fisher Scientific, Inc.). Stained
sections were observed under a fluorescence microscope (Biozero
BZ-9000; Keyence Corporation) and images were captured. Expression
of CD68 in the liver tissues from ten fields per section at x400
magnification was measured as fluorescence intensity using ImageJ
software, version 1.50i (National Institutes of Health). The
results were expressed as the average of CD68 fluorescence
intensity per field.
Quantitative real-time RT-PCR
Total RNA was extracted from mouse tissues and
homogenized in TRIzol Reagent (Life Technologies; Thermo Fisher
Scientific, Inc.). Single-stranded cDNA was generated from 1 µg of
total RNA by reverse transcription using a ReverTra Ace qPCR RT kit
(Toyobo Co., Ltd.), according to the manufacturer's instructions.
Quantitative PCR was performed using TB Green Premix Ex Taq II (Tli
RNaseH Plus; Takara Bio, Inc.). Gene-specific primers used for
real-time RT-PCR were designed using Primer 3 software (http://primer3.sourceforge.net/), based on data
from GenBank. The primer sequences are listed in Table I. Data were normalized to the
expression of glyceraldehyde-3-phosphate dehydrogenase in each
sample.
 | Table IPrimers used for reverse
transcription and quantitative PCR reactions. |
Table I
Primers used for reverse
transcription and quantitative PCR reactions.
| Gene | Forward primer
sequence (5'-3') | Reverse primer
sequence (5'-3') |
|---|
| HGF |
GGCTGAAAAGATTGGATCAGG |
CCAGGAACAATGACACCAAGA |
| EGF |
ATGGGAAACAATGTCACGAAC |
CATCTCTCCCAAGCACTGAAC |
| TNFα |
TCTTCTCATTCCTGCTTGTGG |
GATCTGAGTGTGAGGGTCTGG |
| IL-1β |
TACATCAGCACCTCACAAGCA |
CCAGCCCATACTTTAGGAAGA |
| IL-6 |
CAAAGCCAGAGTCCTTCAGAG |
TAGGAGAGCATTGGAAATTGG |
| Fizz1 |
TGCCAATCCAGCTAACTATCC |
CACACCCAGTAGCAGTCATCC |
| MR |
TTTGTCCATTGCACTTTGAGG |
TGCCAGGTTAAAGCAGACTTG |
| SDF-1 |
GCATCAGTGACGGTAAACCAG |
GCACAGTTTGGAGTGTTGAGG |
| CXCR4 |
CTCTGAAGAAGTGGGGTCTGG |
AAGTAGATGGTGGGCAGGAAG |
| CCR2 |
TTACCTCAGTTCATCCACGGC |
CAAGGCTCACCATCATCGTAG |
| Gapdh |
ACATCAAGAAGGTGGTGAAGC |
AAGGTGGAAGAGTGGGAGTTG |
BM transplantation
BM transplantation was performed as previously
described (10). Briefly, recipient
mice (n=3) were treated with clodronate-loaded liposomes (200
µl/mouse; FormuMax Scientific, Inc.) to deplete the tissue
macrophages 48 h before irradiation. Following euthanasia via
cervical dislocation under 4% isoflurane anesthesia, donor BM cells
were harvested from male GFP+ transgenic mice (8 weeks
old) (n=1). Mice were irradiated with 9.8 Gy using an MBR-1505R
X-ray irradiator (Hitachi Medical Co.) fitted with a filter
(copper, 0.5 mm; aluminum, 2 mm); the cumulative radiation dose was
monitored throughout. Irradiated mice received donor BM-derived
mononuclear cells (1x107 cells/200 µl PBS) via injection
in the tail vein. Following euthanasia via cervical dislocation
under 4% isoflurane anesthesia, liver tissues were also
collected.
Statistical analysis
All results are presented as mean ± standard
deviation (SD). All statistical analyses were performed using
GraphPad Prism software, version 8 (GraphPad Software). Data from
two groups were compared using an unpaired two-tailed Student's
t-test, and data from multiple groups were compared using one-way
analysis of variance followed by Tukey's post-hoc test. A P-value
<0.05 was considered statistically significant.
Results
MCT-induced liver injury and liver
repair
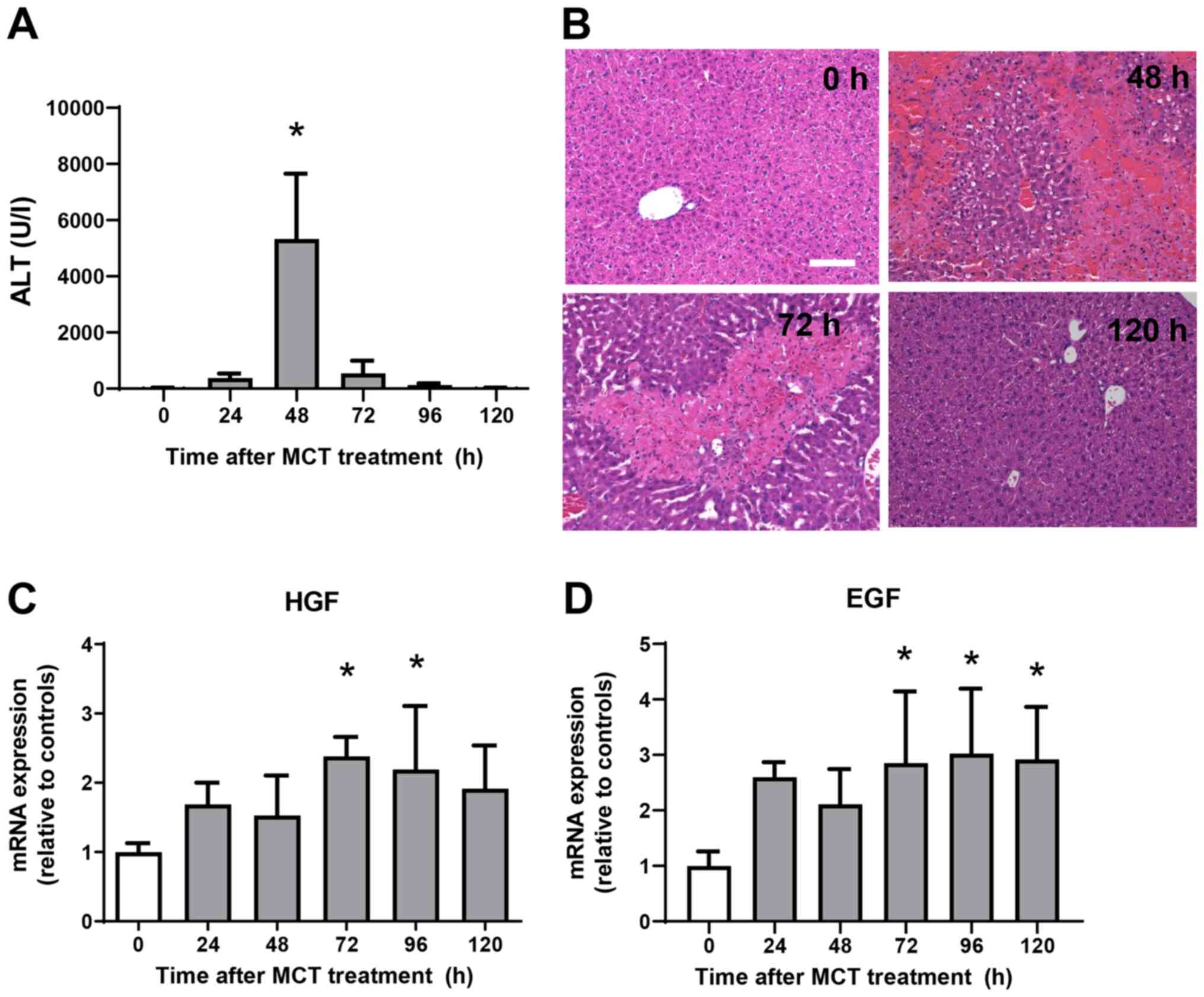
ALT levels were measured from 0 h to 120 h after MCT
administration (Fig. 1A). ALT
levels increased significantly, reaching maximal levels at 48 h
post-MCT treatment before falling again and returning to normal
levels at 120 h post-treatment. Histological analyses of the liver
demonstrated minimal changes at 0 h after MCT treatment (Fig. 1B). At 48 h post-MCT treatment,
significant hemorrhagic necrosis in the centrilobular regions of
the liver was observed. At 72 h post-MCT treatment, hepatic
necrosis around the central veins was clearly localized and the
necrotic area was reduced, which was accompanied by cellular
infiltration of the injured regions. Hepatic necrosis was
diminished and changes were much less obvious at 120 h post-MCT
treatment. These results suggest that MCT-induced liver injury
peaks at 48 h, followed by liver repair from 72 h and resolution at
120 h post-MCT treatment. We also measured the expression of mRNA
encoding tissue repair factors, hepatocyte growth factor (HGF) and
epidermal growth factor (EGF) (Fig.
1C and D) (11). Expression levels of HGF and EGF mRNA
increased during the repair phase (from 72 h to 120 h after MCT
treatment). These findings suggest that the liver recovered from
MCT-induced liver injury, as demonstrated by reduced ALT levels,
diminished area of hepatic necrosis, and increased expression of
growth factors.
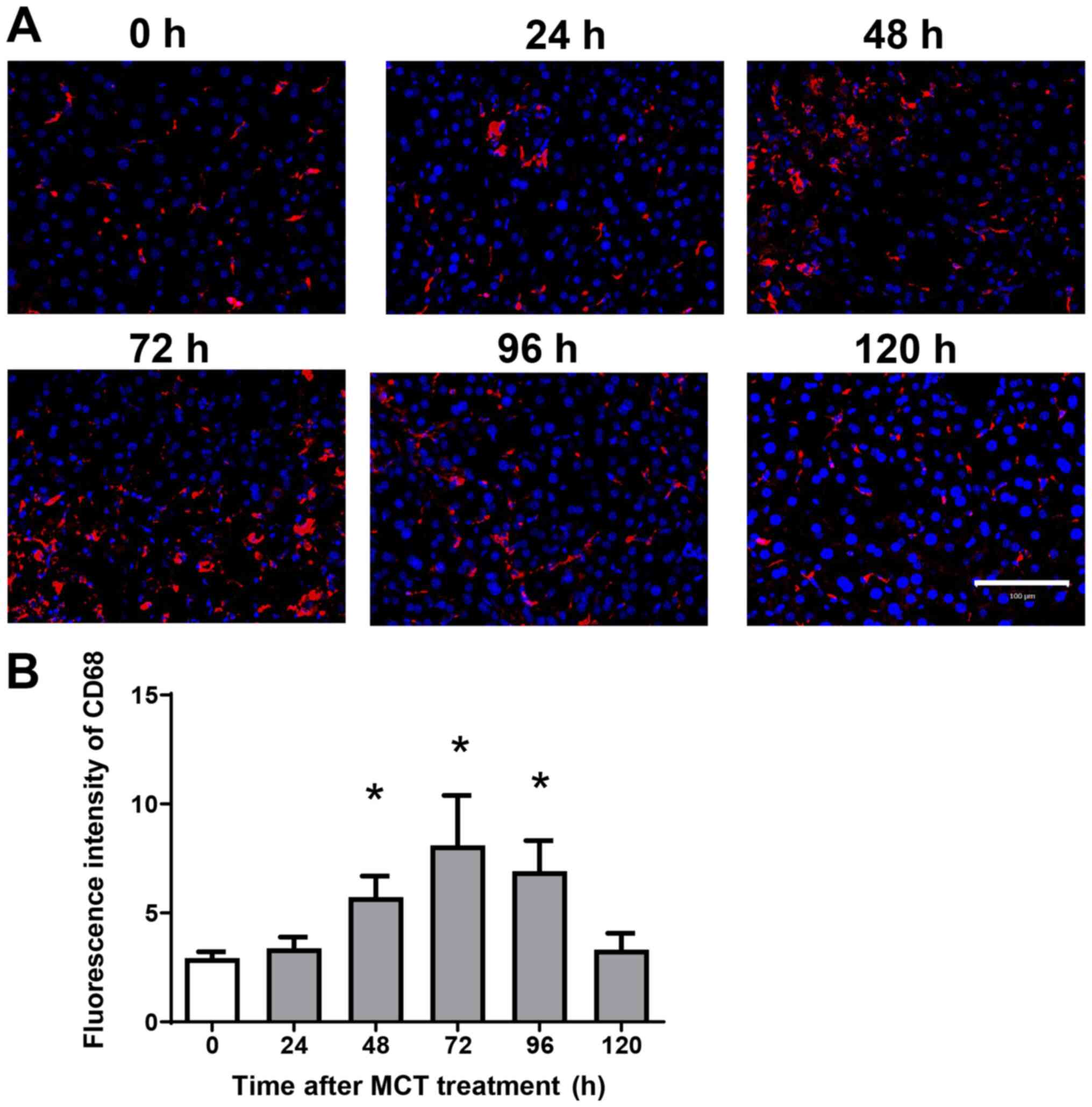
Accumulation of macrophages during MCT
hepatotoxicity
Because hepatic macrophages play an important role
in stimulating liver repair after acute liver injury (ALI), we next
examined the effects of MCT on the accumulation of macrophages in
the liver. Immunofluorescence analysis revealed that
CD68+ cells (macrophages) accumulated in the liver after
MCT treatment (Fig. 2A). The
fluorescence intensity of hepatic CD68 increased, peaking at 72 h
after MCT treatment (Fig. 2B).
These results suggest that accumulation of CD68+ cells
is associated with liver repair after MCT-induced ALI.
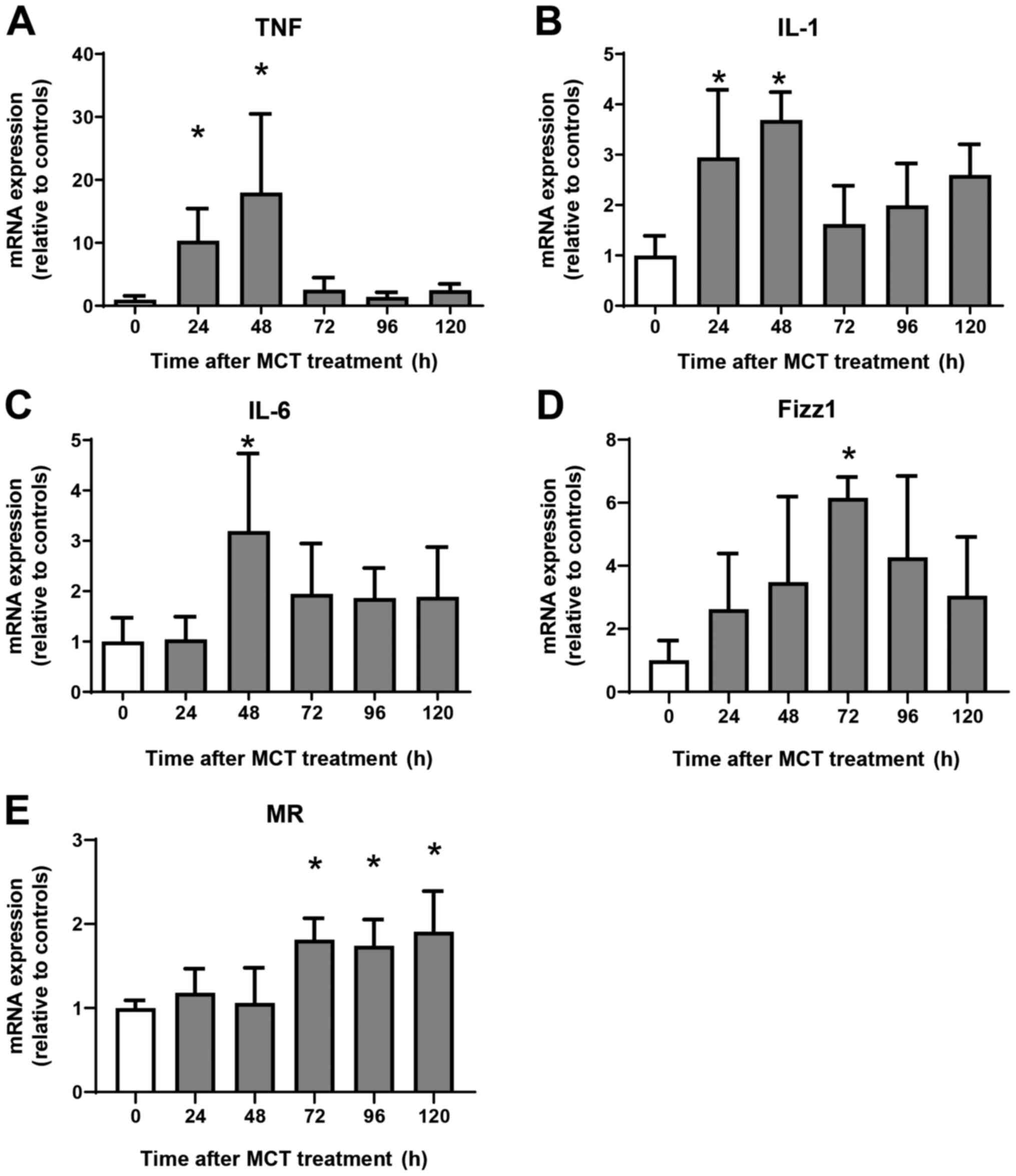
Expression of mRNA encoding genes
associated with a pro-inflammatory phenotype and a reparative
macrophage phenotype
Next, we examined the expression of mRNA encoding
genes of a pro-inflammatory macrophage phenotype, tumor necrosis
factor α (TNFα), interleukin (IL)-1β, and IL-6, and of a reparative
macrophage phenotype, found in inflammatory zone 1 (Fizz1) and
mannose receptor (MR). Expression of mRNA encoding TNFα, IL-1β, and
IL-6 increased at 24 h and 48 h post-MCT treatment, and declined
thereafter (Fig. 3A-C). Although
the expression of mRNA encoding Fizz1 and MR did not change during
the injury phase of MCT-induced hepatotoxicity, expression of Fizz1
increased at 72 h, and that of MR increased at 72, 96, and 120 h
post-treatment (Fig. 3D and
E). Thus, increased expression of
mRNA encoding markers of reparative macrophages is associated with
increased numbers of these macrophages in the liver.
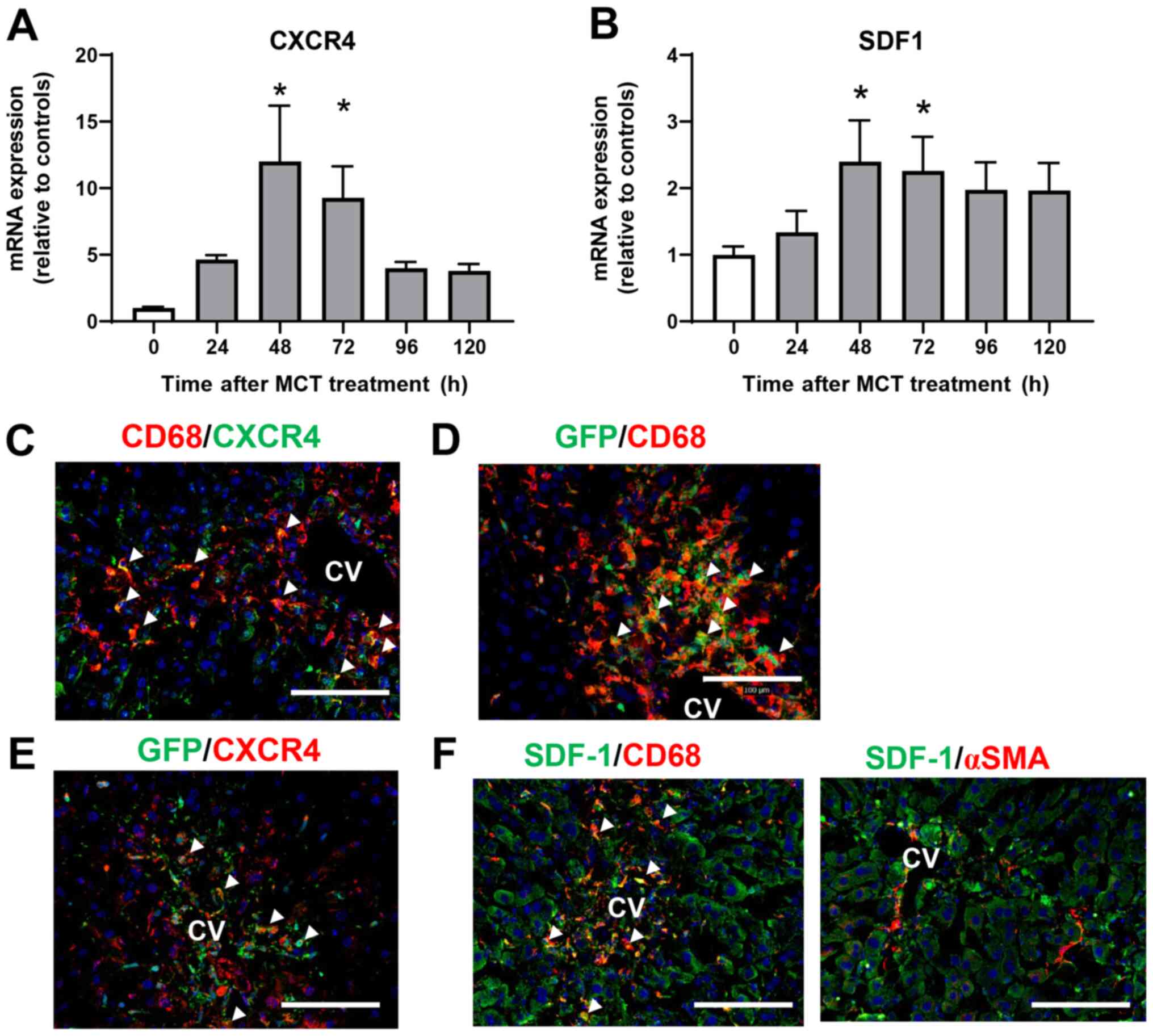
Involvement of SDF-1-CXCR4 in liver
repair after MCT-induced liver injury
To investigate the role of CXCR4 in the accumulation
of macrophages during MCT-induced liver injury, we measured the
expression of CXCR4 mRNA in the liver after MCT treatment.
Expression of CXCR4 mRNA was upregulated at 48 and 72 h after MCT
treatment (Fig. 4A). Expression of
mRNA encoding SDF-1, the ligand of CXCR4, also increased at 48 and
72 h post-MCT treatment (Fig. 4B).
To investigate the cellular source of CXCR4, we performed
immunofluorescence analysis of CXCR4 expression in liver tissues
treated with MCT. Expression of CXCR4 co-localized with
CD68+ cells accumulated in the injured regions at 72 h
after MCT treatment (Fig. 4C),
indicating that macrophages are the main source of CXCR4. We also
examined whether the accumulated macrophages were derived from the
BM. Immunofluorescence analyses of GFP+ BM chimera mice
revealed that both CD68+ cells and GFP+ cells
were extensively accumulated in the injured centrilobular regions
at 72 h post-MCT treatment, and that CD68+ cells
partially co-localized with GFP+ cells (Fig. 4D), indicating that at least some of
the macrophages were recruited from the BM. In addition,
CXCR4+ cells accumulated in the centrilobular regions
also partly co-stained with GFP+ cells in the liver at
72 h after MCT treatment, indicating that some of the
CXCR4+ cells were derived from the BM (Fig. 4E).
To further investigate the cellular source of
intrahepatic SDF-1, we performed immunofluorescence analysis of
SDF-1 in liver tissues treated with MCT for 72 h. SDF-1+
cells were accumulated around the central vein, and expression of
SDF-1 was co-localized with that of CD68, but not with that of αSMA
(Fig. 4F), indicating that
macrophages, and not hepatic stellate cells, are the main source of
SDF-1 during the repair phase of MCT hepatotoxicity.
Blockade of CXCR4 delays liver repair
after MCT-induced liver injury
To examine the functional relevance of CXCR4 during
the repair phase of MCT hepatotoxicity, mice received an i.p.
injection of AMD3100, a specific inhibitor of CXCR4. As shown in
Fig. 5A, ALT levels at 72 h in mice
treated with AMD3100 were higher than those in mice treated with
vehicle. In addition, levels of EGF and HGF mRNA in AMD3100-treated
mice were lower at 72 h post-MCT treatment than those in
vehicle-treated mice (Fig. 5B).
Although the number of CD68+ cells, as indicated by the
fluorescence intensity of CD68 in AMD3100-treated mice was not
different from that in vehicle-treated mice, the level of C-C motif
chemokine receptor 2 (CCR2) mRNA in AMD3100-treated mice was higher
than those in vehicle-treated mice (Fig. 5C). During hepatic inflammation, CCR2
is primarily found in monocyte-derived macrophages, which are
characterized as pro-inflammatory macrophages (9). Regarding CXCR4 expression, there was
no statistical difference in CXCR4 levels between the two
treatments. Furthermore, AMD3100 increased the expression of mRNA
encoding markers of pro-inflammatory macrophages (i.e., TNFα,
IL-1β, and IL-6) and decreased the expression of mRNA encoding
markers of reparative macrophages (i.e., MR, but not Fizz1) at 72 h
after MCT treatment (Fig. 5D).
These results suggest that CXCR4 plays a critical role in promoting
liver repair after MCT administration, and that this repair is
associated with a reduction in the expression of genes related to
pro-inflammatory macrophage markers.
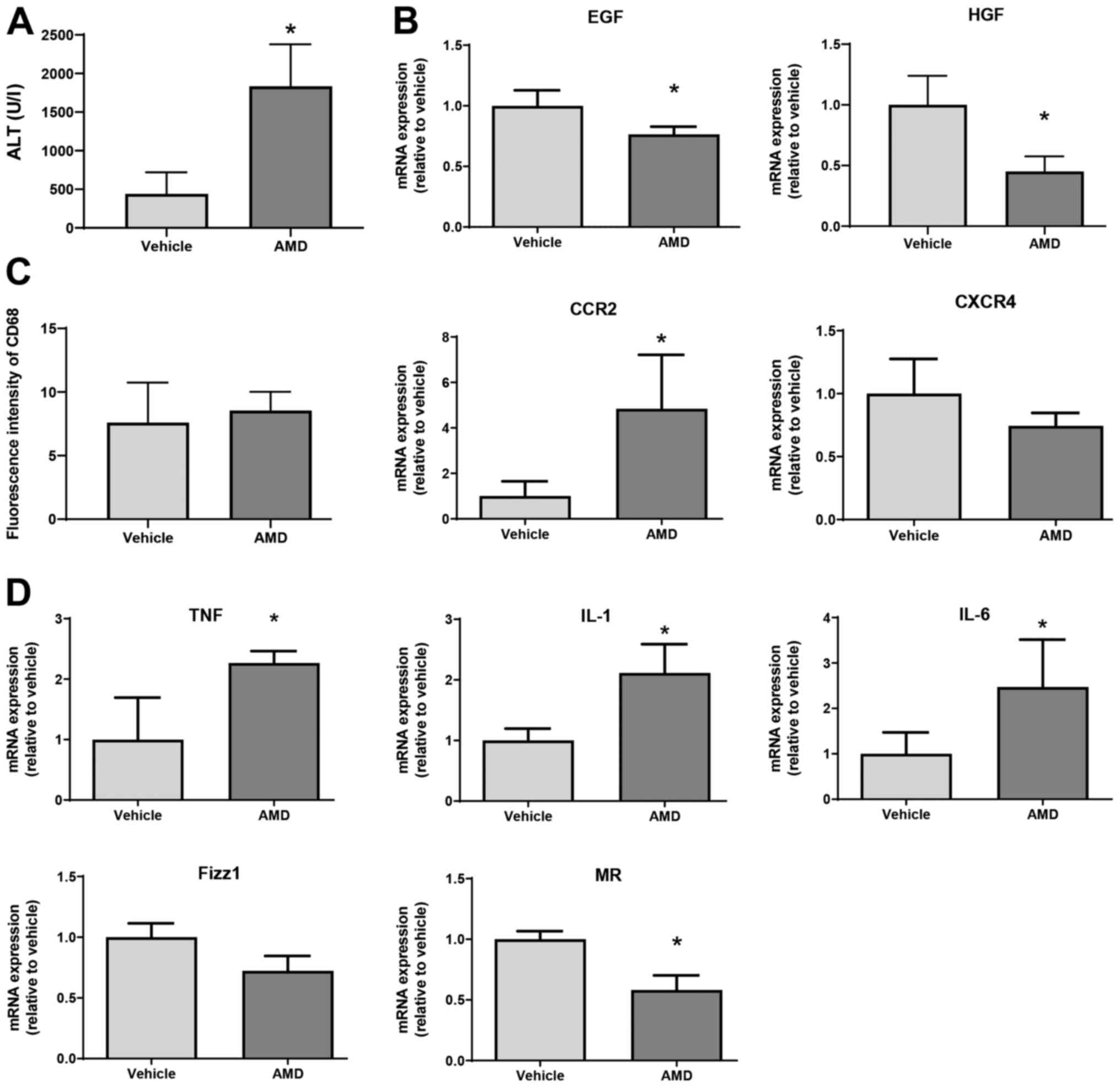 | Figure 5Effect of AMD3100 on liver repair
after MCT treatment. (A) ALT levels in mice treated with AMD3100 or
vehicle at 72 h post-MCT treatment (n=4 mice per group). (B)
Expression of mRNA encoding HGF and EGF in the livers of mice
treated with AMD3100 or vehicle at 72 h post-MCT treatment (n=3-4
mice per group). (C) Fluorescence intensity of CD68 and mRNA
expression of CCR2 and CXCR4 in the livers of mice treated with
AMD3100 or vehicle at 72 h post-MCT treatment (n=4 mice per group).
(D) Expression of mRNA encoding TNFα, IL-1β, IL-6, Fizz1 and MR in
the livers of mice treated with AMD3100 or vehicle at 72 h post-MCT
treatment (n=4-5 mice per group). Data are expressed as the mean ±
SD. *P<0.05 vs. vehicle. MCT, monocrotaline; ALT,
alanine transaminase; HGF, hepatocyte growth factor; EGF, epidermal
growth factor; CCR2, C-C motif chemokine receptor 2; CXCR4, C-X-C
chemokine receptor type 4; TNF, tumor necrosis factor; IL,
interleukin; Fizz1, found in inflammatory zone 1; MR, mannose
receptor. |
SDF-1 facilitates liver repair after
MCT-induced liver injury
Finally, we examined whether SDF-1 affects liver
repair after MCT treatment. SDF-1 reduced ALT levels at 72 h
post-MCT treatment, which was associated with an increased
expression of mRNA encoding EGF and HGF (Fig. 6A and B). The fluorescence intensity of CD68 in
the liver of SDF-1-treated mice was lower than that in
vehicle-treated mice, which was associated with the downregulation
of mRNA encoding CCR2 (Fig. 6C).
However, there was no statistical difference in CXCR4 mRNA levels
between the two treatments. SDF-1 also decreased the expression of
mRNA encoding TNFα, IL-1β, and IL-6, and increased the expression
of mRNA encoding MR (but not Fizz1) (Fig. 6D). These results suggest that
SDF-1/CXCR4 plays a critical role in promoting liver repair after
MCT administration.
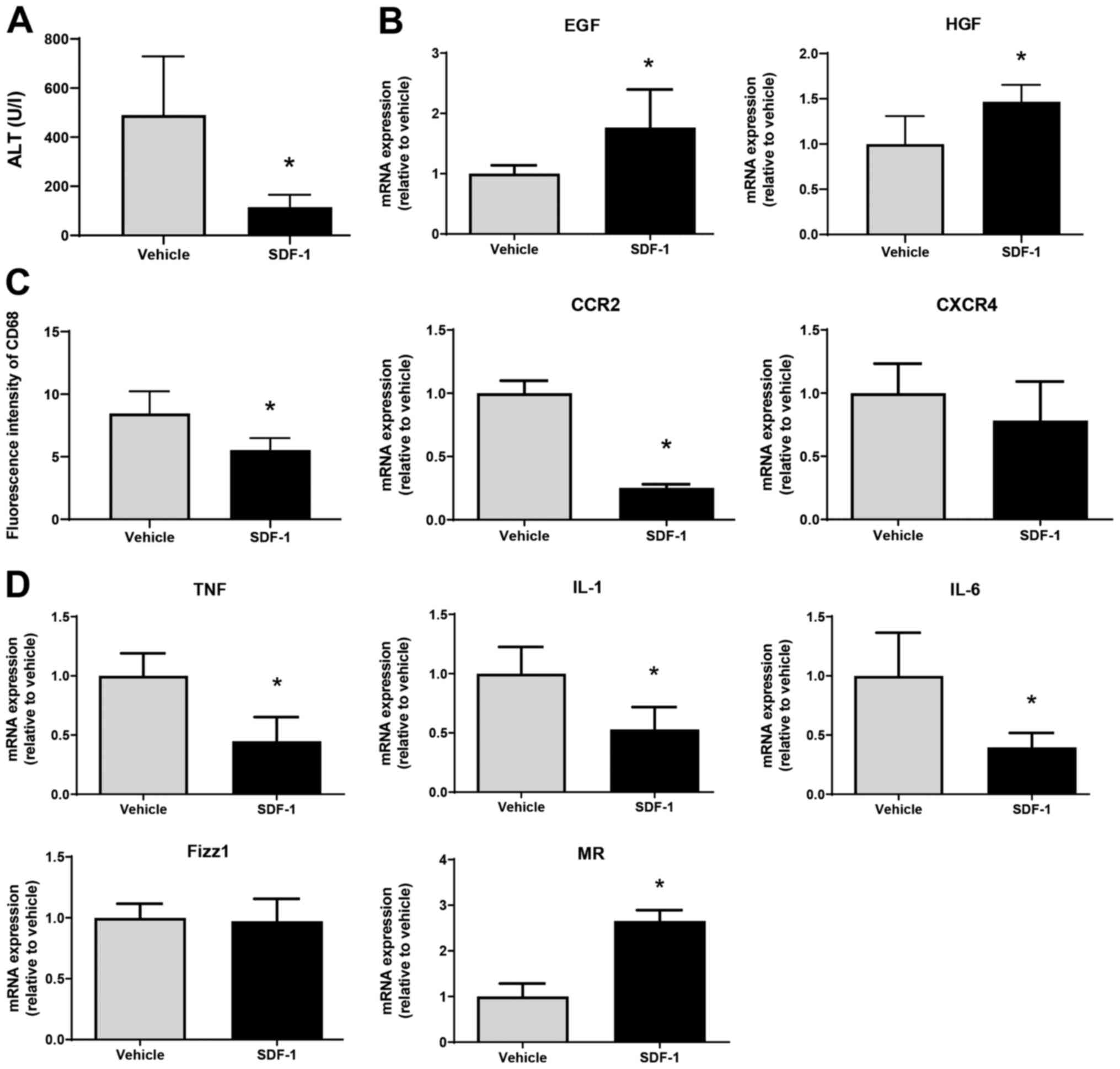 | Figure 6Effects of SDF-1 on liver repair
after MCT treatment. (A) ALT levels in mice treated with SDF-1 or
vehicle at 72 h post-MCT treatment (n=4 mice per group). (B) mRNA
expression of HGF and EGF in the livers of mice treated with SDF-1
or vehicle at 72 h post-MCT treatment (n=4-5 mice per group). (C)
Fluorescence intensity of CD68 and mRNA expression of CCR2 and
CXCR4 in the livers of mice treated with SDF-1 or vehicle at 72 h
post-MCT treatment (n=4-5 mice per group). (D) mRNA expression of
TNFα, IL-1β, IL-6, Fizz1 and MR in the livers of mice treated with
SDF-1 or vehicle at 72 h post-MCT treatment (n=4-5 mice per group).
Data are expressed as the mean ± SD. *P<0.05 vs.
vehicle. SDF-1, stromal cell-derived factor-1; MCT, monocrotaline;
ALT, alanine transaminase; HGF, hepatocyte growth factor; EGF,
epidermal growth factor; CCR2, C-C motif chemokine receptor 2;
CXCR4, C-X-C chemokine receptor type 4; TNF, tumor necrosis factor;
IL, interleukin; Fizz1, found in inflammatory zone 1; MR, mannose
receptor. |
Discussion
The objective of this study was to investigate the
contribution of macrophages to liver repair after MCT-induced liver
injury in a mouse model of SOS. We found that macrophages derived
from the BM accumulated in the liver to repair damaged tissue. This
was associated with increased hepatic expression of SDF-1 and CXCR4
during the repair phase. The SDF-1-CXCR4 axis plays a role in liver
repair by recruiting macrophages with a reparative phenotype.
Initially, ALI induced by MCT was characterized by
damage to the LSEC, including gaps in the surface of the LSEC and
detachment of the LSEC from the sinusoidal wall (4,5). LSEC
injury allows blood components to penetrate into the space of
Disse, resulting in centrilobular hepatocellular damage associated
with the accumulation of platelets and macrophages (12). Of note, we found that hepatic
inflammation induced by MCT administration was resolved and that
severe liver damage was repaired. Significant MCT-induced liver
injury, as evidenced by increased levels of ALT and hepatic
necrosis, were restored to normal levels. This was associated with
increased expression of mRNA encoding TNFα, IL-1β, and IL-6 during
the injury phase of MCT hepatotoxicity and increased expression of
mRNA encoding Fizz1 and MR during the repair phase. In addition,
expression of mRNA encoding hepatic trophic growth factors HGF and
EGF increased during the recovery phase.
Macrophages are key drivers of recovery in liver
tissues damaged by acute injury (9)
caused by chemicals such as acetaminophen (7) and carbon tetrachloride (8). Upon liver injury induced by MCT,
resident Kupffer cells in the injured regions are depleted
(4). Macrophages sense liver
injury, and monocyte-derived macrophages accumulate at the site to
replenish diminished Kupffer cells and repair damaged tissues
(9). The results of the current
study demonstrate that macrophages accumulate in injured regions
during the repair phase of MCT hepatotoxicity, and that accumulated
macrophages are, at least in part, derived from the BM. Based on
the profiles of pro-inflammatory and anti-inflammatory mediators in
the liver after MCT treatment, reparative macrophages contribute to
liver repair from acute MCT-induced liver injury.
It has been reported that, during MCT-induced
inflammation, the co-administration of MCT and lipopolysaccharide
increases ALT levels at 24 h post-treatment, which is associated
with the upregulation of chemokine (C-X-C motif) ligand 16 (CXCL16)
in hepatocytes (23). Inhibition of
the SDF-1-CXCR4 axis suppresses the development of MCT-induced
pulmonary hypertension, which is accompanied by an increase in
CXCR4-expressing cells in the BM (20). These results indicate that
chemokines and their receptors are involved in the progression of
MCT toxicity in the liver and lungs. In addition, the present study
demonstrated that SDF-1-CXCR4 pathway contributes to promoting the
resolution of hepatic inflammation and liver tissue recovery from
acute MCT-induced liver injury. Although C-X-C chemokine receptors
are essential for the regulation of immune cell recruitment to
sites of inflammatory injury, it remains unknown whether C-X-C
chemokine receptors other than CXCR4 play a role in MCT-mediated
inflammation. Further studies are needed to clarify this in
MCT-induced liver injury.
During ALI, SDF-1 plays an important role in the
regeneration by promoting hepatocyte proliferation. The SDF-1-CXCR4
axis is essential for recruiting stem cells from the BM to sites of
injury to promote tissue repair (13). In the liver environment, SDF-1 is
produced by biliary epithelial cells, hepatic stellate cells, and
LSECs (9,24,25).
In this study, immunofluorescence analyses revealed that SDF-1 was
expressed by macrophages, but not by hepatic stellate cells. In
addition, CXCR4, the specific receptor for SDF-1, was expressed by
macrophages, including BM-derived macrophages. Both CXCR4 and SDF-1
were upregulated after MCT treatment and during the repair phase.
Pharmacological inhibition of CXCR4 by AMD3100 aggravated
MCT-induced liver injury, as evidenced by increased ALT levels and
delayed liver repair (indicated by a reduction in hepatic levels of
EGF and HGF). Although the number of accumulated macrophages in the
liver did not differ between mice treated with AMD3100 and vehicle,
increased mRNA levels related to the pro-inflammatory macrophage
phenotype (i.e., TNFα, IL-1β, IL-6, and CCR2), and reduced levels
of markers of a reparative macrophage phenotype (i.e., MR),
suggesting that AMD3100 may promote accumulation of
pro-inflammatory cells. Consistent with this, a previous study
showed that CXCR4 blockade in mice inhibits hepatocyte
proliferation after acetaminophen treatment, indicating that CXCR4
signaling promotes liver regeneration (18). In addition, AMD3100 leads to
sustained hepatic inflammation induced by carbon tetrachloride
injection, along with increased hepatic necrosis and increased
numbers of infiltrating inflammatory cells (17). Furthermore, there was no statistical
difference in CXCR4 levels between AMD3100 and vehicle, indicating
that AMD3100 did not change the hepatic expression of CXCR4 at 72 h
after MCT treatment. These results suggest that AMD3100 inhibits
CXCR4-mediated signal transduction pathways (26) without affecting the expression of
CXCR4 in the livers of mice treated with MCT.
In contrast, administration of SDF-1 accelerated
liver repair after MCT-induced ALI, which was associated with the
downregulation of genes related to a pro-inflammatory macrophage
phenotype and upregulation of genes related to a reparative
macrophage phenotype. SDF-1 administration also decreased the
expression of CCR2, which is primarily found in pro-inflammatory
macrophages (22), suggesting that
SDF-1 reduced the accumulation of CD68+-cells
characterized by pro-inflammatory macrophages. These results
suggest that SDF-1 may be a key driver of liver repair after MCT
administration. Previous studies suggest that binding of SDF-1 to
CXCR4 plays a role in liver repair by recruiting hematopoietic stem
cells to the site of injury in the liver (19). In addition, mice treated with a
CXCR4 inhibitor are more susceptible to chronic liver injury
(17). Similarly, upregulation of
SDF-1 in the liver after partial hepatectomy increases mobilization
of LSEC precursors from the BM to promote liver regeneration in
rats (27). Taken together, these
findings, including our own, indicate an important role for the
SDF-1/CXCR4 axis in liver repair and regeneration in response to
chemical-induced acute or chronic injury or partial
hepatectomy.
Because SDF-1 recruits macrophages through CXCR4
signaling, the administration of SDF-1 would upregulate CXCR4
expression in livers; however, the results showed that the levels
of CXCR4 expression in SDF-1-treated mice did not differ from those
in vehicle-treated mice. The current study demonstrated that SDF-1
treatment attenuated liver injury at 72 h after MCT treatment,
which was associated with reduced accumulation of macrophages in
the liver. Thus, it is speculated that SDF-1 administration did not
upregulate CXCR4 expression at 72 h after MCT treatment.
However, others report that administration of
recombinant SDF-1 decreases the proliferation of hepatocytes after
hepatic I/R injury in mice (28).
Furthermore, they found that treatment of mice with AMD3100
resulted in increased hepatocyte proliferation and reduced necrosis
after hepatic I/R. These results imply that the SDF-1-CXCR4 axis
suppresses liver repair after hepatic I/R. Such discrepancy may be
due to the different models and the different experimental
protocols employed for pharmacological intervention.
In the current study, enhanced liver repair from MCT
hepatotoxicity was associated with increased hepatic levels of EGF
and HGF. Previously, we showed that EGF in macrophages plays a
critical role in facilitating liver repair after ALI induced by
hepatic I/R (29). Upregulation of
HGF is related to liver recovery from acute chemical-induced liver
injury (8). These results suggest
that macrophages accumulate and repair tissues damaged by MCT by
producing EGF and HGF; however, further studies are needed to
confirm this.
In conclusion, this study showed that MCT-induced
liver injury is repaired by the accumulation of macrophages in the
injured region, and that the SDF-1/CXCR4 axis plays an important
role in acute liver injury repair by recruiting reparative
macrophages. Thus, SDF-1 may be useful for improving recovery from
MCT-induced ALI.
Acknowledgements
The authors would like to thank Ms. Michiko Ogino
and Ms. Kyoko Yoshikawa (Department of Pharmacology, Kitasato
University School of Medicine, Sagamihara, Kanagawa 252-0374,
Japan) for their technical assistance.
Funding
Funding: This work was supported by grants from the Japanese
Ministry of Education, Culture, Sports, Science and Technology
(MEXT; grant nos. 19K09156 and 20K17630). This study was also
supported by the Takeda Science Foundation.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FO and YI conceived, designed and performed the
experiments, and wrote the manuscript. SN, NN and TH performed the
experiments. KH and MM performed data analysis and interpretation,
and provided technical support. WK and HA performed data analysis
and interpretation and revised the manuscript critically for
intellectual content. FO and YI confirm the authenticity of all the
raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Animal Experimentation and Ethics Committee of the Kitasato
University School of Medicine (permit no. 2019-036, 2020-103).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
DeLeve LD, Shulman HM and McDonald GB:
Toxic injury to hepatic sinusoids: Sinusoidal obstruction syndrome
(veno-occlusive disease). Semin Liver Dis. 22:27–42.
2002.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Coppell JA, Richardson PG, Soiffer R,
Martin PL, Kernan NA, Chen A, Guinan E, Vogelsang G, Krishnan A,
Giralt S, et al: Hepatic veno-occlusive disease following stem cell
transplantation: Incidence, clinical course, and outcome. Biol
Blood Marrow Transplant. 16:157–168. 2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Valla DC and Cazals-Hatem D: Sinusoidal
obstruction syndrome. Clin Res Hepatol Gastroenterol. 40:378–385.
2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
DeLeve LD, McCuskey RS, Wang X, Hu L,
McCuskey MK, Epstein RB and Kanel GC: Characterization of a
reproducible rat model of hepatic veno-occlusive disease.
Hepatology. 29:1779–1791. 1999.PubMed/NCBI View Article : Google Scholar
|
|
5
|
DeLeve LD, Ito Y, Bethea NW, McCuskey MK,
Wang X and McCuskey RS: Embolization by sinusoidal lining cells
obstructs the microcirculation in rat sinusoidal obstruction
syndrome. Am J Physiol Gastrointest Liver Physiol. 284:G1045–G1052.
2003.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mehendale HM: Tissue repair: An important
determinant of final outcome of toxicant-induced injury. Toxicol
Pathol. 33:41–51. 2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kato T, Ito Y, Hosono K, Suzuki T, Tamaki
H, Minamino T, Kato S, Sakagami H, Shibuya M and Majima M: Vascular
endothelial growth factor receptor-1 signaling promotes liver
repair through restoration of liver microvasculature after
acetaminophen hepatotoxicity. Toxicol Sci. 120:218–229.
2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Minamino T, Ito Y, Ohkubo H, Hosono K,
Suzuki T, Sato T, Ae T, Shibuya A, Sakagami H, Narumiya S, et al:
Thromboxane A(2) receptor signaling promotes liver tissue repair
after toxic injury through the enhancement of macrophage
recruitment. Toxicol Appl Pharmacol. 259:104–114. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Krenkel O and Tacke F: Liver macrophages
in tissue homeostasis and disease. Nat Rev Immunol. 17:306–321.
2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Nishizawa N, Ito Y, Eshima K, Ohkubo H,
Kojo K, Inoue T, Raouf J, Jakobsson PJ, Uematsu S, Akira S, et al:
Inhibition of microsomal prostaglandin E synthase-1 facilitates
liver repair after hepatic injury in mice. J Hepatol. 69:110–120.
2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Nakamoto S, Ito Y, Nishizawa N, Goto T,
Kojo K, Kumamoto Y, Watanabe M, Narumiya S and Majima M: EP3
signaling in dendritic cells promotes liver repair by inducing
IL-13-mediated macrophage differentiation in mice. FASEB J.
34:5610–5627. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Otaka F, Ito Y, Inoue T, Ohkubo H,
Nishizawa N, Kojo K, Betto T, Yamane S, Narumiya S, Koizumi W, et
al: Thromboxane A2 receptor signaling in endothelial cells
attenuates monocrotaline-induced liver injury. Toxicol Appl
Pharmacol. 381(114733)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liepelt A and Tacke F: Stromal
cell-derived factor-1 (SDF-1) as a target in liver diseases. Am J
Physiol Gastrointest Liver Physiol. 311:G203–G209. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jin DK, Shido K, Kopp HG, Petit I,
Shmelkov SV, Young LM, Hooper AT, Amano H, Avecilla ST, Heissig B,
et al: Cytokine-mediated deployment of SDF-1 induces
revascularization through recruitment of CXCR4+
hemangiocytes. Nat Med. 12:557–567. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Amano H, Kato S, Ito Y, Eshima K, Ogawa F,
Takahashi R, Sekiguchi K, Tamaki H, Sakagami H, Shibuya M, et al:
The role of vascular endothelial growth factor receptor-1 signaling
in the recovery from ischemia. PLoS One.
10(e0131445)2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sato T, Amano H, Ito Y, Eshima K, Minamino
T, Ae T, Katada C, Ohno T, Hosono K, Suzuki T, et al: Vascular
endothelial growth factor receptor 1 signaling facilitates gastric
ulcer healing and angiogenesis through the upregulation of
epidermal growth factor expression on
VEGFR1+CXCR4+ cells recruited from bone
marrow. J Gastroenterol. 49:455–469. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Saiman Y, Jiao J, Fiel MI, Friedman SL,
Aloman C and Bansal MB: Inhibition of the CXCL12/CXCR4 chemokine
axis with AMD3100, a CXCR4 small molecule inhibitor, worsens murine
hepatic injury. Hepatol Res. 45:794–803. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Tirone M, Tran NL, Ceriotti C, Gorzanelli
A, Canepari M, Bottinelli R, Raucci A, Di Maggio S, Santiago C,
Mellado M, et al: High mobility group box 1 orchestrates tissue
regeneration via CXCR4. J Exp Med. 215:303–318. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kollet O, Shivtiel S, Chen YQ, Suriawinata
J, Thung SN, Dabeva MD, Kahn J, Spiegel A, Dar A, Samira S, et al:
HGF, SDF-1, and MMP-9 are involved in stress-induced human
CD34+ stem cell recruitment to the liver. J Clin Invest.
112:160–169. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Zhang T, Kawaguchi N, Tsuji K, Hayama E,
Furutani Y, Sugiyama H and Nakanishi T: Silibinin upregulates CXCR4
expression in cultured bone marrow cells (BMCs) especially in
pulmonary arterial hypertension rat model. Cells.
9(1276)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ogawa F, Amano H, Eshima K, Ito Y, Matsui
Y, Hosono K, Kitasato H, Iyoda A, Iwabuchi K, Kumagai Y, et al:
Prostanoid induces premetastatic niche in regional lymph nodes. J
Clin Invest. 124:4882–4894. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Amano H, Mastui Y, Ito Y, Shibata Y, Betto
T, Eshima K, Ogawa F, Satoh Y, Shibuya M and Majima M: The role of
vascular endothelial growth factor receptor 1 tyrosine kinase
signaling in bleomycin-induced pulmonary fibrosis. Biomed
Pharmacother. 117(109067)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hammad MA, Abdel-Bakky MS, Walker LA and
Ashfaq MK: Oxidized low-density lipoprotein and tissue factor are
involved in monocrotaline/lipopolysaccharide-induced
hepatotoxicity. Arch Toxicol. 85:1079–1089. 2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Hong F, Tuyama A, Lee TF, Loke J, Agarwal
R, Cheng X, Garg A, Fiel MI, Schwartz M, Walewski J, et al: Hepatic
stellate cells express functional CXCR4: Role in stromal
cell-derived factor-1alpha-mediated stellate cell activation.
Hepatology. 49:2055–2067. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sawitza I, Kordes C, Reister S and
Häussinger D: The niche of stellate cells within rat liver.
Hepatology. 50:1617–1624. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hatse S, Princen K, Bridger G, De Clercq E
and Schols D: Chemokine receptor inhibition by AMD3100 is strictly
confined to CXCR4. FEBS Lett. 527:255–262. 2002.PubMed/NCBI View Article : Google Scholar
|
|
27
|
DeLeve LD, Wang X and Wang L: VEGF-sdf1
recruitment of CXCR7+ bone marrow progenitors of liver
sinusoidal endothelial cells promotes rat liver regeneration. Am J
Physiol Gastrointest Liver Physiol. 310:G739–G746. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wilson GC, Freeman CM, Kuethe JW, Quillin
RC III, Nojima H, Schuster R, Blanchard J, Edwards MJ, Caldwell CC
and Lentsch AB: CXC chemokine receptor-4 signaling limits
hepatocyte proliferation after hepatic ischemia-reperfusion in
mice. Am J Physiol Gastrointest Liver Physiol. 308:G702–709.
2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ohkubo H, Ito Y, Minamino T, Eshima K,
Kojo K, Okizaki S, Hirata M, Shibuya M, Watanabe M and Majima M:
VEGFR1-positive macrophages facilitate liver repair and sinusoidal
reconstruction after hepatic ischemia/reperfusion injury. PLoS One.
9(e105533)2014.PubMed/NCBI View Article : Google Scholar
|