Introduction
Globally, myocardial infarction (MI) is a leading
cause of morbidity and mortality, and causes progressive
deterioration that ultimately results in heart failure (1). Cardiac fibrosis is a primary event in
MI progression, and is characterized by the transformation of
fibroblasts into myofibroblasts and the production of excessive
extracellular matrix proteins, including collagen I and III, within
the myocardium (2). During the
chronic stages of MI, abnormal cardiac fibrosis inevitably causes
the excessive production of extracellular matrix proteins and
decline in cardiac function (2,3).
Therefore, novel strategies are required to inhibit cardiac
fibrosis to improve heart function in patients with MI.
Sodium-glucose linked transporter (SGLT) 1 and SGLT2
are primary SGLTs that contribute to the reabsorption of
kidney-filtered glucose (4,5). A previous study demonstrated that
SGLT1 was involved in cardioprotection against ischemia-reperfusion
injury (6). By contrast, SGLT2
inhibitors have been reported to decrease blood glucose
independently and reduce the risk of severe heart failure (7). Furtado et al (8) demonstrated that dapagliflozin, an
SGLT2 inhibitor, markedly reduced the risk of both major adverse
cardiovascular events and cardiovascular death/hospitalization for
heart failure in patients with type 2 diabetes mellitus and
previous MI. Moreover, to the best of our knowledge, dapagliflozin
is the only SGLT-2 inhibitor that reduces cardiac necrosis and the
worsening of heart failure (8,9).
Empagliflozin, an SGLT-2 inhibitor, has been studied in a clinical
trial and the results demonstrated reduced cardiovascular mortality
of patients with type 2 diabetes mellitus (10). Furthermore, SGLT2 inhibition with
empagliflozin effectively improved cardiac diastolic function in a
female rodent model of diabetes (11). Ye et al (12) demonstrated that the inhibition of
SGLT-2 reduced NLR family pyrin domain containing 3
(Nlrp3)/apoptosis-associated speck-like protein (ASC) inflammasome
activation and attenuated the development of diabetic
cardiomyopathy in mice. Additionally, dapagliflozin, a selective
SGLT2 inhibitor, served a protective role in cardiac fibrosis in
infarcted rat hearts (11). The
aforementioned previous studies demonstrated that SGLT2 served a
potential role in the pathogenesis of heart disease. However, the
biological function of SGLT2 in cardiac fibrosis is not completely
understood.
MicroRNAs (miRNAs/miRs) have been reported to be
involved in the regulation of cardiac fibrosis. miR-21, miR-34,
miR-199b and miR-208 have been identified to contribute to
myocardial fibrosis and are upregulated in MI (13). By contrast, miR-1, miR-29, miR-133a
and miR-214 are antifibrotic miRNAs (13). However, the mechanism underlying
miRNA-mediated regulation of cardiac fibrosis in MI is not
completely understood.
The present study aimed to investigate the role of
SGLT2 in cardiac fibrosis following MI. Moreover, whether
upregulated SGLT2 levels in cardiac fibrosis following MI are
regulated in a miRNA dependent manner was also investigated.
Materials and methods
Animals, MI model and assessment of
heart function
A total of 65, six to eight-week male Sprague-Dawley
rats (weight, 200-300 g) were purchased from the Academy of
Military Medical Sciences. The rats were kept in a
temperature-controlled room, with a humidity of 40-70%, in a 12 h
light-dark cycle with free access to standard chow and tap water.
All animals were reared in a specific pathogen-free environment at
a comfortable temperature and humidity. All experimental procedures
were approved by the Animal Ethics Committee of the Second
Affiliated Hospital of Wannan Medical College, Wuhu, China
(approval no. DWL-1804-007).
MI was modeled in rats via the permanent ligation of
the left anterior descending branch of the coronary artery with
prolene sutures, as previously described (14). Briefly, rats were anesthetized
intraperitoneally with 40 mg/kg sodium pentobarbital. The thoracic
cavities were opened and the left anterior descending (LAD)
coronary arteries were permanently ligated with a 7-0 polypropylene
suture. In the sham operation group, animals underwent the same
procedure, except the LAD was left untied. Following euthanasia by
anesthetic overload with intraperitoneal 90 mg/kg ketamine and 10
mg/kg xylazine, the infarct zones and far zones of the hearts were
quickly excised for the detection of RNA, protein and fibrosis
levels. The SGLT2 and miR-141 expression was measured at infarct
zones at 1, 2, 3 and 4 weeks post-MI. To evaluate the effect of
SGLT2 on MI in vivo, lentiviruses containing sh-SGLT2 or
sh-NC were obtained from Shanghai GenePharma Co., Ltd. Animals were
divided into the following four groups: i) sham (n=5); ii) MI
(n=5); iii) MI + short hairpin RNA (sh)-negative control (NC, n=5);
and iv) MI + sh-SGLT2 (n=5). Following LAD ligation, 108
PFU of sh-SGLT2, sh-NC or PBS (100 µl) was intramyocardially
injected into the corresponding groups.
Echocardiography was performed to determine cardiac
function at 4 weeks post-MI using a Vevo 2100 system (VisualSonics,
Inc.) with an 80 MHz probe. Left ventricular parameters were
recorded from two-dimensional images using the M-mode interrogation
in the short-axis view.
Masson trichrome staining
At 4 weeks post-MI, heart sections from MI model
rats were excised from an area perpendicular to the axis of the LAD
coronary arteries. Briefly, the tissues were fixed in 4% of
paraformaldehyde for 24 h at room temperature, embedded in paraffin
and 5 µm sections were taken. Sections were then stained using a
Masson's Trichrome stain kit (cat. no. 1004850001; Sigma-Aldrich;
Merck KGaA,) according to the manufacturer's protocol. All of the
images (magnification, x12.5) were captured on a confocal
microscope (Nikon Corporation). Average ratios of the fibrotic
areas to the entire left ventricular cross-sectional area were
analyzed using ImageJ 1.48u software (National Institutes of
Health).
Primary cardiac fibroblasts isolation
and culture
Cardiac fibroblasts were isolated as previously
described (15). Briefly, primary
cardiac fibroblasts were isolated from 1-3 day-old female
Sprague-Dawley rats (n=3). The female neonatal rats used in this
experiment were bred by ourselves, which were co-housed with the
maternal rat since birth. Their parents housing conditions are
aforementioned. Cardiac fibroblasts were cultured in DMEM (Gibco;
Thermo Fisher Scientific, Inc.) containing 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin (Sigma-Aldrich, Merck KGaA) at 37˚C with 5%
CO2. Myocardial fibrosis phenotypes were induced using 20
ng/ml recombinant TGF-β (PeproTech, Inc.) at 37˚C for 72 h, as
previously described (16).
Cell infection and transfection
Lentiviruses containing sh-SGLT2 or sh-NC were
obtained from Shanghai GenePharma Co., Ltd. For lentiviral
infection, primary cardiac fibroblasts were incubated with sh-SGLT2
or sh-NC at an optimal multiplicity of infection of 15 at 37˚C for
12 h. Subsequently, the medium was removed and cells were incubated
in complete medium (DMEM containing 10% FBS, 100 U/ml penicillin
and 100 µg/ml streptomycin) at 37˚C for 72 h. Infection efficiency
was assessed via reverse transcription-quantitative PCR and western
blotting. miR-141 mimics (miR-141 mimics:
5'-UAACACUGUCUGGUAAAGAUGG-3') and scrambled non-coding RNAs
(miR-141 NC: 5'-ACGUGACACGUUCGGAGAATT-3') were purchased from
Ambion (Thermo Fisher Scientific, Inc.). pcDNA3.1 vectors
containing full-length SGLT2 cDNA sequences (pcDNA3.1-SGLT2) were
purchased from Shanghai GenePharma Co., Ltd. Primary cardiac
fibroblasts (3x105) were transfected with 20 pmol
miR-141 mimics, 20 pmol miR-141 NC, 1 µg pcDNA3.1-SGLT2 or 1 µg
empty control vector using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) in serum-free medium.
Control experiments were performed with mock-transfected cells
using the same procedure. After 8 h of transfection at 37˚C, all of
the transfected and mock-transfected primary cardiac fibroblasts
were transferred to DMEM with 10% FBS for an additional 24 h and
incubated at 37˚C. Following this incubation, the subsequent
experiments were conducted.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cardiac tissues and
primary cardiac fibroblasts using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). Total RNA was reverse
transcribed into cDNA using a PrimeScript RT kit (Takara Bio,
Inc.). The temperature protocol used for RT was 37˚C for 15 min and
85˚C for 15 sec. The samples were then kept at 4˚C for immediate
use or -20˚C for long term storage. Subsequently, qPCR was carried
out using the Power SYBR GREEN PCR master mix (Takara Bio, Inc.)
with ABI7300 detector (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The reaction parameters of standard procedure
for two-step amplification were as follows: 95˚C for 10 min, and 40
cycles at 95˚C for 15 sec and 60˚C for 30 sec. The following primer
sequences were used: SGLT2 forward, 5'-GCAGAAGGTCCTGATTGATA-3' and
reverse, 5'-GCGATGACAGAAGCGTAAA-3'; collagen I forward,
5'-CGAGTATGGAAGCGAAGGT-3' and reverse, 5'-CCACAAGCGTGCTGTAGGT-3';
collagen III forward, 5'-CCACCCTGAACTCAAGAGC-3' and reverse,
5'-TGAACTGAAAGCCACCATT-3'; and β-actin forward,
5'-GTAAAGACCTCTATGCCAACA-3' and reverse,
5'-GGACTCATCGTACTCCTGCT-3'; U6 forward, 5'-CTCGCTTCGGCAGCACA-3';
and reverse, 5'-AACGCTTCACGAATTTGCGT-3'. Primers were provided by
Ambion (Thermo Fisher Scientific, Inc.). miR-141 levels were
normalized to U6. mRNA and miRNA expression levels were quantified
using the 2-ΔΔCq method (17) and normalized to the internal
reference genes β-actin and U6, respectively.
Bioinformatics
miRNA binding sites were predicted using TargetScan
(v7.0; targetscan.org) (18-20).
Western blotting
Total protein was extracted from primary cardiac
fibroblasts and cardiac tissues using Laemmli sample buffer
(Bio-Rad Laboratories, Inc.). Protein concentrations were
determined using a BCA protein assay kit (Thermo Fisher Scientific,
Inc.). Proteins (30 µg) were separated using a 10% SDS-PAGE gel and
transferred to PVDF membranes (Roche Diagnostics), which were
blocked with 5% non-fat milk at 25˚C for 1 h. Subsequently, the
membranes were incubated overnight at 4˚C with the following
primary antibodies: Anti-SGLT2 (cat. no. ab37296; 1:1,000, Abcam),
anti-collagen I (cat. no. ab34710; 1:1,000, Abcam), anti-collagen
III (cat. no. ab7778; 1:1,000, Abcam) and anti-β-actin (cat. no.
ab8227; 1:1,000, Abcam). The secondary antibody (HRP-conjugated;
cat. no. ab7090; Abcam) was diluted to a ratio of 1:5,000 and
incubated for 1 h at 25˚C. Protein bands were visualized using
enhanced chemiluminescence (Thermo Fisher Scientific, Inc.).
β-actin was used as the loading control.
Cell viability assay
An MTT assay was performed to assess cell viability,
as previously described (21).
Briefly, primary cardiac fibroblasts (5x103) were seeded
into 96-well plates and treated with designated treatments. After
48 h, 15 µl MTT solution (Sigma-Aldrich; Merck KGaA) was added to
each well and incubated at 37˚C for 4 h. Subsequently, 200 µl DMSO
was added into each well to dissolve the formazan. Absorbance was
measured at a wavelength of 490 nm using a microplate reader (Tecan
Austria GmbH).
Luciferase reporter assay
SGLT2 3'-untranslated region (UTR) sequences were
inserted into pmiR-RB-REPORT vectors (Guangzhou Ribobio Co., Ltd.).
SGLT2 3'-UTR-mutants were generated in which 6 complementary
binding site nucleotides were altered. 293T cells (4x105
cells) were co-transfected with SGLT23'-UTR-wild-type (1 µg) or
SGLT2 3'-UTR-mutant (1 µg) and miR-141 mimics (40 pmol) or miR-141
NC (40 pmol) using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.). After 48 h, transfected cells were
collected and luciferase activity was detected using a Luciferase
Reporter kit (Promega Corporation). Firefly luciferase activity was
normalized to Renilla luciferase activity.
Statistical analysis
Statistical analysis was carried out using GraphPad
Prism software (version 5.01; GraphPad Software, Inc.). Data are
presented as the mean ± SD. All experiments were performed at least
in triplicate. Comparisons among groups were analyzed using a
one-way ANOVA and Tukey's post hoc tests with GraphPad Prism
software version 7.0 (GraphPad Software, Inc.). Comparisons between
two groups were analyzed using an unpaired Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
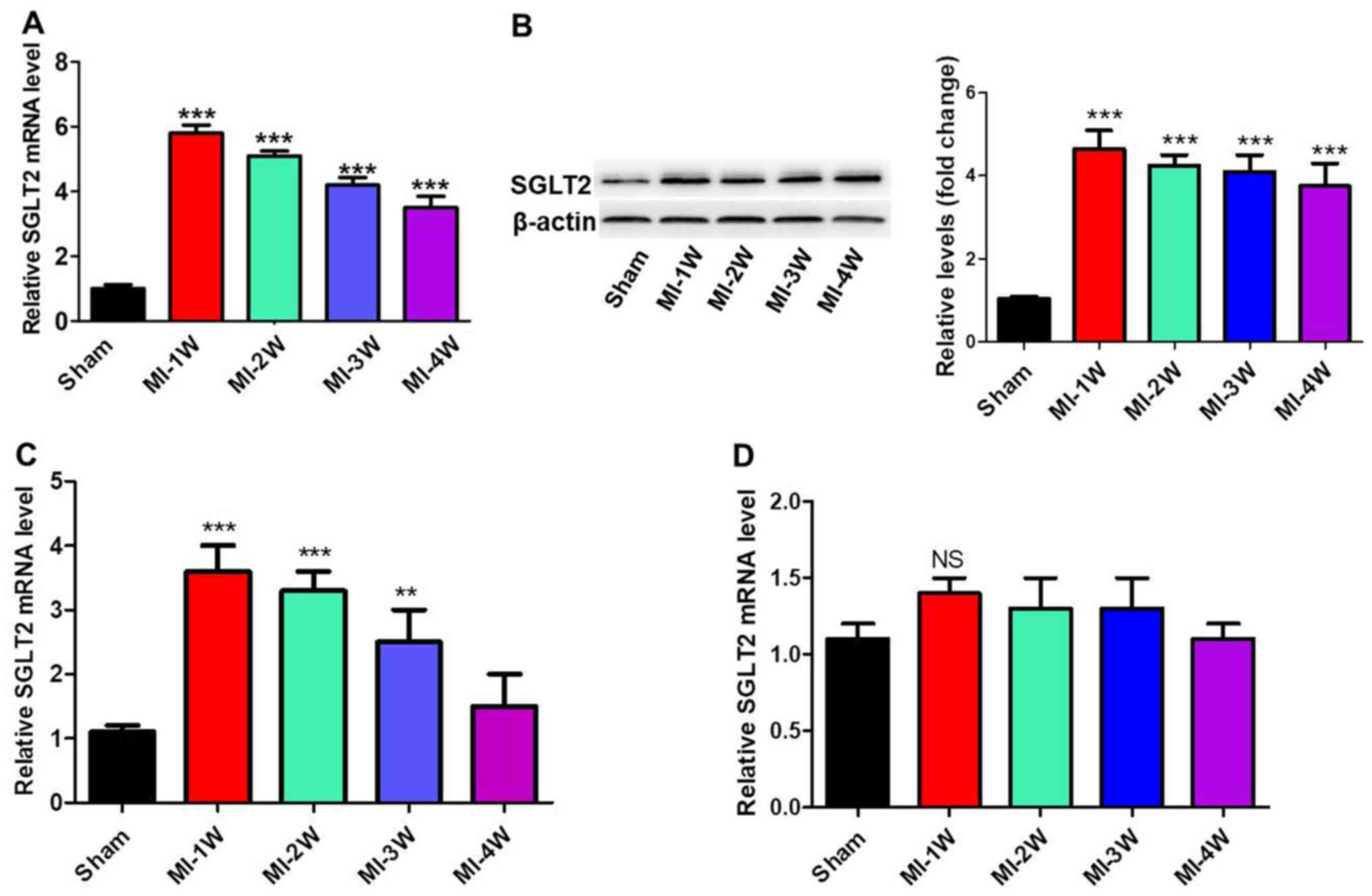
SGLT2 expression is increased in the
infarct myocardium post-MI in rats
To investigate the role of SGLT2 in the regulation
of cardiac fibrosis in vivo, MI rat models were established.
mRNA and protein expression levels of SGLT2 in cardiac tissues at
infarct zones at 1, 2, 3 and 4 weeks post-MI were analyzed via
RT-qPCR and western blotting (Fig.
1A and B). SGLT2 expression
levels in MI model rats were significantly increased at week 1
post-MI compared with sham rats, with levels gradually decreasing
with time. Furthermore, SGLT2 mRNA expression levels in the border
and far zones, the zone of heart ventricular near the atril area,
were analyzed. SGLT2 expression levels in the border zone (Fig. 1C) were similar to the expression
levels in the infarct zone. In contrast, SGLT2 expression levels
were not changed in the far zones of infarcts at 1, 2, 3 and 4
weeks post-MI (Fig. 1D). The
results indicated that SGLT2 served an important role in MI.
SGLT2 knockdown improves cardiac
function following MI
MI model rats were treated with sh-SGLT2 or sh-NC
lentiviruses to demonstrate the effect of SGLT2 on rat cardiac
function following MI. sh-SGLT2 significantly reduced SGLT2
expression levels in MI tissues compared with the sh-NC group
(Fig. 2A and B). Echocardiography was performed to
evaluate left ventricular function in the different groups
(Fig. 2C). Left ventricular mass
indices, including left ventricular ejection fractions (Fig. 2D), left ventricular end diastolic
volume (Fig. 2E), left ventricular
end systolic volume (Fig. 2F),
maximum left ventricular change in pressure over time [(-)/(+) left
ventricular diastolic pressure/dt maximum; Fig. 2G and H], heart rate (Fig. 2I) and E/A (ratio of the peak early
transmitral flow velocity to peak late transmitral flow velocity;
Fig. 2J) were analyzed to determine
cardiac function. The results indicated that sh-SGLT2 enhanced
heart function following MI.
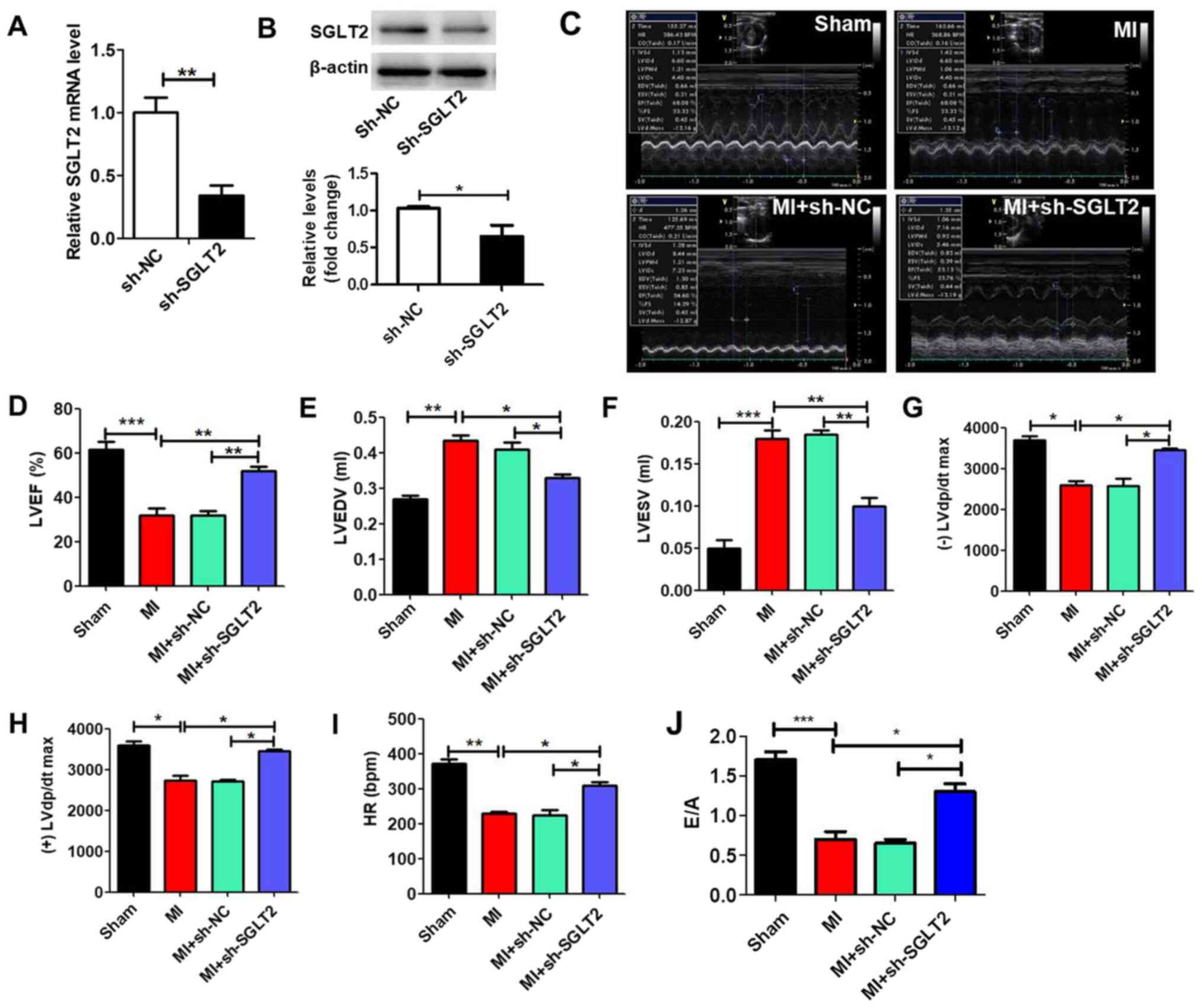 | Figure 2SGLT2 knockdown improves rat heart
function following MI in vivo. SGLT2 (A) mRNA and (B)
protein expression levels in infarcted areas treated with sh-SGLT2
or sh-NC (n=5). (C) Echocardiography results of rats in the
different groups (n=5). At 4 weeks post-MI, ventricular parameters
were measured and analyzed by echocardiography, including (D) LVEF,
(E) LVEDV, (F) LVESV, (G) (-) LVdp/dtmax, (H) (+) LVdp/dtmax, (I)
HR and (J) E/A ratio (n=5). *P<0.05,
**P<0.01, ***P<0.001. SGLT2,
sodium-glucose linked transporter 2; MI, myocardial infarction; sh,
short hairpin RNA; NC, negative control; LVEF, left ventricular
ejection fractions; LVEDV, left ventricular end diastolic volume;
LVESV, left ventricular end systolic volume; (-)/(+) LVdp/dtmax,
the maximum left ventricular change in pressure/time; HR, heart
rate; E/A ratio, ratio of the peak early transmitral flow velocity
to peak late transmitral flow velocity. |
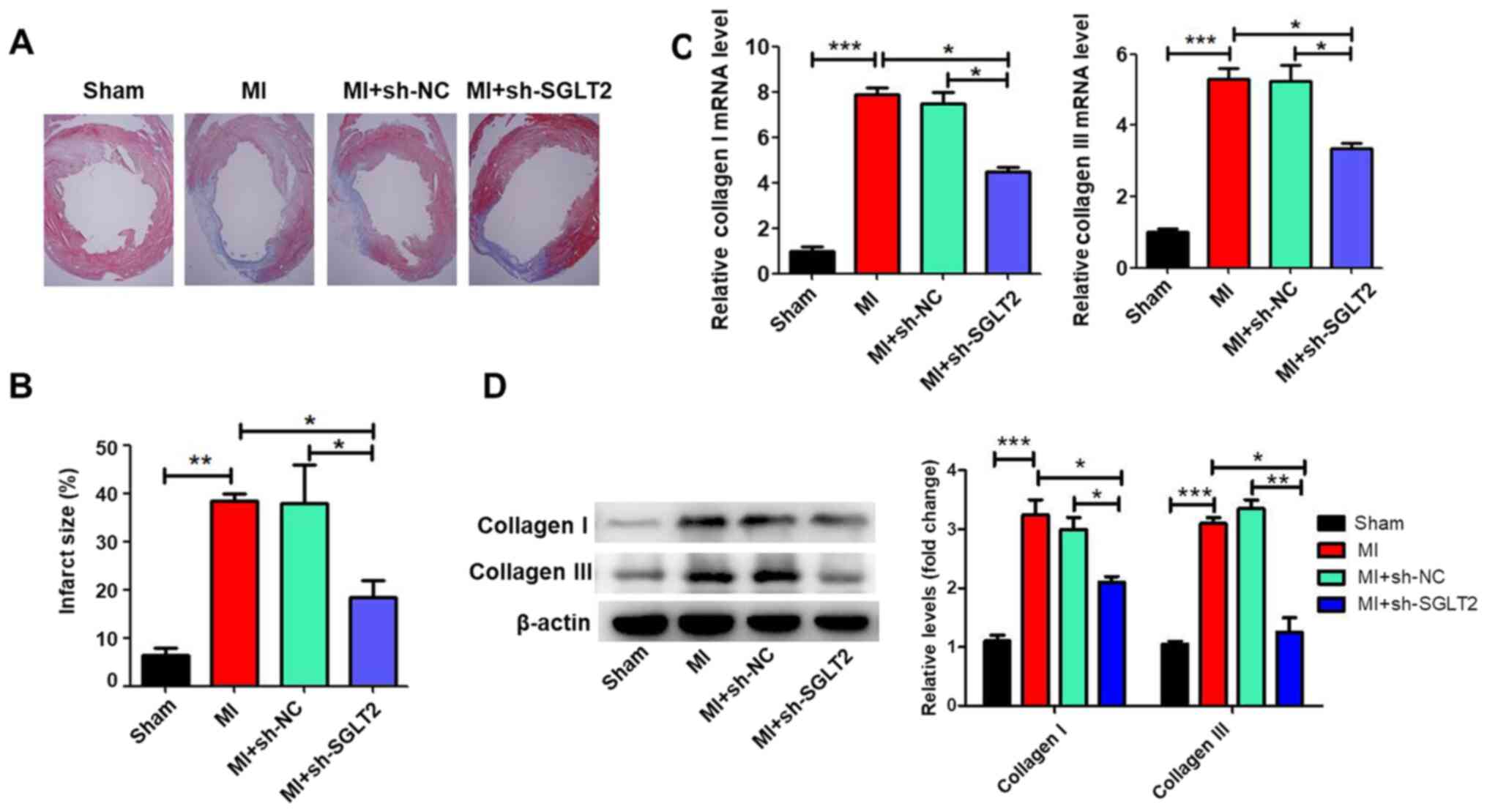
SGLT2 knockdown attenuates cardiac
fibrosis following MI
Masson's trichrome staining was performed to assess
the effects of SGLT2 on cardiac fibrosis of infarcted hearts.
Treatment with sh-SGLT2 decreased the fibrotic region (the blue
region) and significantly decreased the infarct size compared with
the MI or sh-NC-treated control groups (Fig. 3A and B). Compared with the sham group, MI
significantly increased the expression of collagen I and collagen
III (Fig. 3C and D), which was reversed by sh-SGLT2.
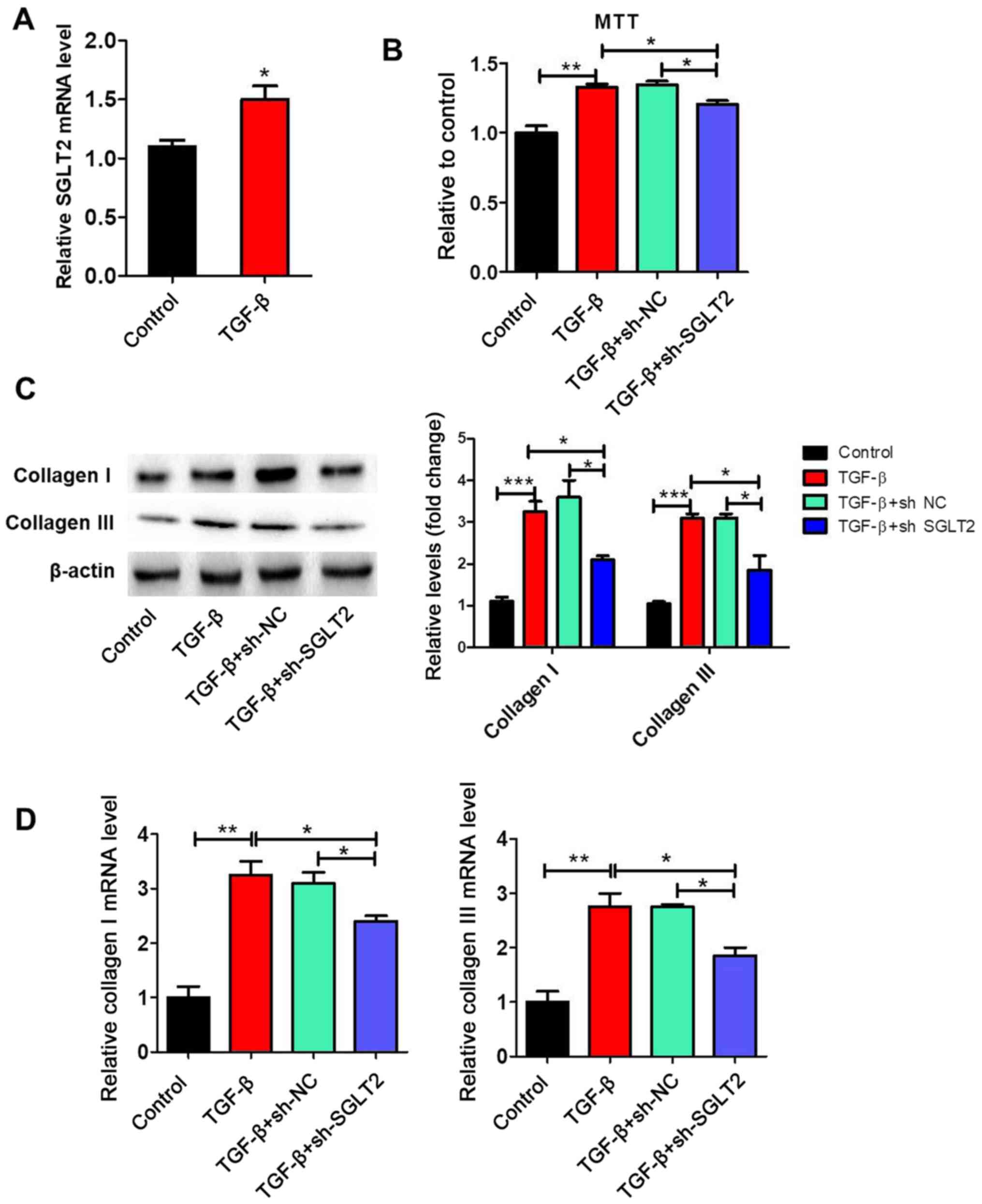
SGLT2 knockdown inhibits TGFβ-induced
proliferation and collagen synthesis in cardiac fibroblasts
SGLT2 expression levels were significantly increased
by TGFβ treatment compared with the control group (Fig. 4A). To further clarify the effect of
SGLT2 on cardiac fibroblasts proliferation, primary cardiac
fibroblasts were stimulated with TGFβ in vitro. The MTT
assay indicated that TGFβ significantly increased proliferation by
>1.3 fold compared with the control group (Fig. 4B). TGFβ-induced proliferation was
significantly decreased by sh-SGLT2, but not by sh-NC. The protein
and mRNA expression levels of collagen I and collagen III in
cardiac fibroblasts were assessed via western blotting and RT-qPCR,
respectively. The protein (Fig. 4C)
and mRNA (Fig. 4D) expression
levels of collagen I and collagen III were significantly
upregulated in the TGFβ group compared with the control group.
Furthermore, TGFβ-induced collagen I and collagen III expression
was significantly reversed by sh-SGLT2.
SGLT2 is targeted by miR-141
To identify why SGLT2 exhibited aberrant expression
in the cardiac tissue infarct zones following MI, the present study
hypothesized that dysregulated miRNAs may regulate SGLT2 expression
during MI. A previous study reported that miR-141 was decreased in
diabetic mice myocardium and cardiac fibroblasts treated with
angiotensin II (22). TargetScan
(v7.0; targetscan.org) was used to predict
miR-141 binding sites in the 3'-UTR of SGLT2 (Fig. 5A). miR-141 expression levels in MI
model rats were significantly decreased at 1 week post-MI compared
with sham rats, but gradually increased with time (Fig. 5B). The results of the luciferase
assay demonstrated that miR-141 overexpression significantly
decreased the luciferase activity of the wild-type 3'-UTR compared
with miR-141 NC. By contrast, miR-141 overexpression did not
significantly alter the luciferase activity of mutant 3'UTR
compared with miR-141 NC (Fig. 5C).
Treatment with miR-141 mimics significantly increased miR-141
expression levels compared with miR-141 NC (Fig. 5D). Moreover, miR-141 overexpression
significantly inhibited the mRNA (Fig.
5E) and protein (Fig. 5F)
expression levels of SGLT2 compared with miR-141 NC.
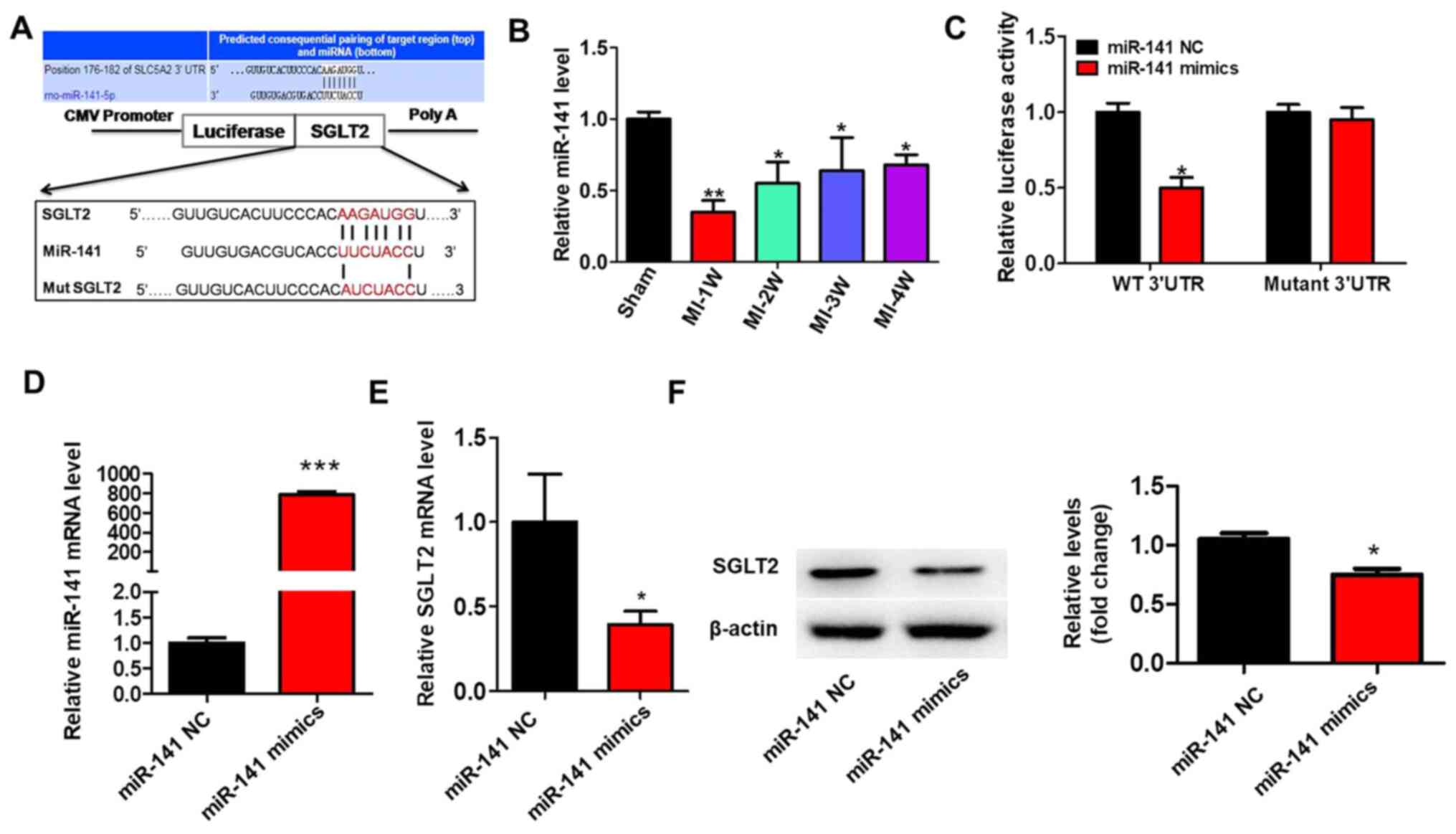 | Figure 5SGLT2 is targeted by miR-141. (A) The
binding site between the 3'-UTR of SGLT2 mRNA and miR-141. SGLT2
3'-UTR-mutants were generated in which 6 complementary binding site
nucleotides were altered. (B) miR-141 expression levels in the
infarcted areas of MI model rats at indicated times. (C) Relative
luciferase activities were determined by conducting a luciferase
reporter assay. (D) Transfection efficiency of miR-141 mimics.
SGLT2 (E) protein and (F) mRNA expression levels in primary cardiac
fibroblasts transfected with miR-141 mimics. *P<0.05,
**P<0.01, ***P<0.001. SGLT2,
sodium-glucose linked transporter 2; miR, microRNA; UTR,
untranslated region; miR, microRNA; W, week; WT, wild-type; MUT,
mutant; NC, negative control. |
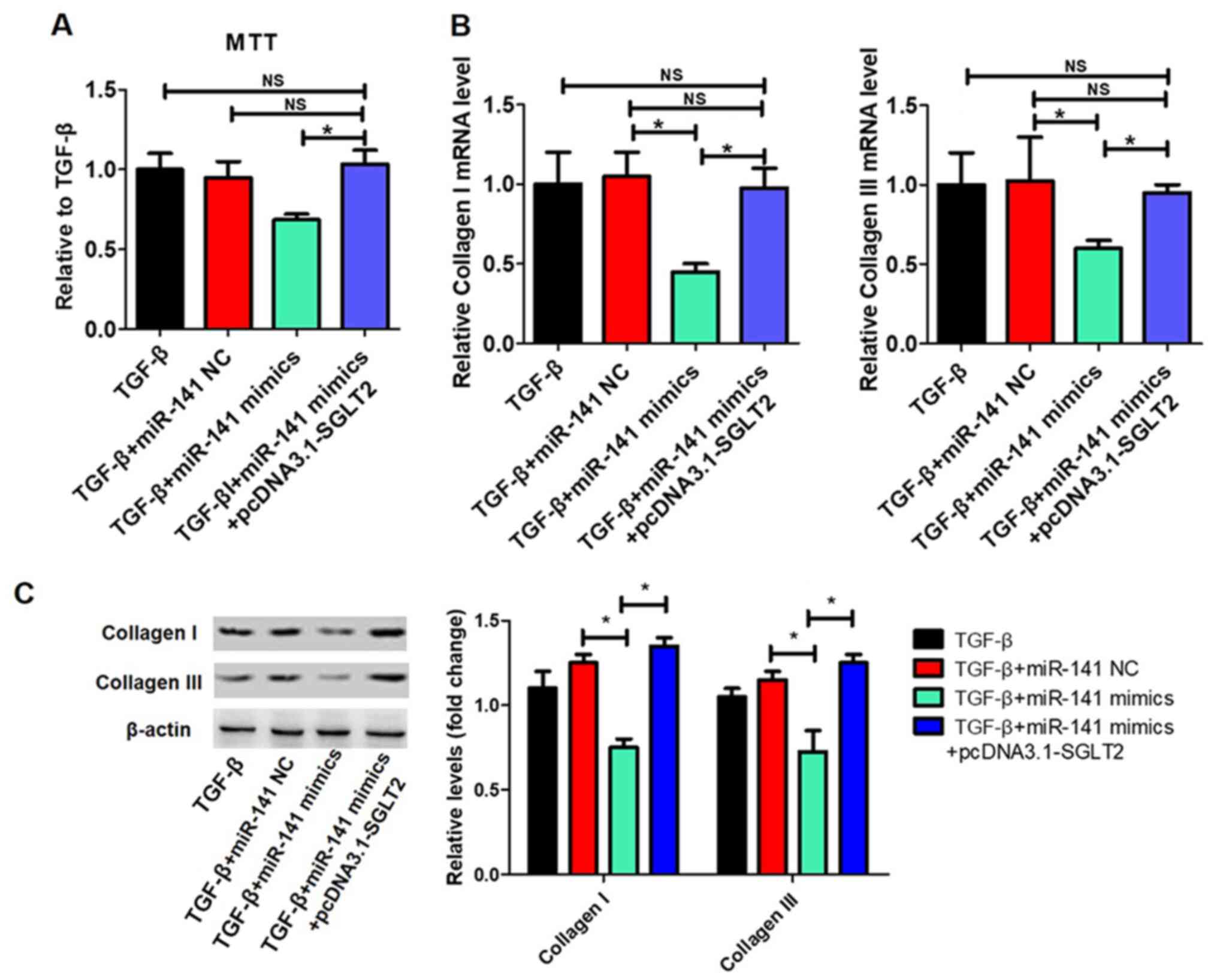
TGFβ-induced proliferation and
collagen synthesis in cardiac fibroblasts is regulated by the
miR-141/SGLT2 axis
The effects of the miR-141/SGLT2 axis on
TGFβ-induced proliferation and collagen synthesis in cardiac
fibroblasts were examined. The transfection efficiency of
pcDNA3.1-SGLT2 is presented in Fig.
6A. miR-141 overexpression significantly inhibited TGFβ-induced
cardiac fibroblast proliferation, which was reversed by SGLT2
overexpression (Fig. 6B).
Additionally, miR-141 overexpression significantly decreased
TGFβ-induced collagen I and collagen III expression levels, whereas
SGLT2 overexpression significantly reversed miR-141
overexpression-mediated effects (Fig.
6C).
Discussion
As a hallmark of MI, cardiac fibrosis is one of the
most important factors that leads to heart failure (23,24).
Fibrosis is characterized by the accumulation of excess collagen,
which causes cardiomyocyte dysfunction, abnormal differentiation of
cardiac fibroblasts and interstitial fibrosis (25). Current antifibrotic drugs slow the
progression of cardiac fibroblasts, but do not prevent or reverse
progression (26). Therefore, it is
crucial to identify specific targets for alternative therapeutic
strategies.
SGLT2 inhibitors have been widely studied to improve
cardiac diseases (10-12).
Empagliflozin, an SGLT2 inhibitor, reduced cardiovascular
mortality, as well as the occurrence of nonfatal MI and nonfatal
strokes in patients with type 2 diabetes mellitus (10). Furthermore, empagliflozin or
dapagliflozin, another SGLT-2 inhibitor, effectively improved
cardiac diastolic function in a female rat model of diabetes
(11). Additionally, SGLT-2
inhibition reduced the activation of the Nlrp3/ASC inflammasome and
attenuated the development of diabetic cardiomyopathy in mice
(12). The present study aimed to
determine whether SGLT2 alleviated effects on the progression of
cardiac fibrosis. The results demonstrated that SGLT2 levels in MI
model rats were significantly increased at 1 week post-MI compared
with sham rats, and continued to gradually decrease with time.
Furthermore, SGLT2 knockdown improved cardiac function and
attenuated cardiac fibrosis following MI in vivo compared
with MI or sh-NC groups. SGLT2 knockdown also inhibited
TGFβ-induced proliferation and collagen synthesis in cardiac
fibroblasts in vitro. Okada et al (27) demonstrated that dapagliflozin, an
SGLT2 inhibitor, influenced interactions between SGLT2 and collagen
I and IV, and established that discoidin domain receptor I served
an important role in the adherence of collagen I and IV. Moreover,
the expression levels of NLRP3, TNFα and caspase-1 were reduced in
mice treated with dapagliflozin and the phosphorylated-adenosine
monophosphate activated protein kinase (AMPK)/total-AMPK ratio was
enhanced (12). The results of the
aforementioned studies were similar to the results of the present
study, which indicated that SGLT2 was associated with collagen
deposition, collectively suggesting that SGLT2 served a key role in
the pathogenesis of cardiac fibrosis.
miRNAs are 22-23 nucleotides noncoding RNA molecules
that serve important roles in cardiovascular health and disease
(28). For example, miR-98
downregulated TGFβ receptor 1 expression, ameliorated
TGF-β1-induced differentiation and inhibited collagen production of
cardiac fibroblasts (29).
Downregulation of miR-29 effectively increased the expression of
collagens COL1A1, COL1A2 and COL3A1 in vitro and in
vivo (30). Li et al
(31) reported that miR-130a was
increased in angiotensin II-infused mice, and promoted the
expression of profibrotic genes and differentiation of myofibroblasts
by inhibiting peroxisome proliferator activated receptor γ
expression (31). The present study
demonstrated that miR-141 levels in MI model rats decreased at 1
week post-MI compared with sham rats, and gradually increased with
time. It has been reported that miR-141 levels were decreased in
diabetic mice myocardium and cardiac fibroblasts treated with
angiotension II (22). The results
of the present study indicated that miR-141 overexpression
significantly inhibited TGFβ-induced proliferation and collagen
synthesis in cardiac fibroblasts. Therefore, miR-141 may serve an
inhibitory role in cardiac fibrosis.
Bioinformatics analysis revealed that miR-141 bound
to the 3'-UTR of SGLT2. The results of luciferase and western
blotting assays suggested that miR-141 directly regulated the
expression of SGLT2. Additionally, SGLT2 overexpression reversed
miR-141-mediated reductions of TGFβ-induced proliferation and
collagen I and collagen III expression levels in cardiac
fibroblasts. Therefore, the present study suggested that there was
an association between miR-141 and SGLT2 in the pathogenesis of
cardiac fibrosis.
In summary, the present study indicated that SGLT2
expression was upregulated in cardiac fibrosis, and that SGLT2
knockdown reduced cardiac fibrosis and improved cardiac function
following MI. Additionally, the results suggested that SGLT2 was
regulated by miR-141 in the pathogenesis of cardiac fibrosis.
Therefore, the results of the present study provided evidence that
the miR-141/SGLT2 axis may serve as a novel target for the
treatment of cardiac fibrosis.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CZ and GL conceived, designed and performed the
experiments. SF analyzed the data. GL wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Animal Ethics Committee of the Second Affiliated Hospital of Wannan
Medical College, Wuhu, China (approval no. DWL-1804-007).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mcmanus DD, Gore J, Yarzebski J, Spencer
F, Lessard D and Goldberg RJ: Recent trends in the incidence,
treatment, and outcomes of patients with STEMI and NSTEMI. Am J
Med. 124:40–47. 2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Pontecorboli G, Ventura RMFI, Carlosena A,
Benito EM, Pratgonzales S, Padeletti L and Mont L: Use of
delayed-enhancement magnetic resonance imaging for fibrosis
detection in the atria: A review. Europace. 19:180–189.
2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Nagalingam RS, Safi HA and Czubryt MP:
Gaining myocytes or losing fibroblasts: Challenges in cardiac
fibroblast reprogramming for infarct repair. J Mol Cell Cardiol.
93:108–114. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Tahrani AA, Barnett AH and Bailey CJ: SGLT
inhibitors in management of diabetes. Lancet Diabetes Endocrinol.
1:140–151. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wells RG, Mohandas TK and Hediger MA:
Localization of the Na+/glucose cotransporter gene SGLT2 to human
chromosome 16 close to the centromere. Genomics. 17:787–789.
1993.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kashiwagi Y, Nagoshi T, Yoshino T, Tanaka
TD, Ito K, Harada T, Takahashi H, Ikegami M, Anzawa R and Yoshimura
M: Expression of SGLT1 in human hearts and impairment of cardiac
glucose uptake by phlorizin during ischemia-reperfusion injury in
mice. PLoS One. 10(e0130605)2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Koepsell H: The Na(+)-D-glucose
cotransporters SGLT1 and SGLT2 are targets for the treatment of
diabetes and cancer. Pharmacol Ther. 170:148–165. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Furtado RHM, Bonaca MP, Raz I, Zelniker
TA, Mosenzon O, Cahn A, Kuder J, Murphy SA, Bhatt DL, Leiter LA, et
al: Dapagliflozin and cardiovascular outcomes in patients with type
2 diabetes mellitus and previous myocardial infarction.
Circulation. 139:2516–2527. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Reid J, Rana K, Niman S, Sheikh-Ali M,
Lewis T, Choksi RR and Goldfaden RF: Sodium-glucose cotransporter-2
(SGLT-2) inhibitors for cardiovascular disease prevention. Am J
Cardiovasc Drugs. 20:419–429. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zinman B, Wanner C, Lachin JM, Fitchett D,
Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ,
et al: Empagliflozin, cardiovascular outcomes, and mortality in
type 2 diabetes. N Engl J Med. 373:2117–2128. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Habibi J, Aroor AR, Sowers JR, Jia G,
Hayden MR, Garro M, Barron BJ, Mayoux E, Rector RS, Whaley-Connell
A and DeMarco VG: Sodium glucose transporter 2 (SGLT2) inhibition
with empagliflozin improves cardiac diastolic function in a female
rodent model of diabetes. Cardiovasc Diabetol. 16(9)2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ye Y, Bajaj M, Yang HC, Perez-Polo JR and
Birnbaum Y: SGLT-2 inhibition with dapagliflozin reduces the
activation of the Nlrp3/ASC inflammasome and attenuates the
development of diabetic cardiomyopathy in mice with type 2
diabetes. Further augmentation of the effects with saxagliptin, a
DPP4 inhibitor. Cardiovasc Drugs Ther. 31:119–132. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Piccoli MT, Bar C and Thum T: Non-coding
RNAs as modulators of the cardiac fibroblast phenotype. J Mol Cell
Cardiol. 92:75–81. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang H, Zhang X, Li Y, Ma Y, Zhang Y, Liu
Z, Zhou J, Lin Q, Wang Y, Duan C and Wang C: Improved myocardial
performance in infarcted rat heart by co-injection of basic
fibroblast growth factor with temperature-responsive chitosan
hydrogel. J Heart Lung Transplant. 29:881–887. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Yuan J, Chen H, Ge D, Xu Y, Xu H, Yang Y,
Gu M, Zhou Y, Zhu J, Ge T, et al: Mir-21 promotes cardiac fibrosis
after myocardial infarction via targeting Smad7. Cell Physiol
Biochem. 42:2207–2219. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hao K, Lei W, Wu H, Wu J, Yang Z, Yan S,
Lu XA, Li J, Xia X, Han X, et al: LncRNA-Safe contributes to
cardiac fibrosis through Safe-Sfrp2-HuR complex in mouse myocardial
infarction. Theranostics. 9:7282–7297. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wang H, Qi C and Wan D: MicroRNA-377-3p
targeting MMP-16 inhibits ovarian cancer cell growth, invasion, and
interstitial transition. Ann Transl Med. 9(124)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Guo J, Gan Q, Gan C, Zhang X, Ma X and
Dong M: LncRNA MIR205HG regulates melanomagenesis via the
miR-299-3p/VEGFA axis. Aging (Albany NY). 13:5297–5311.
2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wu H, Zhou X, Wang X, Cheng W, Hu X, Wang
Y, Luo B, Huang W and Gu J: miR-34a in extracellular vesicles from
bone marrow mesenchymal stem cells reduces rheumatoid arthritis
inflammation via the cyclin I/ATM/ATR/p53 axis. J Cell Mol Med.
25:1896–1910. 2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Qu X, Du Y, Shu Y, Gao M, Sun F, Luo S,
Yang T, Zhan L, Yuan Y, Chu W, et al: MIAT is a pro-fibrotic long
non-coding RNA governing cardiac fibrosis in post-infarct
myocardium. Sci Rep. 7(42657)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhou B and Yu JW: A novel identified
circular RNA, circRNA_010567, promotes myocardial fibrosis via
suppressing miR-141 by targeting TGF-β1. Biochem Biophys Res
Commun. 487:769–775. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Moore-Morris T, Guimaraes-Camboa N, Yutzey
KE, Puceat M and Evans SM: Cardiac fibroblasts: From development to
heart failure. J Mol Med (Berl). 93:823–830. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Leask A: Getting to the heart of the
matter new insights into cardiac fibrosis. Circ Res. 116:1269–1276.
2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Schelbert EB, Fonarow GC, Bonow RO, Butler
J and Gheorghiade M: Therapeutic targets in heart failure:
Refocusing on the myocardial interstitium. J Am Coll Cardiol.
63:2188–2198. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Okada J, Yamada E, Saito T, Yokoo H, Osaki
A, Shimoda Y, Ozawa A, Nakajima Y, Pessin JE, Okada S and Yamada M:
Dapagliflozin inhibits cell adhesion to collagen I and IV and
increases ectodomain proteolytic cleavage of DDR1 by increasing
ADAM10 activity. Molecules. 25(495)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Grimaldi V, De Pascale MR, Zullo A,
Soricelli A, Infante T, Mancini FP and Napoli C: Evidence of
epigenetic tags in cardiac fibrosis. J Cardiol. 69:401–408.
2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cheng R, Dang R, Zhou Y, Ding M and Hua H:
MicroRNA-98 inhibits TGF-β1-induced differentiation and collagen
production of cardiac fibroblasts by targeting TGFBR1. Human Cell.
30:192–200. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Van Rooij E, Sutherland LB, Thatcher JE,
Dimaio JM, Naseem RH, Marshall WS, Hill JA and Olson EN:
Dysregulation of microRNAs after myocardial infarction reveals a
role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA.
105:13027–13032. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li L, Bounds KR, Chatterjee P and Gupta S:
MicroRNA-130a, a potential antifibrotic target in cardiac fibrosis.
J Am Heart Assoc. 6(e006763)2017.PubMed/NCBI View Article : Google Scholar
|