Introduction
Down syndrome (DS) or trisomy 21 is a chromosomal
disorder that is caused by the total or partial trisomy of
chromosome 21(1), which is
associated with delayed nervous development, intellectual
disability and a characteristic facial appearance. All affected
individuals also experience cognitive delays and weak muscle tone
(hypotonia) and they usually present with immune system problems
(2). Furthermore, individuals with
DS have been discovered to have an increased risk of developing
Alzheimer's disease and leukemia (3).
Studies have demonstrated that neurological and
immune abnormalities in DS commonly occur in the early
developmental stage. At present, basic research on DS relies
heavily on clinically obtained peripheral blood samples. However,
these samples cannot meet the requirements of basic research,
particularly when it comes to the study of early developmental
mechanisms. Since 2006, induced pluripotent stem cells (iPSCs) have
been considered an invaluable tool for disease modeling and
regenerative therapies (4). iPSCs
share most of the characteristics of human embryonic stem cells
(ESCs); they are pluripotent and self-renew indefinitely, and have
the potential to differentiate into any cell type in the body,
including those pertinent to psychiatric disorders (5,6).
Furthermore, iPSCs were indicated to have similar genetic and
epigenetic features to those of ESCs (7,8).
Therefore, iPSCs enable the investigation of the influence of the
epigenetic status on the early developmental stage and provide a
resource for stem cell-based therapies.
Long non-coding RNAs (lncRNAs) are non-protein
coding transcripts that are >200 nucleotides long and are
members of the family of ncRNAs. Although lncRNAs are not
translated into protein, lncRNAs have been reported to serve a
highly important regulatory role in the pathogenesis of numerous
types of diseases (9). For
instance, lncRNAs, as crucial gene antisense transcripts, have been
indicated to drive the pathophysiology associated with Alzheimer's
disease and regulators of oncogenes during the development of
leukemia (10-12).
Since patients with DS frequently develop Alzheimer's disease and
leukemia, lncRNAs may be potential regulators in DS. A previous
study by our group reported that lncRNAs were abnormally expressed
in DS (13), suggesting that
lncRNAs may be crucial factors in DS; however, the mechanistic
roles of lncRNAs remain to be fully elucidated.
As an initial framework for understanding the
function of lncRNAs, lncRNAs are typically divided into two types:
Cis-acting and trans-acting, based on the local or distant
regulation of gene expression, respectively (14,15).
It has been suggested that the ability of lncRNA to regulate the
expression levels of nearby genes in cis may be attributable to the
mature lncRNA transcript or the fact that they do not rely on
lncRNAs themselves to have a regulatory role (16). In trans-acting genes, lncRNAs have
been indicated to interact with proteins and/or other RNA molecules
to regulate the expression of target genes (16). Although an abnormal expression of
lncRNAs has been observed in DS, the regulatory pattern of lncRNAs
remains to be elucidated. To understand the potential function and
regulatory pattern of lncRNAs in DS, the aim of the present study
was to investigate the putative cis- or trans-acting targets of
lncRNAs in DS and normal iPSCs. RNA sequencing (RNA-seq) was
performed to analyze the genome-wide transcriptomic changes in DS.
Subsequently, the regulatory mechanisms of differentially expressed
(DE) lncRNAs were predicted and analyzed.
Materials and methods
Sample preparation, RNA isolation and
library preparation
Normal iPSCs and DS-iPSCs were generated from dermal
fibroblast cells, which were purchased from the American Type
Culture Collection (ACS-1011: Normal, newborn, male; ACS-1033: DS,
newborn, male). For all experiments, three biological replicates
were performed and ≥2 technical replicates were used within each
biological replicate. Total RNA was extracted from the iPSCs using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The
RNA concentration and purity were measured with a NanoDrop ND-1000
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.). An Abiotin-labeled specific probe [Ribo-Zero™ ribosomal
(r)RNA Removal kit] was used to remove the rRNA from the extracted
total RNA. Subsequently, the complementary (c)DNA strand was
synthesized using random primers and reverse transcriptase using
the TruSeq Stranded kit (cat. no. PC-121-2001; Illumina, Inc.). The
second-strand cDNA was subsequently synthesized and double-stranded
cDNA was generated. The double-stranded cDNA underwent
end-repair/dA-tail and adaptor ligation (17). The products were purified using
magnetic beads to create the final cDNA library. High-throughput
sequencing of the cDNA library was performed using Illumina HiSeq
2000 platform (Illumina, Inc.).
Sequencing and quality analysis
The clean reads were mapped to the reference genome
sequence using HISAT tools (18)
and the transcripts were assembled with StringTie (19). Following transcript reconstitution,
Cuffcompare (20) was used to
compare these transcripts with known mRNA and lncRNA to obtain
information on their positional relationship. To distinguish the
mRNAs and lncRNAs, three pieces of software [CPC (http://cpc.cbi.pku.edu.cn), txCdsPredic (http://hgdownload.soe.ucsc.edu/admin/exe/) and CNCI
(http://www.bioinfo.org/software/cnci)] (20,21)
were used to predict the coding ability of the transcripts and all
transcripts met the following requirements: i) Fragments per
kilo-base of exon per million fragments mapped (FPKM) ≥0.5; ii)
coverage >1; iii) length >200. In addition, the estimation
accuracy for genes with low expression was improved by Cuffmerge
(22) and the combined transcripts
were used as final results for subsequent analysis. Following the
analysis of all transcripts annotated as mRNA or lncRNA, Bowtie2
was used to compare clean reads to the reference sequence (23), and the FPKM were then calculated as
the expression levels of genes and transcripts using RSEM software
(v1.3.1; http://deweylab.biostat.wisc.edu/rsem) (24). The results obtained were used for
the subsequent analysis.
Identification of DE lncRNAs and
mRNAs
The differential expression levels of lncRNAs and
mRNAs were calculated based on the normalized FPKM using the DEGseq
package (25). q<0.001 and |log2
fold change (FC)| >1 were set as the threshold values for
significant differential expression.
lncRNA target gene prediction and
enrichment analysis
To determine the correlation between lncRNAs and
mRNAs, Spearman's rank correlation coefficient and Pearson's
correlation coefficient were calculated and the strength and the
direction of the correlations were thereby obtained, with
Spearman_correlations and Pearson_correlations ≥0.6 considered to
indicate a significant correlation. The sliding window strategy was
used to search cis-acting target genes 10 kb upstream and 20 kb
downstream of mRNAs (26). Above
this range, RNAplex (http://www.tbi.univie.ac.at/~htafer/) was used to
analyze the binding energy of lncRNA to mRNA (27). A binding energy of E-value ≤30 was
considered to be trans-acting. To predict the cis-acting results,
the different lncRNA-mRNA modules were counted, including lncRNAs
located within 10 kb upstream or 20 kb downstream of mRNA, and the
overlap between lncRNA and mRNA was determined. The overlap may be
divided into different subclasses, including lnc-Overlap-mRNA and
lnc-AntiOverlap-mRNA (28,29). The classification criteria are
presented in Table SI. These cis
target genes were subjected to Gene Ontology (GO; http://geneontology.org) functional term and Kyoto
Encyclopedia of Genes and Genomes (KEGG; https://www.genome.jp/kegg/) signaling pathway
enrichment analysis.
Validation of DE lncRNAs and
mRNAs
In order to verify the functions related to lncRNAs
in DS-iPSCs and normal iPSCs, lncRNAs and mRNAs were selected based
on the results of lncRNA target gene prediction and enrichment
analysis. Subsequently, several lncRNA-mRNA pairs were screened
using RT-qPCR to demonstrate the reliability of the analysis. RNA
extraction was performed following using TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was synthesized
using random primers and the PrimeScript RT Reagent kit (cat. no.
RR047A; Takara Bio, Inc.). RT-qPCR was performed with the One-Step
TB Green PrimeScript RT-PCR Kit (cat. no. RR066A; Takara Bio, Inc.)
following the manufacturer's instruction. RT-qPCR parameters were
as follows: 95˚C for 2 min, 40 cycles of 95˚C for 5 sec and 60˚C
for 34 min. Primers for the RT-qPCR were designed using Primer Bank
(https://pga.mgh.harvard.edu/primerbank/) or Primer 5.0
software. The GAPDH gene was used to standardize the expression
levels. The sequences of PCR were as follows: GAPDH forward,
5'-CGGAGTCAACGGATTTGGTCGTAT-3' and reverse,
5'-AGCCTTCTCCATGGTGGTGAAGAC-3'; NONHSAT009060.2 forward,
5'-CCTGGCTTCTGGTCAAAC-3' and reverse, 5'-AAGGCAACTCAGTCACTAACAC-3';
NONHSAT022318.2 forward, 5'-TCAGTTCAAGGCAACACTGC-3' and reverse
5'-AGGTGGCACTGACCATATCC-3'; ACTN3 forward,
5'-GTACCGCAACGTCAACGTG-3' and reverse, 5'-CGTAGTCGATGAGGTCAGGG-3';
and SRGAP2C-F forward, 5'-ATTGGGCAGCTGAGCATACA-3', and reverse,
5'-TTGGGTCCAGTAACGTATTCCA-3'. Each type of iPSCs included three
samples and all reactions were performed three times for each
sample. The results of the RT-qPCR for verification were analyzed
using the 2-ΔΔCq method (30). Student's t-test was performed on
RT-qPCR data and P<0.05 was considered significant.
Cytoplasmic and nuclear
fractionation
The Minute (TM) Cytoplasmic and Nuclear
Fractionation kits (Invent; cat. no. SC-003) were used for the
separation of nuclear and plasma iPSCs. Concurrently, GAPDH was
used as the internal reference gene for the nucleus and X-inactive
specific transcript (XIST) was used as the internal reference for
the cytoplasm (31). The expression
location and relative expression levels of the lncRNAs were
determined by RT-qPCR. RT-qPCR was performed as aforementioned.
Statistical analysis
A total of 48 samples were designed for the
experiment (4 genes x2 samples x2 subcellular locations x3
technical repetitions). The RT-qPCR results for each gene were
subjected to one-way ANOVA and for significance, Tukey's honestly
significant differences test was performed using SPSS (version
16.0; SPSS, Inc.). For all tests, a P<0.05 was set for
statistical significance.
Results
Identification of DE genes in iPSCs by
RNA-Seq
To identify lncRNAs and mRNAs expressed in DS, six
cDNA libraries were constructed using DS-iPCs and normal iPSCs. A
total of 763,789,996 raw reads were obtained. Following the removal
of reads containing N ratios >10%, low-quality reads and adapter
fragments, 699,869,860 clean reads were obtained. Following
comparison of the data with the reference genome, 85,191 lncRNA
transcripts and 40,726 mRNAs were identified using bioinformatics
analysis. Compared to mRNAs, more lncRNAs were observed to be
significantly DE (P<0.05; Fig.
1A), which suggested the functional importance of lncRNAs in
DS. RNA-Seq analysis of lncRNA transcription (4,285 upregulated and
5,239 downregulated; Fig. 1B and
C) and mRNA transcription (4,017
upregulated and 4,432 downregulated) demonstrated that differential
gene expression was present in DS-iPSCs. All DE lncRNAs were well
annotated with known chromosomal locations; DE lncRNAs were
distributed across chromosomes without any locational preference
(Fig. 1D).
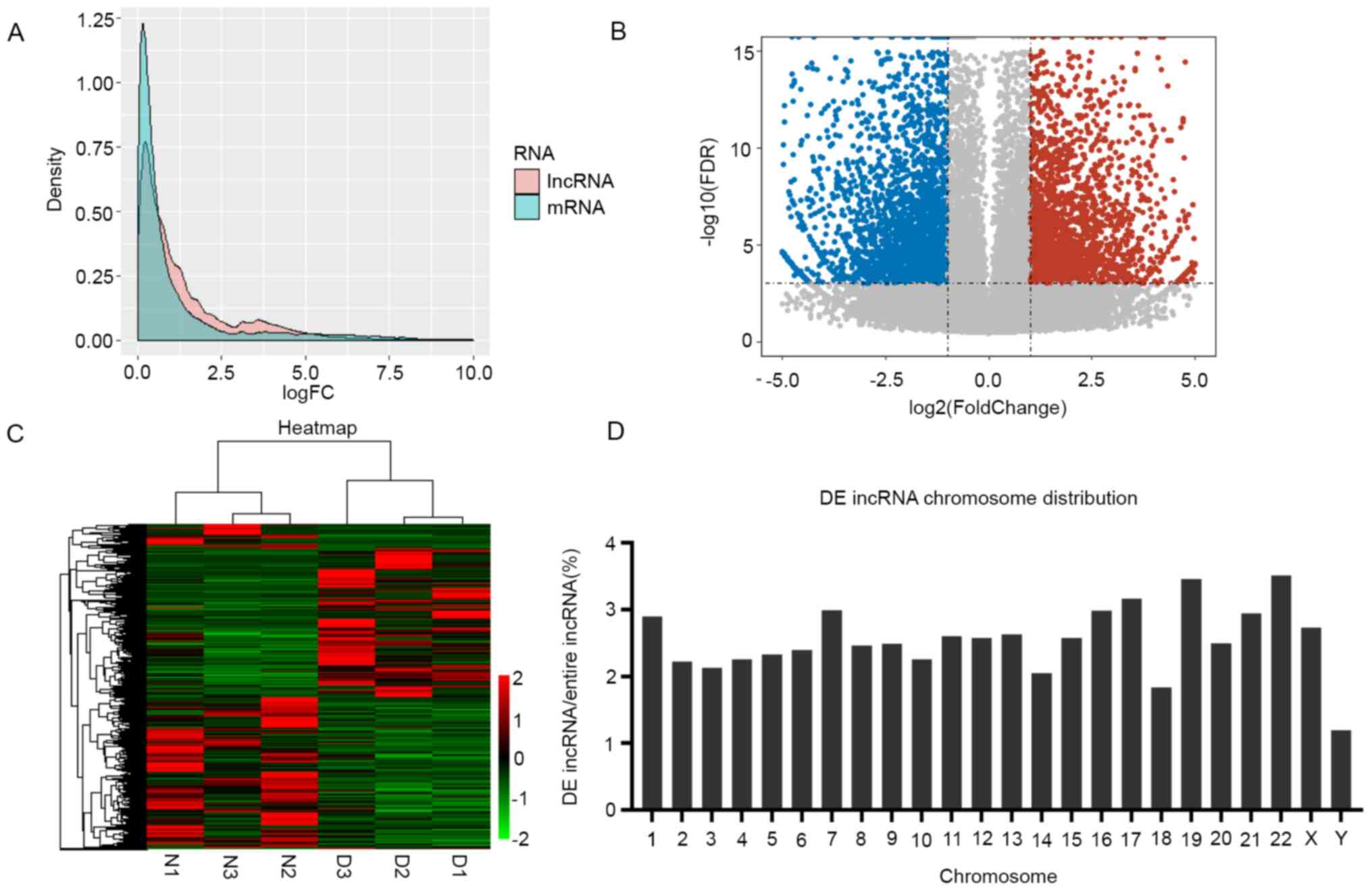 | Figure 1Transcriptional patterns of lncRNAs
in DS-iPSCs. (A) Density plot demonstrating the FC values for the
expression levels of lncRNA and protein-coding genes. Pink and blue
represent the FC of lncRNAs and mRNAs, respectively. The
differential expression of lncRNAs was more significant than that
of mRNAs. (B) Volcano plot displaying the 4,285 upregulated and
5,239 downregulated lncRNAs. The horizontal line represents
P<0.001 in log10 scale and the vertical lines represent the
upregulation and downregulation by two-FC. The red and blue dots
represent the upregulated and downregulated lncRNAs, respectively,
and non-significantly DE genes are represented by black circles.
(C) Heatmap indicating the DE lncRNAs in the DS and control
samples. Each column represents one sample and each row one lncRNA.
The relative expression levels of lncRNAs are depicted according to
the color scale. Red indicates upregulation and green
downregulation, whereas the three left columns represent the
control samples and the three right columns the DS samples. The DE
lncRNAs are clearly self-segregated into clusters, N1, N2 and N3
represent the three biological replications as normal control, and
D1, D2 and D3 are represent the three biological replications from
the DS patient. (D) Proportion of DE lncRNAs in each chromosome.
The ordinate indicates the percentage of DE lncRNAs in each
chromosome. lncRNA, long non-coding RNA; DS, Down syndrome; iPSCs,
induced pluripotent stem cells; DE, differentially expressed; FDR,
false discovery rate; FC, fold change. |
Target gene prediction of DE
lncRNAs
lncRNA expression levels have a significant impact
on neighboring (cis) or distal (trans) protein-coding genes.
LncRNA-mRNA interactions are useful to clarify the biological
functions of DE lncRNAs. To determine the lncRNAs and their
potential functions in DS, the potential interactions between the
lncRNA and mRNA transcripts were investigated. The
cis-/trans-acting target analysis was performed using the
association of the expression profiles of DE-lncRNAs and DE-mRNAs.
The mRNAs adjacent to the lncRNAs were screened as cis-acting
target genes. Trans-acting was determined by calculating the
binding energy. First, target genes were predicted in the whole
genome and 30,118 lncRNA-mRNA pairs were obtained. The lncRNA-mRNA
pairs were screened using the DE mRNAs and lncRNAs. In total, 1,305
lncRNA-mRNA pairs involved in cis and trans regulation were
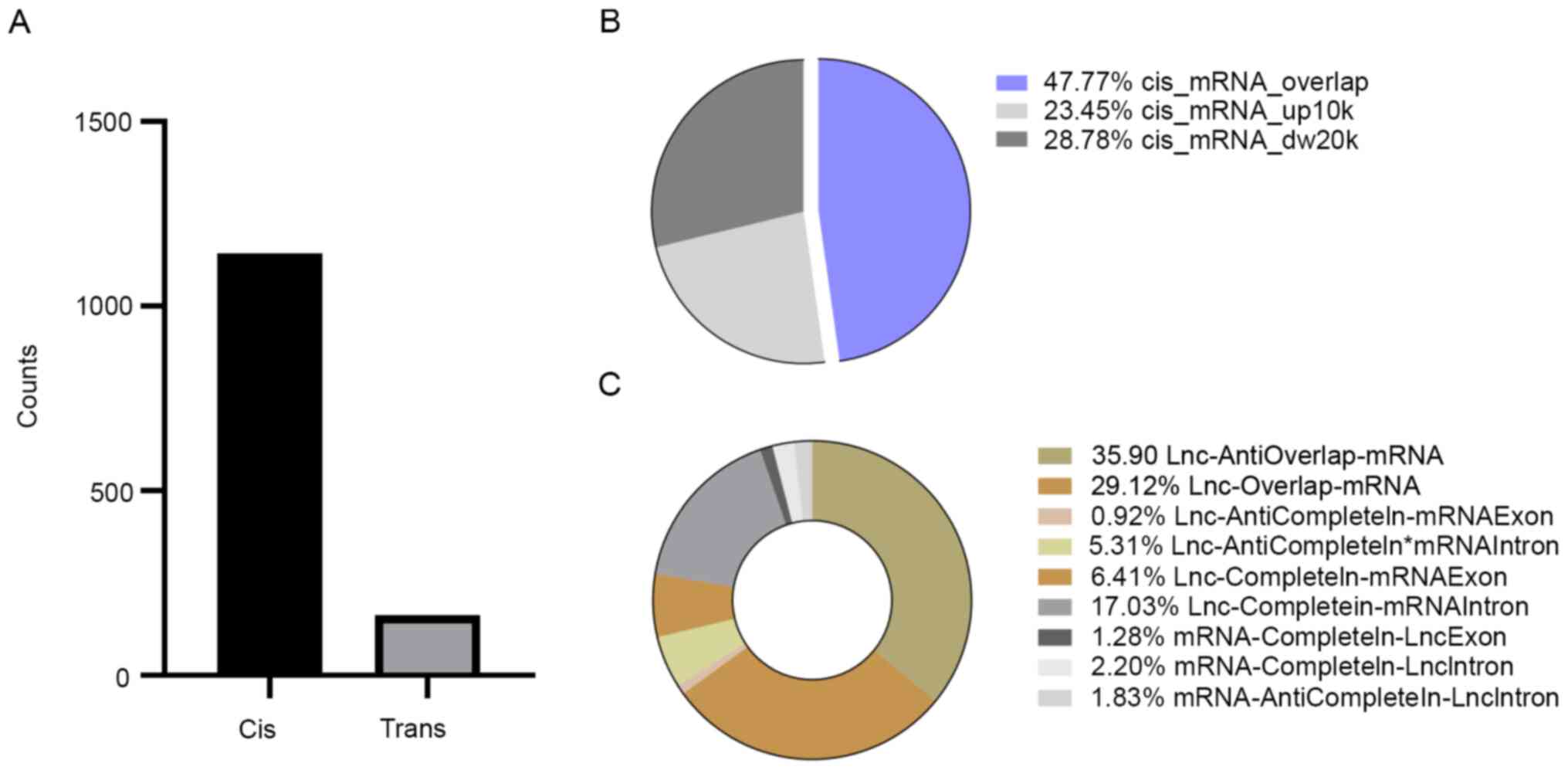
identified. Altogether, 1,143 cis-acting lncRNA-mRNA interactions
involving 883 DE mRNAs and 162 trans-acting lncRNA-mRNA
interactions involving 118 DE mRNAs were identified (Fig. 2A). Overall, the influence of
cis-acting of lncRNAs was discovered to be more important than
trans-acting, indicating that cis-acting lncRNAs may have a more
significant role in DS (P<0.01).
In cis-acting, most interactions were overlapping,
with 546 lncRNA-mRNA pairs identified; the remaining lncRNA-mRNA
pairs were mapped 10 kb upstream (268 pairs) and in 20 kb
downstream, with 329 lncRNA-mRNA pairs (Fig. 2B). It was revealed that >60% of
the overlapping interaction were accounted for by the
lncRNA-anti-overlap-mRNA (35.90%), lncRNA-overlap-mRNA (29.12%) and
lncRNA-completein-mRNA intron (17.03%; Fig. 2C), which may be an important part of
cis-acting in DS. The results indicated that cis-acting may not
necessarily rely on a promoter or enhancer but also on the gene
body.
Functional analysis of the DE lncRNAs
in DS
To gain further insight into the function of
identified lncRNAs, GO functional term enrichment and KEGG
signaling pathway enrichment analysis were used to understand the
biological functions and signaling pathways of the target genes of
each lncRNA module. The biological pathways of the targets and
their upstream/downstream relationship may help determine the
potential molecular mechanisms of lncRNAs and contribute to the
design experiments to further verify the pathogenesis of lncRNA in
DS (32). To further investigate
the potential function of DE lncRNAs, GO functional term enrichment
analysis and KEGG signaling pathway enrichment analysis were also
performed. A total of 397 DE cis-acting target genes were obtained
following the conversion of the transcript ID into the gene ID and
the removal of duplicates. To further reveal the biological pathway
information of the potential targets of these DE lncRNAs in DS,
those DE protein-coding genes were subjected to GO functional term
enrichment analysis. Representative GO terms significantly enriched
by DE mRNAs were identified based on a P-value (P≤0.01, manually
removing redundant functional terms). Functional analysis
demonstrated that cis target genes of lncRNAs were enriched in 183
GO terms, which were involved in nervous and muscle system
development (Fig. 3A). The
corresponding potential target genes were subjected to KEGG
signaling pathway enrichment analysis. The results revealed that
these DE target genes were able to be mapped to 23 signaling
pathways; among these signaling pathways, 15 significantly enriched
KEGG signaling pathways were determined (P<0.05; Fig. 3B), including the ‘Neurotrophin
signaling pathway’ and ‘MAPK signaling pathway’. These results
suggested that DE lncRNAs may act through their target genes to
regulate the nervous, muscle and immune systems during the early
development of DS. Therefore, these results indicated that lncRNAs
may function in cis mode on neighboring DS genes to influence
nervous and muscle development. To further validate the present
results, two lncRNA-mRNA pairs were selected for RT-qPCR
verification. Detailed lncRNA-mRNA information was provided in
Table SII. lncRNAs with a
significant differential expression [logFC ≥|2|; false discovery
rates (q) ≤0.001, (False discovery rates were calculated using the
q-value conversion algorithm)] and their target genes that were
enriched in neural and muscle biological pathways were selected for
verification. The DS phenotype may be used to associate enrichment
results with specific genes. Therefore, two mRNAs were selected:
SLIT-ROBO Rho GTPase activating protein 2C (SRGAP2C) and actin-α3
(ACTN3), which were enriched in the biological process term of
nerve and muscle development. Significant differences were
discovered in both the expression levels of the mRNAs and
corresponding lncRNAs (logFC ≥|2|, P≤0.01; Fig. 3C).
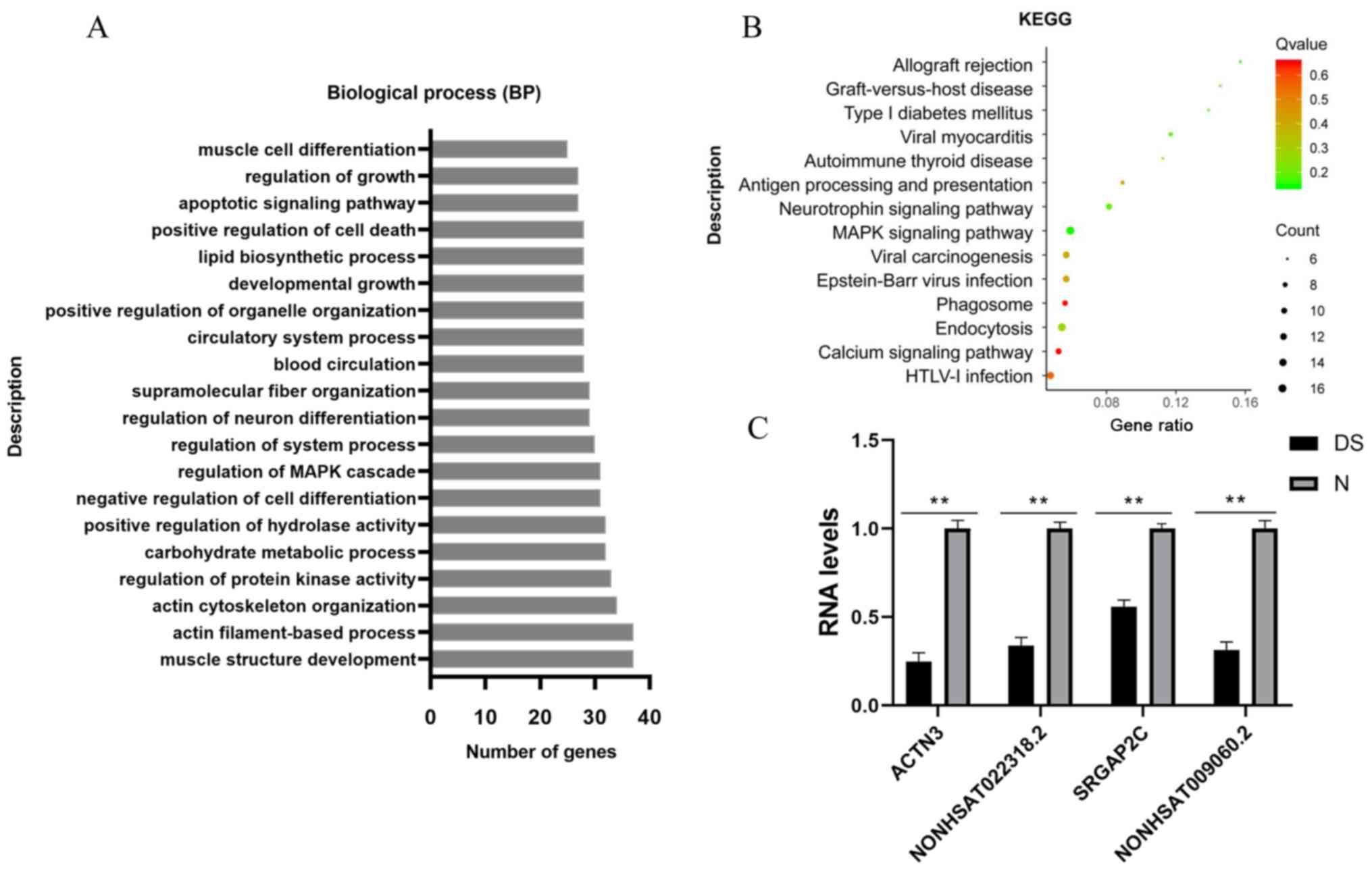 | Figure 3Target genes were screened by
enrichment and quantitative analysis. (A) Analysis of the top 20
overrepresented GO terms of lncRNAs, including GO:0061061, muscle
structure development; GO:0042692, muscle cell differentiation;
GO:0051961, negative regulation of nervous system development; and
GO:0045664, regulation of neuron differentiation. (B) KEGG
signaling pathway enrichment analysis of the corresponding cis
potential target genes of lncRNAs in DS. The enrichment factor was
calculated by the number of enriched genes and the P-value. (C)
Validation of the two pairs of lncRNA-mRNA using RT-qPCR. Relative
expression levels of selected mRNAs SRGAP2C and ACTN3 and lncRNAs
NONHSAT009060.2 and NONHSAT022318.2 in DS (n=3), as compared with
the controls (n=3). The results of the reverse RT-qPCR analysis
were consistent with the RNA-sequencing data. Bar graphs represent
the mean ± standard deviation. All reactions were repeated three
times for each mRNA or lncRNA. GAPDH was used as the internal
control. **P<0.01 vs. normal controls. BP, biological
process; GO, Gene Ontology; lncRNA, long non-coding RNA; KEGG,
Kyoto Encyclopedia of Genes and Genomes; DS, Down syndrome; N,
normal control; ACTN3, actin-α3; SRGAP2C, SLIT-ROBO Rho GTPase
activating protein 2C. |
Nuclear and cytoplasmic location of
the target lncRNAs
The subcellular localization of an lncRNA is closely
associated with its biological mechanism; thus, a correlation
analysis between the localization and expression levels of lncRNAs
in iPSCs was performed. In addition, nuclear fractionation analysis
was performed and the expression levels of NONHSAT009060.2 and
NONHSAT022318.2 in the nucleus and cytoplasm were analyzed. Nuclear
segregation was assayed using XIST, while the cytoplasmic
segregation was analyzed using GAPDH as the housekeeping gene. The
results clearly suggested that NONHSAT009060.2 and NONHSAT022318.2
were expressed in both the cytoplasm and nucleus, with
NONHSAT009060.2 exhibiting a moderate preference for the nucleus
(Fig. S1A and B). Since lncRNAs located in the nucleus
play a major role in transcriptional regulation and lncRNAs located
in the cytoplasm mainly play a role in post-transcriptional
regulation (Fig. S1), this
indicated that the expression assessment and identification of the
lncRNAs was valid. Since the subcellular localization of a lncRNA
is not dependent on the cell type (33), the model is applicable to all human
cells.
Discussion
DS is a complex syndrome mediated by numerous genes
(34). Epigenetic modifications,
such as DNA methylation and lncRNAs, are necessary mechanisms to
regulate gene expression levels. A diverse range of mechanisms have
been reported for the lncRNA-mediated epigenetic modulation of gene
expression. Previous studies have indicated that lncRNA may mainly
serve through cis- and trans-regulatory functions to regulate the
expression levels of target genes (35,36),
which is the major regulatory pattern of lncRNA in higher
organisms.
A previous study by our group indicated that lncRNA
expression profiles were significantly dysregulated in DS-iPSCs
(13). The present study also
showed that the differential expression levels of lncRNAs were more
significant than that of mRNAs, indicating that lncRNAs play an
important role in the regulation process; however, the exact manner
in which lncRNAs are involved in the pathogenesis of DS has
remained elusive. To further investigate the roles of epigenetic
alterations on biological processes and signaling pathways in DS, a
comprehensive analysis of lncRNA expression levels and potential
regulatory patterns in normal iPSCs and DS-iPSCs was performed. The
results revealed that the cis-acting role of lncRNAs had an
important impact in DS. The cis-acting pattern was classified
according to the positional relationship between lncRNAs and mRNAs.
The present classification results demonstrated that ~1/2 of the
lncRNAs-mRNAs involved in cis-acting belonged to lncRNAs with
overlapping mRNAs and the lncRNAs and mRNAs exhibited the highest
antisense overlap. Previous studies have also demonstrated that
lncRNAs regulated the coding gene expression levels through the
genomic position effect (37).
Antisense lncRNAs transcribed against and overlapping with the
protein-coding genes have been indicated to regulate their
protein-coding counterparts through multiple mechanisms (38). Several antisense lncRNAs have been
suggested to regulate the shear and degradation of mRNA through
complementary pairing with mRNA following transcription, affecting
the expression levels of mRNA (39,40).
The present results also validated the results of a previous study
indicating that antisense transcription may be far more extensive
than previously anticipated (41).
It should be noted that cis-acting antisense intronic RNAs
have a regulatory function (42),
which may stabilize protein-coding transcripts or regulate their
alternative splicing. The most abundant wholly intronic antisense
RNAs are transcribed from introns of genes related to the
regulation of transcription (43).
Cis-acting antisense intronic lncRNAs were also observed in the
present data, which provided a clue to their functional relevance.
This observation offers a novel avenue for future studies.
To precisely clarify the function of DE target
genes, GO term enrichment and KEGG signaling pathway enrichment
analysis was performed to investigate the potential biological
signaling pathways and functions involved. The results of the
present study revealed that the cis-acting targets of DE lncRNAs
were mainly enriched in the biological process term ‘nervous and
muscle growth and development’, which may be associated with the
mental impairment and low muscle tone of patients with DS. The
functions of lncRNAs have remained to be fully elucidated and the
present study provided comprehensive novel insight into the
regulatory patterns of mRNAs and lncRNAs in DS by analyzing
expression profiles.
The present study also provided a novel perspective
for pathological processes in DS, namely that cis-acting lncRNAs
have a significant role in controlling/regulating the expression
levels of the target genes and thus modulating the clinical
phenotype of DS. For instance, SRGAP2C was discovered to act
antagonistically on these proteins during the development of
cortical neurons, regulating the development of excitatory and
inhibitory synapses of the cortical pyramidal neurons in
vivo, protracting the maturation and increasing the density of
excitatory and inhibitory synapses and indirectly increasing
neuronal migration. SRGAP2C has also been implicated in cognition,
learning and memory (44). Nerve
disorders in patients with DS are currently among the most critical
research hotspots. The results of the present study suggested that
the abnormal expression levels of neurodevelopment-related genes
caused by dysregulated lncRNA expression may be an important cause
of nervous system damage occurring during the early developmental
stage. ACTN3 is primarily expressed in skeletal muscle and
functions as a structural component of the sarcomeric Z line, which
has been indicated to be associated with vertical craniofacial
skeletal patterns (45). In
addition, previous studies have reported that the lack of ACTN3 led
to decreased muscle strength (46).
It is well-known that craniofacial dysplasia frequently occurs in
patients with DS (47) and that the
downregulation of ACTN3 in the early developmental stage may be a
possible factor for the onset of these clinical symptoms in DS.
In conclusion, in the present study, bioinformatics
analysis was performed to preliminarily predict the functions of
lncRNAs and their interactions with mRNAs in DS. Further studies
should be performed to determine the interactions between lncRNAs
and the target genes mentioned above. Elucidation of the precise
transcriptional regulatory role of lncRNAs in DS may help
understand the pathogenesis of DS and promote the diagnosis and
development of novel therapeutics for this disease.
Supplementary Material
Nuclear to cytoplasmic ratio of target
genes. (A) In normal-iPSCs, the reverse RT-qPCR results
demonstrated that the expression levels of NONHSAT009060.2 in the
nucleus accounted for 78.90% of the total and those of
NONHSAT022318.2 accounted for 28.78%. (B) In DS-iPSCs, the results
were similar to those presented above. NONHSAT009060.2 was
indicated to be localized in the nucleus (73.16%), while only a
small fraction of NONHSAT022318.2 was expressed in the nucleus.
*P<0.05; **P<0.01. ns, no significance;
DS, Down syndrome; iPSCs, induced pluripotent stem cells; XIST,
X-inactive specific transcript.
Overlap classification
instructions.
Other information, such as the
position between lncRNA and mRNA.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by grants from the National Key
Research and Development Program of China (grant nos.
2019YFA0801402, 2016YFC1000503 and 2016YFC0905102), the National
Natural Science Foundation of China (grant nos. 81971421 and
81471485), Key Disciplines of Top Priority in Shanghai (grant no.
2017ZZ02019), Shanghai Key Clinical Specialty Project (grant no.
shslczdzk05705) and the Experimental Animals Project of Shanghai
Municipality (grant nos. 18140901600 and 18140901601).
Availability of data and materials
The sequencing data of all samples were deposited in
the Sequence Read Archive (SRA), under accession no. SRP289173. The
sequencing data set is comprised of 6 samples, three replicates for
each of the two groups: The normal control group and DS group,
including the following:
Normal control 1 (N1) https://www.ncbi.nlm.nih.gov/sra/SRX9382487
Normal control 2 (N2) https://www.ncbi.nlm.nih.gov/sra/SRX9382488
Normal control 3 (N3) https://www.ncbi.nlm.nih.gov/sra/SRX9382489
DS 1 (D1) https://www.ncbi.nlm.nih.gov/sra/SRX9382484
DS 2 (D2) https://www.ncbi.nlm.nih.gov/sra/SRX9382485
DS 3 (D3) https://www.ncbi.nlm.nih.gov/sra/SRX9382486
Authors' contributions
WM, as the first author, contributed to the
experimental design, drafting of the manuscript and the analysis
and interpretation of the data for the study. YL completed the cell
culture experiments. WM and HM performed RT-qPCR. ZR reviewed and
edited the manuscript. JY and ZR are the corresponding authors and
contributed to the experimental conception and design, the critical
revision of the manuscript and the final approval of the manuscript
to be published. WM and JY were responsible for checking and
approving the authenticity of the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Patterson D: Molecular genetic analysis of
Down syndrome. Hum Genet. 126:195–214. 2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Korenberg JR, Chen XN, Schipper R, Sun Z,
Gonsky R, Gerwehr S, Carpenter N, Daumer C, Dignan P, Disteche C,
et al: Down syndrome phenotypes: The consequences of chromosomal
imbalance. Proc Natl Acad Sci USA. 91:4997–5001. 1994.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Weijerman ME and de Winter JP: Clinical
practice. The care of children with Down syndrome. Eur J Pediatr.
169:1445–1452. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bilic J and Izpisua Belmonte JC: Concise
review: Induced pluripotent stem cells versus embryonic stem cells:
Close enough or yet too far apart? Stem Cells. 30:33–41.
2012.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Hoffmann A, Ziller M and Spengler D:
Progress in iPSC- based modeling of psychiatric disorders. Int J
Mol Sci. 20(4896)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bar-Nur O, Russ HA, Efrat S and Benvenisty
N: Epigenetic memory and preferential lineage-specific
differentiation in induced pluripotent stem cells derived from
human pancreatic islet beta cells. Cell Stem Cell. 9:17–23.
2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan
P, Kim J, Aryee MJ, Ji H, Ehrlich LI, et al: Epigenetic memory in
induced pluripotent stem cells. Nature. 467:285–290.
2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Harries LW: Long non-coding RNAs and human
disease. Biochem Soc Trans. 40:902–906. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ng SY, Lin L, Soh BS and Stanton LW: Long
noncoding RNAs in development and disease of the central nervous
system. Trends Genet. 29:461–468. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Emmrich S, Streltsov A, Schmidt F,
Thangapandi VR, Reinhardt D and Klusmann JH: LincRNAs MONC and
MIR100HG act as oncogenes in acute megakaryoblastic leukemia. Mol
Cancer. 13(171)2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Faghihi MA, Modarresi F, Khalil AM, Wood
DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G III, Kenny PJ,
Wahlestedt C, et al: Expression of a noncoding RNA is elevated in
Alzheimer's disease and drives rapid feed-forward regulation of
beta-secretase. Nat Med. 14:723–730. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Qiu JJ, Liu YN, Ren ZR and Yan JB:
Dysfunctions of mitochondria in close association with strong
perturbation of long noncoding RNAs expression in down syndrome.
Int J Biochem Cell Biol. 92:115–120. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Rinn JL and Chang HY: Genome regulation by
long noncoding RNAs. Annu Rev Biochem. 81:145–166. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Quinn JJ and Chang HY: Unique features of
long non-coding RNA biogenesis and function. Nat Rev Genet.
17:47–62. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kopp F and Mendell JT: Functional
classification and experimental dissection of long noncoding RNAs.
Cell. 172:393–407. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Pei S, Minhajuddin M, Adane B, Khan N,
Stevens BM, Mack SC, Lai S, Rich JN, Inguva A, Shannon KM, et al:
AMPK/FIS1-mediated mitophagy is required for self-renewal of human
AML stem cells. Cell Stem Cell. 23:86–100.e6. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kim D, Langmead B and Salzberg SL: HISAT:
A fast spliced aligner with low memory requirements. Nat Methods.
12:357–360. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Pertea M, Pertea GM, Antonescu CM, Chang
TC, Mendell JT and Salzberg SL: StringTie enables improved
reconstruction of a transcriptome from RNA-seq reads. Nat
Biotechnol. 33:290–295. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Trapnell C, Williams BA, Pertea G,
Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ and Pachter
L: Transcript assembly and quantification by RNA-Seq reveals
unannotated transcripts and isoform switching during cell
differentiation. Nat Biotechnol. 28:511–515. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ,
Wei L and Gao G: CPC: Assess the protein-coding potential of
transcripts using sequence features and support vector machine.
Nucleic Acids Res. 35:W345–W349. 2007.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and cufflinks. Nat Protoc. 7:562–578.
2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Langmead B and Salzberg SL: Fast
gapped-read alignment with Bowtie 2. Nat Methods. 9:357–359.
2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Li B and Dewey CN: RSEM: Accurate
transcript quantification from RNA-Seq data with or without a
reference genome. BMC Bioinformatics. 12(323)2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang L, Feng Z, Wang X, Wang X and Zhang
X: DEGseq: An R package for identifying differentially expressed
genes from RNA-seq data. Bioinformatics. 26:136–138.
2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Narzisi G, O'Rawe JA, Iossifov I, Fang H,
Lee YH, Wang Z, Wu Y, Lyon GJ, Wigler M and Schatz MC: Accurate de
novo and transmitted indel detection in exome-capture data using
microassembly. Nat Methods. 11:1033–1036. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Tafer H, Amman F, Eggenhofer F, Stadler PF
and Hofacker IL: Fast accessibility-based prediction of RNA-RNA
interactions. Bioinformatics. 27:1934–1940. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Knauss JL and Sun T: Regulatory mechanisms
of long noncoding RNAs in vertebrate central nervous system
development and function. Neuroscience. 235:200–214.
2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kornienko AE, Guenzl PM, Barlow DP and
Pauler FM: Gene regulation by the act of long non-coding RNA
transcription. BMC Biol. 11(59)2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Brown CJ, Hendrich BD, Rupert JL,
Lafrenière RG, Xing Y, Lawrence J and Willard HF: The human XIST
gene: Analysis of a 17 kb inactive X-specific RNA that contains
conserved repeats and is highly localized within the nucleus. Cell.
71:527–542. 1992.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhou Q, Wan Q, Jiang Y, Liu J, Qiang L and
Sun L: A landscape of murine long non-coding RNAs reveals the
leading transcriptome alterations in adipose tissue during aging.
Cell Rep. 31(107694)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gudenas BL and Wang L: Prediction of
LncRNA subcellular localization with deep learning from sequence
features. Sci Rep. 8(16385)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Letourneau A, Santoni FA, Bonilla X,
Sailani MR, Gonzalez D, Kind J, Chevalier C, Thurman R, Sandstrom
RS, Hibaoui Y, et al: Domains of genome-wide gene expression
dysregulation in Down's syndrome. Nature. 508:345–350.
2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Luo S, Lu JY, Liu L, Yin Y, Chen C, Han X,
Wu B, Xu R, Liu W, Yan P, et al: Divergent lncRNAs regulate gene
expression and lineage differentiation in pluripotent cells. Cell
Stem Cell. 18:637–652. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S,
Jiang Z, Xu J, Liu Q and Cao X: The STAT3-binding long noncoding
RNA lnc-DC controls human dendritic cell differentiation. Science.
344:310–313. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ørom UA, Derrien T, Beringer M, Gumireddy
K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q,
et al: Long noncoding RNAs with enhancer-like function in human
cells. Cell. 143:46–58. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Magistri M, Faghihi MA, St Laurent G III
and Wahlestedt C: Regulation of chromatin structure by long
noncoding RNAs: Focus on natural antisense transcripts. Trends
Genet. 28:389–396. 2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Paralkar VR, Taborda CC, Huang P, Yao Y,
Kossenkov AV, Prasad R, Luan J, Davies JOJ, Hughes JR, Hardison RC,
et al: Unlinking an lncRNA from its associated cis element. Mol
Cell. 62:104–110. 2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Engreitz JM, Haines JE, Perez EM, Munson
G, Chen J, Kane M, McDonel PE, Guttman M and Lander ES: Local
regulation of gene expression by lncRNA promoters, transcription
and splicing. Nature. 539:452–455. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Katayama S, Tomaru Y, Kasukawa T, Waki K,
Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, et
al: Antisense transcription in the mammalian transcriptome.
Science. 309:1564–1566. 2005.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yan MD, Hong CC, Lai GM, Cheng AL, Lin YW
and Chuang SE: Identification and characterization of a novel gene
Saf transcribed from the opposite strand of Fas. Hum Mol Genet.
14:1465–1474. 2005.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Nakaya HI, Amaral PP, Louro R, Lopes A,
Fachel AA, Moreira YB, El-Jundi TA, da Silva AM, Reis EM and
Verjovski-Almeida S: Genome mapping and expression analyses of
human intronic noncoding RNAs reveal tissue-specific patterns and
enrichment in genes related to regulation of transcription. Genome
Biol. 8(R43)2007.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Fossati M, Pizzarelli R, Schmidt ER,
Kupferman JV, Stroebel D, Polleux F and Charrier C: SRGAP2 and its
human-specific paralog co-regulate the development of excitatory
and inhibitory synapses. Neuron. 91:356–369. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Cunha A, Nelson-Filho P, Marañón-Vásquez
GA, Ramos AGC, Dantas B, Sebastiani AM, Silvério F, Omori MA,
Rodrigues AS, Teixeira EC, et al: Genetic variants in ACTN3 and
MYO1H are associated with sagittal and vertical craniofacial
skeletal patterns. Arch Oral Biol. 97:85–90. 2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Clarkson PM, Devaney JM, Dressman HG,
Thompson PD, Hubal MJ, Urso M, Price TB, Angelopoulos TJ, Gordon
PM, Moyna NM, et al: ACTN3 genotype is associated with increases in
muscle strength in response to resistance training in women. J Appl
Physiol (1985). 99:154–163. 2005.PubMed/NCBI View Article : Google Scholar
|
|
47
|
van Marrewijk DJ, van Stiphout MA,
Reuland-Bosma W, Bronkhorst EM and Ongkosuwito EM: The relationship
between craniofacial development and hypodontia in patients with
Down syndrome. Eur J Orthod. 38:178–183. 2016.PubMed/NCBI View Article : Google Scholar
|