Introduction
Chronic obstructive pulmonary disease (COPD), which
is a progressive degenerative lung disease, is a preventable and
treatable disease characterized by incomplete reversibility and
airflow limitation, including emphysema and chronic bronchitis
(1-3).
It is currently the fourth leading cause of death worldwide, just
behind ischemic heart disease and cerebrovascular disease, and an
increasing number of individuals are diagnosed with the disease
(4). Therefore, new and effective
treatment strategies targeting COPD are urgently needed. Patients
with COPD have difficulty breathing and present with airflow
obstruction, which is often accompanied by discomforts, such as
coughing and sputum production (5,6).
However, the pathogenesis is not yet fully understood. The
occurrence of COPD is the result of the combined effects of genetic
and environmental pathogenic factors. Cigarette smoke is the most
common risk factor for COPD. The rates of respiratory disease, lung
function impairment and mortality of smokers are significantly
higher compared with non-smokers (7,8).
Smoking cessation has been shown to effectively delay the
progressive decline of lung function.
Endothelial cells (ECs), as the basic unit of blood
vessels, and play key roles in physiological metabolism. Under
harmful conditions, the regulatory mechanisms become disrupted,
which may be one of the main initiating factors for various
diseases (9,10). The pathogenesis of acute lung injury
includes alveolitis and tissue recovery, and the vascular response
is the central link in the occurrence of inflammation (11). Previous data suggested that abnormal
apoptosis serves as a significant factor contributing to the
destruction of pulmonary tissue in COPD (12). Both animal and human studies
confirmed that endothelial apoptosis plays a key role in the
pathogenesis of COPD (13,14). It was reported that human pulmonary
microvascular endothelial cells (HPMECs) play important roles in
the progression of COPD, and the present study further investigated
COPD using HPMECs as a model (15).
Long non-coding RNAs (lncRNAs), a class of
non-coding RNAs that are >200 nucleotides in length, participate
in the structure and function of chromatin, and promote a series of
biological functions (16,17). An increasing number of studies have
suggested that lncRNAs are associated with a number of human
diseases, such as cancer, cardiovascular diseases, gynecological
diseases and inflammatory diseases (18,19).
Studies have suggested that lncRNAs participate in regulating the
process of COPD (20). Previously,
taurine-upregulated gene 1 (TUG1), one of the most commonly
examined lncRNAs, was shown to be upregulated in patients with
COPD, and TUG1 knockdown reduced inflammation and airway remodeling
in an animal model and reversed inflammation and collagen
deposition induced by cigarette smoke extract (CSE) in vitro
(21,22). However, the mechanisms and roles of
TUG1 in COPD remains unclear.
It was hypothesized that TUG1 may be involved in
COPD pathogenesis through regulating EC apoptosis. The present
study investigated whether TUG1 played a role in cell apoptosis of
HPMECs induced by CSE, and further studied its molecular mechanism
in order to provide a more theoretical basis and novel strategies
for the treatment of COPD.
Materials and methods
Cell culture
HPMECs were obtained from the American Type Culture
Collection and cultured in DMEM (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin/streptomycin at 5% CO2 and 37˚C.
CSE preparation
CSE preparation was performed as described
previously (23). Smoke from 10
cigarettes (Chengdu Cigarette Factory) was bubbled through DMEM,
and this solution was considered 100% CSE. Then the 100% CSE
solution was frozen and stored at -80˚C until required. CSE was
diluted with DMEM medium to a final concentration of 1% for the
present study.
Apoptosis assay
HPMECs were treated with 1% CSE for 24 h, and then
collected by trypsinization, washed in PBS and resuspended in 1X
binding buffer at a density of 1x106 cells/ml. A total
of 100 µl cell suspension was then transferred to a 5 ml tube, and
stained with 5 µl Annexin V/FITC and PI solution, according to the
manufacturer's protocol (BD Biosciences). Stained cells were
analyzed by flow cytometry (FCM) using a BD FACSCalibur flow
cytometer (BD Biosciences) within 1 h. All the experiments were
performed at least three times. Data were analyzed using FlowJo
software (version 7.2.4; FlowJo LLC).
Western blot analysis
HPMECs were treated with 1% CSE for 24 h and
dissolved in RIPA buffer (Beyotime Institute of Biotechnology)
after washing in cold PBS. The supernatant was centrifuged at
10,000 x g at 4˚C for 15 min to obtain total protein. Protein
concentration was measured using a BCA protein assay kit (Beyotime
Institute of Biotechnology). Equal amounts of protein (40 µg/lane)
were separated by 10% SDS-PAGE. Separated proteins were transferred
to PVDF membranes (Thermo Fisher Scientific, Inc.), which were then
blocked at room temperature for 1 h in PBS-Tween-20 (PBS-T)
solution containing 5% non-fat milk. Subsequently, membranes were
incubated with cleaved caspase-3 (cat. no. ab32042; 1:1,000;
Abcam), caspase-3 (cat. no. ab32351; 1:1,000; Abcam), Bcl-2 like 11
axis (BCL2L11; cat. no. ab32158; 1:1,000; Abcam) or GAPDH (cat. no.
sc-47724; 1:500; Santa Cruz Biotechnology, Inc.) antibodies at 4˚C
overnight. Membranes were then washed three times with PBS-T and
incubated with the Goat Anti-Mouse IgG Antibody (cat. no. bc-0296G;
1:1,000; BIOSS) or Goat Anti-Rabbit IgG Antibody (cat. no.
bc-0295G; 1:1,000; BIOSS) for 1 h at room temperature. ECL
substrate (Thermo Fisher Scientific, Inc.) was used to visualize
the protein bands according to the manufacturer's protocol. Band
densities were quantified using Gel-Pro Analyzer Densitometry
software (version 6.3; Media Cybernetics, Inc.). All the
experiments were performed at least three times.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total cellular RNA was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and reverse transcribed into cDNA using a cDNA Synthesis kit
according to the manufacturer's protocol (Invitrogen; Thermo Fisher
Scientific, Inc.). The relative expression of genes was quantified
using a Prism 7000 Real-Time PCR system with SYBR qPCR MasterMix
(Vazyme Biotech Co., Ltd.) in accordance with the manufacturer's
protocol. Primers were purchased from Sangon Biotech Co., Ltd., and
primer sequences were as follows: GAPDH forward,
5'-CTTTGGTATCGTGGAAGGACTC-3' and reverse,
5'-GTAGAGGCAGGGATGATGTTCT-3'; U6 forward,
5'-GCTTCGGCAGCACATATACTAAAAT-3' and reverse,
5'-CGCTTCACGAATTTGCGTGTCAT-3'; lncRNA TUG1 forward,
5'-GACCGTCCAATGACCTTCCT-3' and reverse, 5'-TGGCTGAATGCTTCTTGGGT-3';
miR-9a-5p forward, 5'-GCGGCGGTCTTTGGTTATCTAG-3' and reverse,
5'-ATCCAGTGCAGGGTCCGAGG-3'; and BCL2L11 forward,
5'-CACCAGCACCATAGAAGAA-3' and reverse, 5'-ATAAGGAGCAGGCACAGA-3'.
The following thermocycling conditions were used for qPCR: Initial
denaturation for 5 min at 95˚C; followed by 35 cycles of 95˚C for
10 sec and 60˚C for 30 sec. U6 or GAPDH were used as the internal
controls. The relative mRNA expression levels of lncRNA TUG1,
miR-9a-5p and BCL2L11 were calculated using the 2-ΔΔCq
method (24). All samples were
analyzed at least in triplicate.
miRNA target analysis and dual
luciferase reporter assay
The association between miR-9a-5p and BCL2L11 was
identified using TargetScan version 7.1 (targetscan.org/vert_71). The 3'-untranslated region
products of BCL2L11 containing the target sequence of miR-9a-5p
were obtained by RT-qPCR and fused into a pmirGLO vector (Promega
Corporation) to construct the BCL2L11-wild-type (BCL2L11-WT) and
BCL2L11-mutated-type (BCL2L11-MUT) reporter vectors. The results
showed that BCL2L11 is a potential target of miR-9a-5p. 293 cells
(ATCC® CRL-1573; ATCC), that were cultured >24 h,
were transfected with BCL2L11-WT or BCL2L11-MUT and miR-9a-5p mimic
or mimic control using Lipofectamine® 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) for 48 h. Luciferase
activity was measured using a dual luciferase reporter assay system
(Promega Corporation) according to the manufacturer's instructions.
Renilla luciferase activity was used as a control. All the
experiments were performed at least three times.
Cell transfection
Normal HPMECs were inoculated in 6-well plates
overnight and transfected with control-small interfering RNA
(siRNA), TUG1-siRNA, inhibitor control, miR-9a-5p inhibitor,
TUG1-siRNA + inhibitor control, TUG1-siRNA + miR-9a-5p inhibitor,
mimic control, miR-9a-5p mimic, control-plasmid, BCL2L11-plasmid,
miR-9a-5p mimic + control-plasmid or miR-9a-5p mimic +
BCL2L11-plasmid (Shanghai GenePharma Co., Ltd.) using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). A total of 48 h after transfection, cells were
collected, and the transfection efficiency was assessed using
RT-qPCR. For the in vitro analysis, 48 h after cell
transfection, the cells were subjected to 1% CSE for 24 h. Cells
without any treatment were used as the control.
Caspase-3 activity assay
The activities of caspase-3 were measured using a
colorimetric assay kit (cat. no. C1115; Beyotime Institute of
Biotechnology) in accordance with the manufacturer's
instructions.
Statistical analysis
Data are presented as the mean ± standard deviation
of at least three independent tests. Statistical comparisons
between groups were assessed using a Student's t-test or a one-way
ANOVA followed by a Tukey's post hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
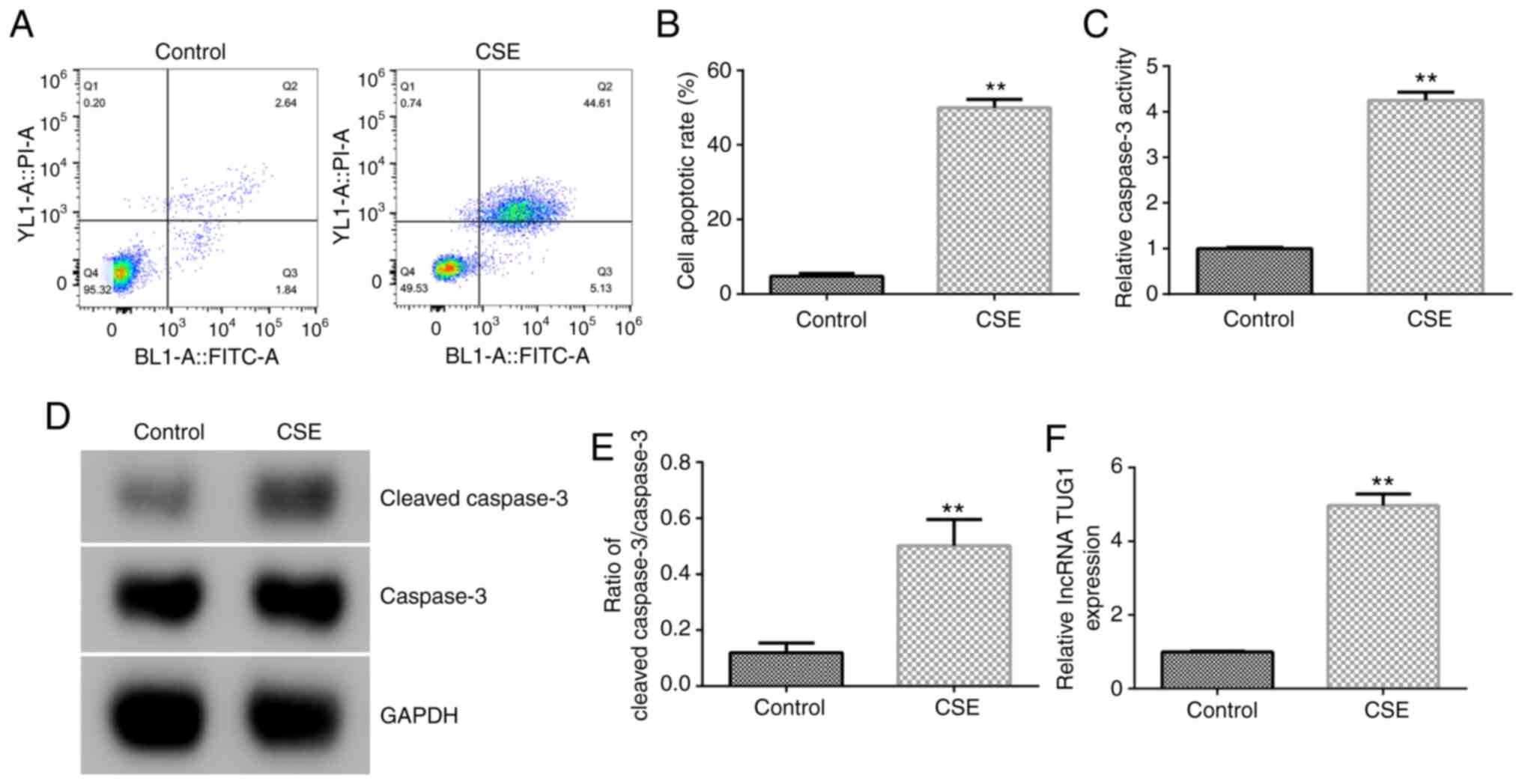
CSE influences HPMEC growth
Cell apoptosis of HPMECs treated with 1% CSE for 24
h was detected by FCM. The results showed that CSE significantly
induced cell apoptosis in HPMECs (Fig.
1A and B). Compared with the
control group, 1% CSE markedly increased caspase-3 activity,
cleaved caspase-3 protein expression and the
cleaved-caspase-3/caspase-3 ratio (Fig.
1C-E). Moreover, lncRNA TUG1 expression in HPMECs was
significantly increased with 1% CSE treatment compared with the
control group (Fig. 1F). The
results confirmed that lncRNA TUG1 was significantly upregulated in
1% CSE induced HPMECs.
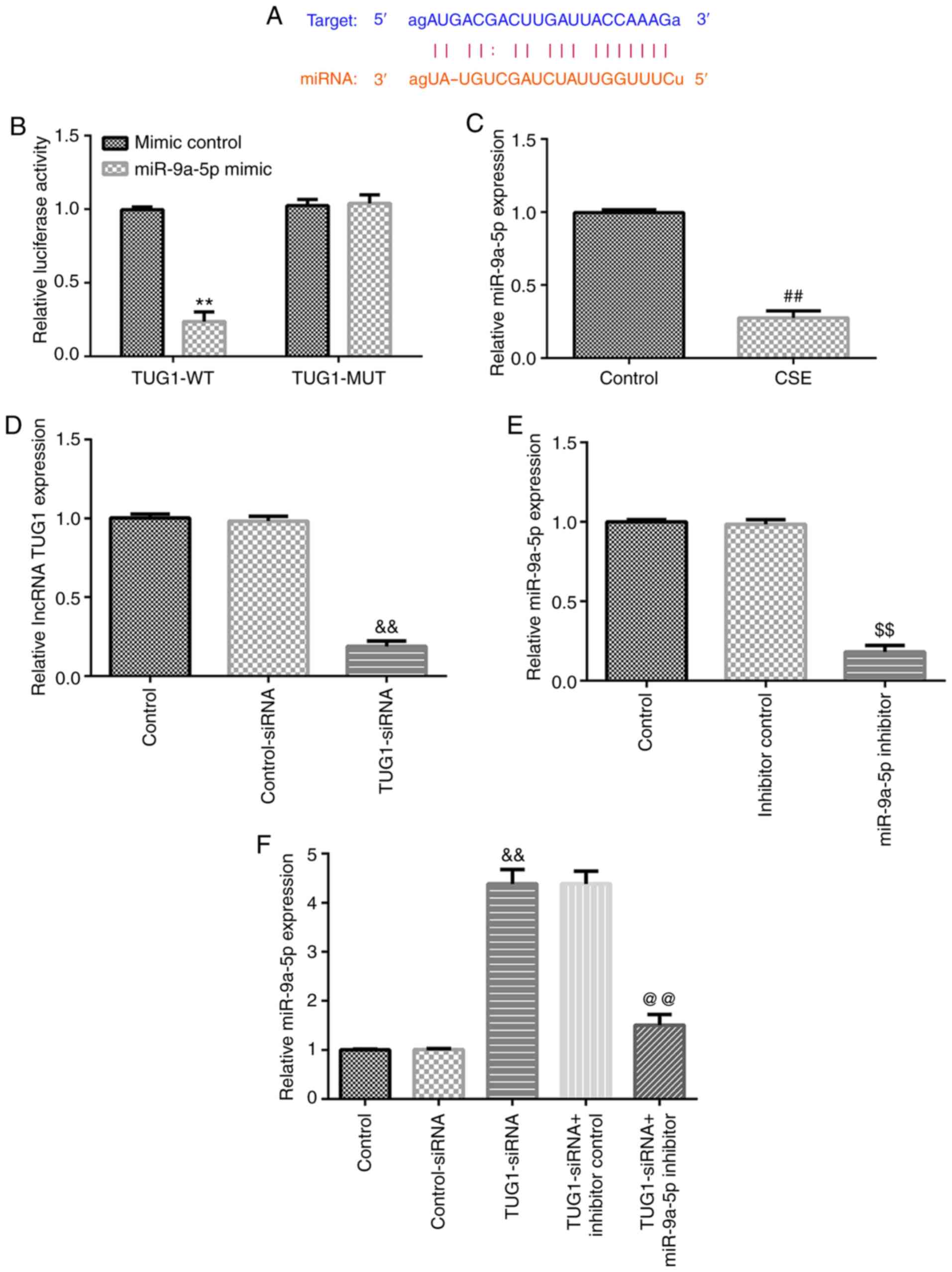
TUG1 negatively regulates miR-9a-5p
expression in HPMECs
A previous study showed the presence of a binding
site between TUG1 and miR-9a-5p (25) (Fig.
2A). In the present study, the targeted relationship between
TUG1 and miR-9a-5p was verified using a dual luciferase reporter
assay (Fig. 2B). Moreover,
miR-9a-5p expression in HPMECs was significantly decreased
following the 1% CSE treatment compared with the control group
(Fig. 2C). To further assess the
regulatory relationship between TUG1 and miR-9a-5p, HPMECs were
transfected with different plasmids, and the transfection
efficiency was determined using RT-qPCR. As shown in Fig. 2D and E, TUG1-siRNA and miR-9a-5p inhibitor
significantly reduced the expression of TUG1 and miR-9a-5p,
respectively. Compared with the control-siRNA group, TUG1-siRNA
significantly increased miR-9a-5p expression in HPMECs, which was
reversed by co-transfection with miR-9a-5p inhibitor (Fig. 2F). The data indicated that TUG1
negatively regulated miR-9a-5p expression in HPMECs.
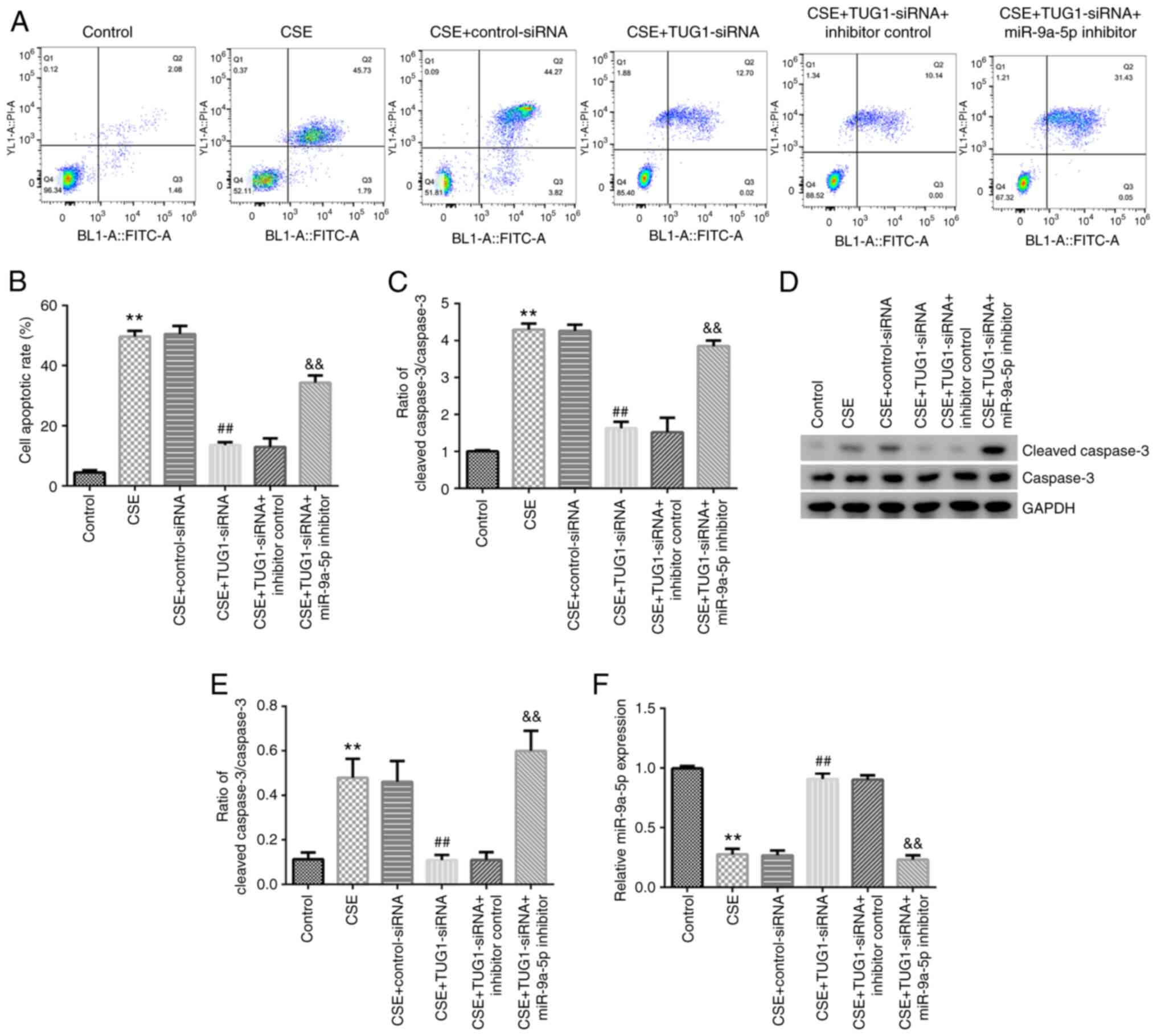
TUG1 inhibits cell apoptosis induced
by CSE in HPMECs by regulating miR-9a-5p
As shown in Fig.
3A-F, 1% CSE significantly induced cell apoptosis, increased
caspase-3 activity, enhanced cleaved caspase-3 protein expression,
increased the cleaved caspase-3/caspase-3 ratio and downregulated
miR-9a-5p expression in HPMECs. Moreover, compared with the CSE +
control-siRNA group, TUG1-siRNA significantly reduced cell
apoptosis, caspase-3 activity, cleaved caspase-3 protein expression
and the cleaved caspase-3/caspase-3 ratio, and upregulated
miR-9a-5p expression in HPMECs. All these changes were reversed by
miR-9a-5p inhibitor co-transfection. The above results suggested
that TUG1 inhibited cell apoptosis induced by CSE in HPMECs by
regulating miR-9a-5p.
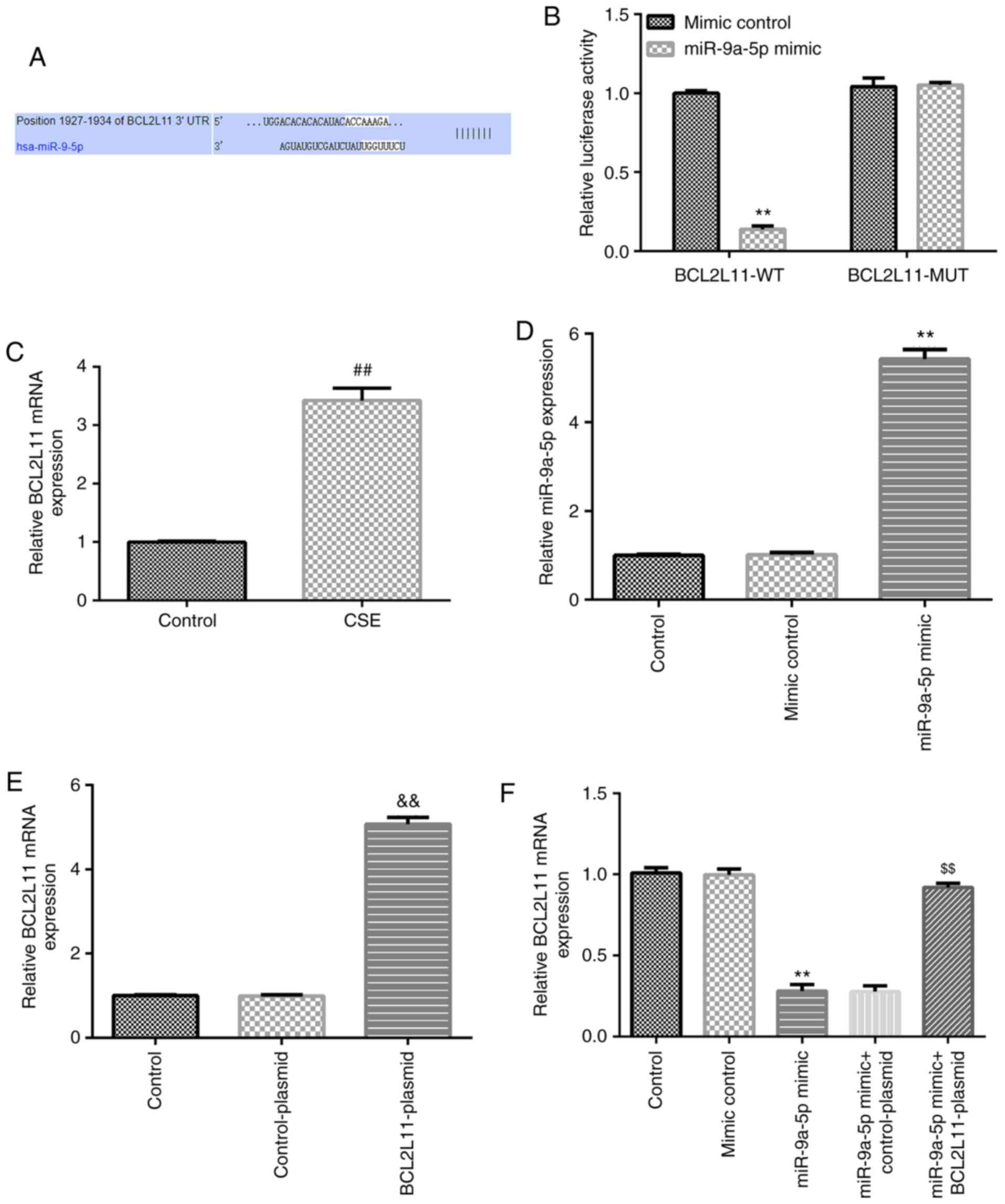
BCL2L11 is a direct target gene of
miR-9a-5p
TargetScan analysis showed the presence of a binding
site between BCL2L11 and miR-9a-5p (Fig. 4A), which was verified by a dual
luciferase reporter assay (Fig.
4B). Moreover, the mRNA expression of BCL2L11 in HPMECs was
significantly increased with 1% CSE treatment compared with the
control group (Fig. 4C). Further
results showed that miR-9a-5p expression in the miR-9a-5p mimic
group was higher compared with the mimic control group (Fig. 4D). BCL2L11-plasmid significantly
increased BCL2L11 mRNA expression in HPMECs (Fig. 4E). Compared with the mimic control
group, miR-9a-5p mimic significantly decreased BCL2L11 mRNA
expression in HPMECs, which was reversed by the co-transfection
with the BCL2L11-plasmid (Fig. 4F).
Thus, it was shown that BCL2L11 was the direct target gene of
miR-9a-5p, and it was negatively regulated by miR-9a-5p.
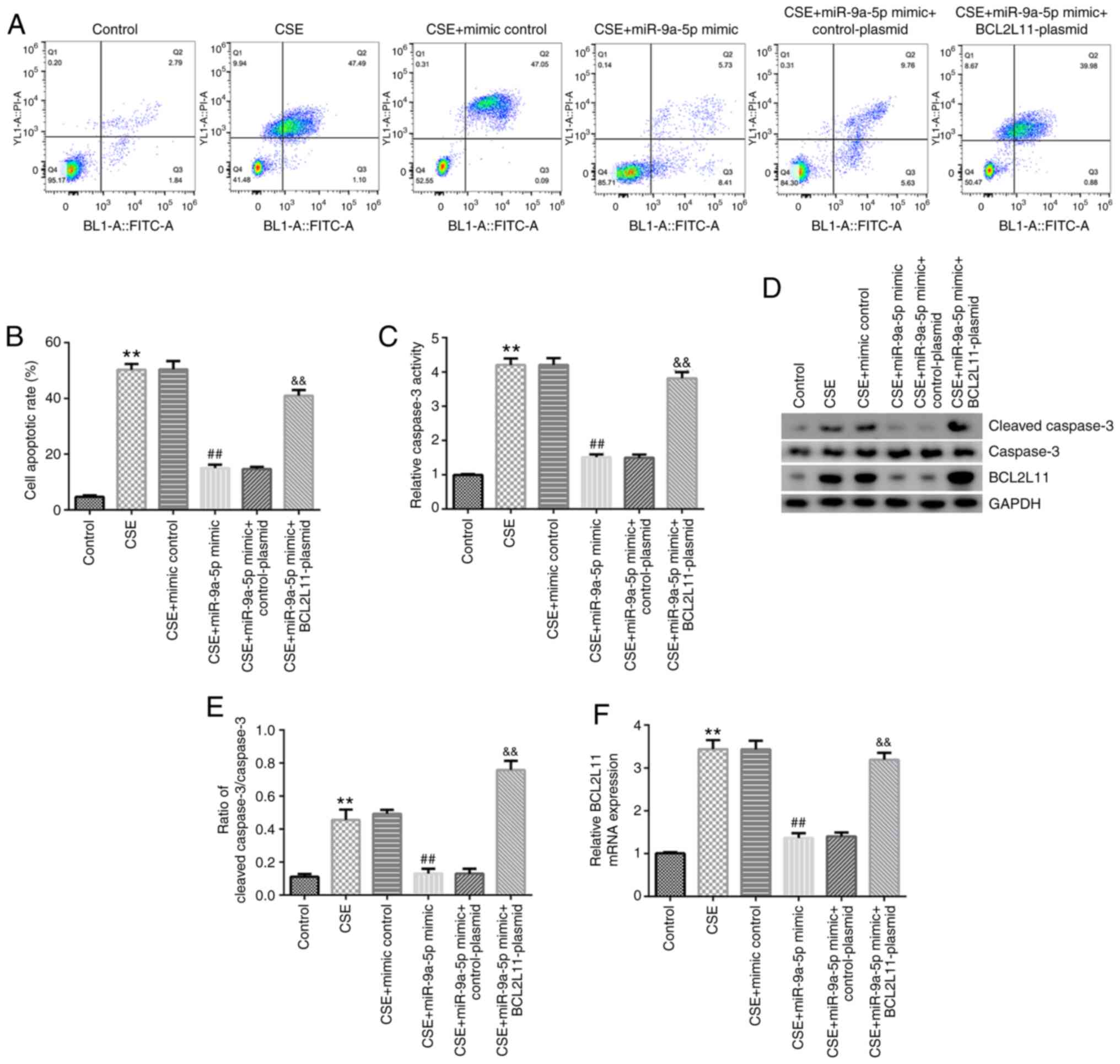
miR-9a-5p inhibits cell apoptosis
induced by CSE in HPMECs by regulating BCL2L11
Cells were treated with 1% CSE following
transfection. Cell apoptosis, caspase-3 activity, cleaved caspase-3
expression, the cleaved caspase-3/caspase-3 ratio and BCL2L11
expression were detected. The results showed that CSE significantly
induced cell apoptosis, increased caspase-3 activity, cleaved
caspase-3 expression and the cleaved caspase-3/caspase-3 ratio, and
enhanced BCL2L11 protein and mRNA expression in HPMECs. Similarly,
compared with the CSE + mimic control group, miR-9a-5p mimic
inhibited cell apoptosis, decreased caspase-3 activity, cleaved
caspase-3 expression and the cleaved caspase-3/caspase-3 ratio, and
reduced BCL2L11 protein and mRNA expression in HPMECs. These
changes were significantly reversed by BCL2L11-plasmid
co-transfection (Fig. 5A-F). In
conclusion, miR-9a-5p inhibited cell apoptosis induced by CSE in
HPMECs by regulating BCL2L11.
Discussion
COPD is a chronic lung disease characterized by
incomplete reversible airflow limitation. Common complications
include chronic respiratory failure, spontaneous pneumothorax and
chronic pulmonary heart disease (4,26). The
global incidence of COPD amongst individuals >40 years of age is
9-10%. COPD not only causes respiratory impairment, but also
affects circulatory function and can be life-threatening in severe
cases (27). HPMEC growth is
associated with the process of COPD. HPMECs were used in the
present study, and it was found that 1% CSE induced cell apoptosis
and increased lncRNA TUG1 expression in HPMECs, consistent with
previous reports. The results showed that increased lncRNA TUG1
levels could be used as a diagnostic marker for COPD.
Furthermore, the present study investigated the
mechanisms of lncRNA TUG1 in COPD. It was previously reported that
there is a binding site between TUG1 and miR-9a-5p (25), and this association was verified in
the present study. In addition, a negative regulatory mechanism was
found between lncRNA TUG1 and miR-9a-5p in HPMECs. Endothelial
apoptosis serves a key role in the pathogenesis of COPD (13,14).
The lncRNA TUG1/miR-9a-5p axis has also been reported to serve a
significant role in the regulation of cell apoptosis (25). Thus, it was hypothesized that TUG1
may be involved in COPD pathogenesis through regulating EC
apoptosis. Therefore, whether TUG1 affected cell apoptosis in
HPMECs via regulation of miR-9a-5p was determined, and several
experiments in HPMECs were performed following knockdown of TUG1 or
miR-9a-5p expression. FCM analysis indicated that knocking down
TUG1 reduced cell apoptosis induced by CSE in HPMECs, and the
effects were reversed by downregulating miR-9a-5p. Similarly, the
present study predicted and verified that BCL2L11 is the direct
target gene of miR-9a-5p. miR-9a-5p was found to negatively
regulate BCL2L11 expression in HPMECs. BCL2L11, a member of the
BCL-2 family, is located in the outer membrane of mitochondria and
it serves as an important regulator of apoptosis (28,29).
Thus, miR-9a-5p may regulate HPMECs apoptosis via negatively
regulating BCL2L11 expression. The FCM results showed that
miR-9a-5p overexpression relieved cell apoptosis induced by CSE in
HPMECs, and the effect was reversed by upregulating BCL2L11. The
caspase family participates in mediating cell apoptosis, and
caspase-3 is a key regulatory molecule involved in numerous
pathways in apoptotic signal transduction (30,31).
The present study detected caspase-3 activity and expression. The
results indicated that TUG1, miR-9a-5p and BCL2L11 affected HPMEC
apoptosis by regulating caspase-3.
In conclusion, the present study found that lncRNA
TUG1 serves a role in cell apoptosis induced by CSE in HPMECs via
modulating a miR-9a-5p/BCL2L11 axis; the mechanism identified in
the present study is shown in Fig.
S1. These findings suggested that TUG1 was a potential
effective target for COPD treatment.
Supplementary Material
Molecular mechanism by which lncRNA
TUG1 regulates human pulmonary microvascular endothelial cell
apoptosis induced by CSE. lncRNA TUG1, long non-coding RNA
taurine-upregulated gene 1; CSE, cigarette smoke extract; BCL2L11,
Bcl-2 like 11; HPMEC, human pulmonary microvascular endothelial
cell; miR, microRNA.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XC and MM conceived and designed the current study,
as well as acquired, analyzed and interpreted the data, and
prepared the manuscript. YS and XJ contributed to the acquisition
and analysis of the data. ZY contributed to acquisition and
analysis of the data, and prepared the manuscript. All authors read
and approved the final manuscript, and confirm the authenticity of
all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rabe KF and Watz H: Chronic obstructive
pulmonary disease. Lancet. 389:1931–1940. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Duffy SP and Criner GJ: Chronic
obstructive pulmonary disease: Evaluation and management. Med Clin
North Am. 103:453–461. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lareau SC, Fahy B, Meek P and Wang A:
Chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care
Med. 199:P1–P2. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Berg K and Wright JL: The pathology of
chronic obstructive pulmonary disease: Progress in the 20th and
21st centuries. Arch Pathol Lab Med. 140:1423–1428. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sorathia L: Palliative care in chronic
obstructive pulmonary disease. Med Clin North Am. 103:517–526.
2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
van Geffen WH, Kerstjens HAM and Slebos
DJ: Emerging bronchoscopic treatments for chronic obstructive
pulmonary disease. Pharmacol Ther. 179:96–101. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Aghapour M, Raee P, Moghaddam SJ, Hiemstra
PS and Heijink IH: Airway epithelial barrier dysfunction in chronic
obstructive pulmonary disease: Role of cigarette smoke exposure. Am
J Respir Cell Mol Biol. 58:157–169. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Postma DS, Bush A and van den Berge M:
Risk factors and early origins of chronic obstructive pulmonary
disease. Lancet. 385:899–909. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Barnes PJ: Inflammatory mechanisms in
patients with chronic obstructive pulmonary disease. J Allergy Clin
Immunol. 138:16–27. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sohal SS: Epithelial and endothelial cell
plasticity in chronic obstructive pulmonary disease (COPD). Respir
Investig. 55:104–113. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Butt Y, Kurdowska A and Allen TC: Acute
lung injury: A clinical and molecular review. Arch Pathol Lab Med.
140:345–350. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Siganaki M, Koutsopoulos AV, Neofytou E,
Vlachaki E, Psarrou M, Soulitzis N, Pentilas N, Schiza S, Siafakas
NM and Tzortzaki EG: Deregulation of apoptosis mediators' p53 and
bcl2 in lung tissue of COPD patients. Respir Res.
11(46)2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chen Y, Luo H, Kang N, Guan C, Long Y, Cao
J, Shen Q, Li J, Yang M, Peng H and Chen P: Beraprost sodium
attenuates cigarette smoke extract-induced apoptosis in vascular
endothelial cells. Mol Biol Rep. 39:10447–10457. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yang M, Chen P, Peng H, Zhang H, Chen Y,
Cai S, Lu Q and Guan C: Cigarette smoke extract induces aberrant
cytochrome-c oxidase subunit II methylation and apoptosis in
human umbilical vascular endothelial cells. Am J Physiol Cell
Physiol. 308:C378–C384. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sun Y, An N, Li J, Xia J, Tian Y, Zhao P,
Liu X, Huang H, Gao J and Zhang X: miRNA-206 regulates human
pulmonary microvascular endothelial cell apoptosis via targeting in
chronic obstructive pulmonary disease. J Cell Biochem.
120:6223–6236. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hombach S and Kretz M: Non-coding RNAs:
Classification, biology and functioning. Adv Exp Med Biol.
937:3–17. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Novak J, Vašků JB and Souček M: Long
non-coding RNAs in the pathophysiology of atherosclerosis. Vnitr
Lek. 64:77–82. 2018.PubMed/NCBI(In Czech).
|
|
18
|
Beermann J, Piccoli MT, Viereck J and Thum
T: Non-coding RNAs in development and disease: Background,
mechanisms, and therapeutic approaches. Physiol Rev. 96:1297–1325.
2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kok FO and Baker AH: The function of long
non-coding RNAs in vascular biology and disease. Vascul Pharmacol.
114:23–30. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang H, Sun D, Li D, Zheng Z, Xu J, Liang
X, Zhang C, Wang S, Wang J and Lu W: Long non-coding RNA expression
patterns in lung tissues of chronic cigarette smoke induced COPD
mouse model. Sci Rep. 8(7609)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Tang W, Shen Z, Guo J and Sun S: Screening
of long non-coding RNA and TUG1 inhibits proliferation with TGF-β
induction in patients with COPD. Int J Chron Obstruct Pulmon Dis.
11:2951–2964. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gu W, Yuan Y, Wang L, Yang H, Li S, Tang Z
and Li Q: Long non-coding RNA TUG1 promotes airway remodelling by
suppressing the miR-145-5p/DUSP6 axis in cigarette smoke-induced
COPD. J Cell Mol Med. 23:7200–7209. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li D, Hu J, Wang T, Zhang X, Liu L, Wang
H, Wu Y, Xu D and Wen F: Silymarin attenuates cigarette smoke
extract-induced inflammation via simultaneous inhibition of
autophagy and ERK/p38 MAPK pathway in human bronchial epithelial
cells. Sci Rep. 6(37751)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yang D, Yu J, Liu HB, Yan XQ, Hu J, Yu Y,
Guo J, Yuan Y and Du ZM: The long non-coding RNA TUG1-miR-9a-5p
axis contributes to ischemic injuries by promoting cardiomyocyte
apoptosis via targeting KLF5. Cell Death Dis.
10(908)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Riley CM and Sciurba FC: Diagnosis and
outpatient management of chronic obstructive pulmonarydisease: A
review. JAMA. 321:786–797. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lin YH, Tsai CL, Tsao LI and Jeng C: Acute
exacerbations of chronic obstructive pulmonary disease (COPD)
experiences among COPD patients with comorbid gastrooesophageal
reflux disease. J Clin Nurs. 28:1925–1935. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Concannon CG, Tuffy LP, Weisová P, Bonner
HP, Dávila D, Bonner C, Devocelle MC, Strasser A, Ward MW and Prehn
JH: AMP kinase-mediated activation of the BH3-only protein Bim
couples energy depletion to stress-induced apoptosis. J Cell Biol.
189:83–94. 2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kilbride SM, Farrelly AM, Bonner C, Ward
MW, Nyhan KC, Concannon CG, Wollheim CB, Byrne MM and Prehn JH:
AMP-activated protein kinase mediates apoptosis in response to
bioenergetic stress through activation of the pro-apoptotic Bcl-2
homology domain-3-only protein BMF. J Biol Chem. 285:36199–36206.
2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Crowley LC and Waterhouse NJ: Detecting
cleaved caspase-3 in apoptotic cells by flow cytometry. Cold Spring
Harb Protoc: Nov 1, 2016 (Epub ahead of print). doi:
10.1101/pdb.prot087312.
|
|
31
|
Rogers C, Fernandes-Alnemri T, Mayes L,
Alnemri D, Cingolani G and Alnemri ES: Cleavage of DFNA5 by
caspase-3 during apoptosis mediates progression to secondary
necrotic/pyroptotic cell death. Nat Commun. 8(14128)2017.PubMed/NCBI View Article : Google Scholar
|