Introduction
Acute lung injury (ALI) is a common life-threatening
inflammatory condition that is associated with high morbidity and
mortality rates in addition to high costs (1), with >3 million ARDS cases and
75,000 deaths annually worldwide (2,3). The
pathogenesis of ALI is highly complex. A number of studies have
suggested that pulmonary edema and the hyperactivation of the
innate immune response are closely associated with the onset of ALI
(4,5). Environmental and internal factors such
as infection and sepsis (6), can
mediate tissue inflammation and subsequent damage by inducing the
secretion of inflammatory factors, including interleukin (IL)-1β,
IL-6, IL-8 and tumor necrosis factor-α (TNF-α). Dysregulation of
this process can eventually lead to alveolar-capillary barrier
disruption and pulmonary edema (7,8).
MicroRNAs (miRNAs or miRs) are a type of small, non-coding,
single-stranded RNAs that are 17-24 nucleotides in length and can
modulate the expression of target genes on a post-transcriptional
level (9,10). It has been reported that miRs serve
an important role in the occurrence and progression of ALI
(11). Toll-like receptor 4 (TLR4)
is a member of the conserved type I transmembrane proteins, which
was first identified in microbes in 1980(12). TLR4 mainly mediates the recognition
of pathogens to initiate the inflammation signaling pathway
(13). Several members of these
proteins have since been found in mammals, including and humans
(14,15). Among them, TLR4 is a receptor that
can recognize lipopolysaccharides (LPS) and plays a pivotal role in
mediating lung injury (16). Upon
activation, this upregulated state can then be modulated by several
miRs, including miR-140, miR-181a and miR-182, via a negative
feedback mechanism to inhibit inflammation (17-19)
Ventilation therapy approaches, such as mechanical
ventilation with the use of low tidal volumes, have been developed
for treating ALI (19). This method
is designed to protect the lungs from excessive mechanical burden
(19). It has also been documented
that intratracheal installation of primary rat alveolar type II
(ATII) cells in a rat ALI model can attenuate lung injury (20). However, the mechanistic details
underlying the pathogenesis of ALI has not been fully elucidated.
It was previously found that upregulation miR-16 can inhibit the
inflammatory response in a ALI model of mouse (21). Therefore, the present study was
undertaken to investigate whether miR-16 upregulation can attenuate
the ALI-induced inflammatory responses. The role of miR-16 was
investigated in a LPS-induced rat model of ALI, which aimed to
uncover its underlying mechanisms of action and determine whether
it is a potential target for the clinical therapy of ALI.
Materials and methods
Bioinformatics analysis
TargetScan database (release no. 7.2; https://www.targetscan.org/vert_72/) was used to
calculate the prediction of targets of miR-16.
Animals
In total, 10 8-week-old female Sprague-Dawley rats
weighing 240±20 g were purchased from Beijing Vital River
Laboratory Animal Technology Co., Ltd and were kept in 23±2˚C with
a 12-h light/dark cycle, 55-60% humidity, with ad libitum
access to food and water. All the animal protocols (approval no.
2019-KY-005) were approved by The Ethics Committee of Zhengzhou
University, with all animal experiments performed according to the
National Institutes of Health Guidelines for the Care and Use of
Laboratory Animals (GB/T 35892-2018).
Establishment of LPS-induced lung
injury rat model
LPS (Sigma-Aldrich; Merck KGaA) was diluted in PBS
to a final concentration of 10 mg/ml. Rats were randomly divided
into two groups. Rats in the LPS group (n=5) were administered LPS
(0.1 mg/kg) through tracheal intubation after anesthesia using
isoflurane inhalation at a dose of 4 and 2% for induction and
maintenance, respectively, at a gas flow rate of 0.25 l/min
(22). Rats in the control group
were treated with the same volume of PBS alone. At 6 h following
the establishment of the LPS-induced lung injury model, all rats
were euthanized with intraperitoneal injection of 200 mg/kg sodium
pentobarbital followed by cervical dislocation. Subsequently,
peripheral blood and the lung tissues were collected from all
rats.
Cell culture and transfection
The NHBE (cat. no. PCS-300-010) normal human
bronchial epithelial cell line and 293T cells were purchased from
the American Type Culture Collection. Cells were cultured in DMEM
(Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (Thermo
Fisher Scientific, Inc.) and 1% penicillin at 37˚C in a humified
atmosphere of 5% CO2. miR-16 expression was tested in
NHBE cells which was treated with LPS (0, 1, 2, 3, 4, 8 and 16
µg/ml) for 12 h at 37˚C by reverse transcription-quantitative PCR
(RT-qPCR). In addition, miR-16 expression was tested in NHBE cells
that were incubated with 2 µg/ml LPS for a range of indicated time
durations (0, 6, 12, 24, 36 and 48 h) at 37˚C by RT-qPCR.
The group method of NHBE cells as follows: i) miR-NC
group; ii) miR-NC + LPS group, where the cells were transfected
with miR-NC and then treated with 2 µg/ml LPS; iii) miR-16 mimic
group, where cells were transfected with the miR-16 mimic; iv)
miR-16 mimic + LPS group, where cells were transfected with the
miR-16 mimic and then treated with 2 µg/ml LPS; v) si-NC group; vi)
si-NC + LPS group, where the NHBE cells were transfected with si-NC
and then treated with 2 µg/ml LPS; vii) si-TLR4 group; viii)
si-TLR4 + LPS group, where NHBE cells were transfected with si-TLR4
and then treated with 2 µg/ml LPS; and ix) miR-16 mimic + pcDNA
TLR4 + LPS group, where cells were transfected with miR-16 mimic
and pcDNA-TLR4 and then treated with 2 µg/ml LPS. All transfection
experiments lasted 48 h.
Specific small interfering RNAs (siRNAs) targeting
TLR4, miR-16 mimics and negative control (NC) mimics (50 nM;
Shanghai GenePharma Co., Ltd.) were transfected into NHBE cells for
48 h using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocols.
The sequences were as follows: MiR-16 mimics sense,
5'-UAGCAGCACGUAAAUAUUGGCG-3' and antisense,
5'-CGCCAAUAUUUACGUGCUGCUA-3'; miR-NC sense,
UUCUCCGAACGUGUCACGUTT-3' and antisense,
5'-ACGUGACACGUUCGGAGAATT-3'; TLR4 siRNA sense,
5'-GGACAGCUUAUAACCUUAATT-3' and antisense,
5'-UUAAGGUUAUAAGCUGUCCTT-3' and siRNA-NC sense,
5'-UUCUCCGAACGUGUCACGUTT-3' and antisense,
5'-ACGUGACACGUUCGGAGAATT-3'. Total RNA was extracted from NHBE
cells using the TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) before the cDNA was obtained by reverse
transcription using the random primer (cat. no. B300818-0100;
Sangon Biotech Co., Ltd.) and 5X AMV RT Buffer (cat. no.
B610020-0500; Sangon Biotech Co., Ltd.), using the temperature
protocol of 42˚C for 60 min and 72˚C for 10 min. The TLR4 target
was obtained by PCR (Direct PCR Kit; cat. no. B639289; Sangon
Biotech Co., Ltd.) using TLR4-specific primers (TLR4 forward,
5'-AAAAGCTTGCCACCATGATGTCTGCCTCGCGCCTGG-3' and reverse,
5'-AAGGATCCTCAGATAGATGTTGCTTCCTGC-3'). The temperature protocol was
95˚C for 30 sec, followed by 40 cycles of 95˚C for 5 sec and 60˚C
for 34 sec. The whole sequence of TLR4 was then sub-cloned into the
pcDNA.3.1 vector (Shanghai GenePharma Co., Ltd.) via the
HindⅢ and BamHⅠ restriction sites to construct the
TLR4 overexpression plasmid, which was also transfected into NHBE
cells using Lipofectamine 2000(17).
Histological analysis
For histological analysis, the lung tissues from
PBS- or LPS-treated rats were fixed with 4% paraformaldehyde
overnight at room temperature and embedded with paraffin.
Subsequently, 4-µm sections of the lung tissues were dehydrated in
an ascending ethanol gradient and then xylene and were stained with
hematoxylin and eosin (H&E) (23). At room temperature, the samples were
stained with hematoxylin for 5 min and eosin for 1 min. Each slide
was observed under light microscopy at x200 magnification.
Representative images for each group are shown.
ELISA
The secretion levels of TNF-α (cat. no.
E-EL-R0019c), IL-1β (cat. no. E-EL-H0149c) and IL-6 (cat. no.
E-EL-H0102c) were determined using the corresponding ELISA kits
(Elabscience Biotechnology Co., Ltd.) in peripheral blood samples
of rats and cell culture medium of NHBE cells. All procedures were
performed independently in triplicate according to manufacturer's
protocol.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the lung tissues of
rats or NHBE cells using the TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). For miRNA analysis,
cDNA was synthesized by M-MLV reverse transcriptase with a special
reverse transcription primer for miRNAs (PrimeScript™ 1st Strand
cDNA Synthesis Kit; cat. no. 6110A; Takara Bio, Inc.) using the
protocol of 30˚C 10 min, 42˚C 60 min and 95˚C for 5 min. For mRNA
analysis, cDNA was synthesized using a PrimeScript™ RT reagent kit
(cat. no. RR037A; Takara Bio, Inc.) using the protocol of 16˚C for
30 min, 42˚C for 30 min and 85˚C 10 min. For miRNAs, qPCR was
performed using Hairpin-it™ miRNAs qPCR Quantitation Kit (cat. no.
QPM-010, Shanghai GenePharma Co., Ltd.). The primer sequences of
miR-16 used were the following: Forward, 5'-CGCGCTAGCAGCACGTAAAT-3'
and reverse, 5'-GTGCAGGGTCCGAGGT-3'; U6 forward,
5'-CTCGCTTCGGCAGCACA-3' and reverse, 5'-AACGCTTCACGAATTTGCGT-3'.
For mRNA, qPCR was performed using the LightCycler® RNA
Master SYBR Green I (cat. no. 3064760001; Roche Diagnostics). The
sequences of the primers were as follows: TLR4 forward,
5'-GGCTCCTGATGCAAGATGCCCCT-3' and reverse,
5'-CTGCCTTGAATACCTTCACACGT-3'; TNF-α forward,
5'-CTGGGACAGTGACCTGGACT-3' and reverse, 5'-GCACCTCAGGGAAGAGTCTG-3';
IL-1β forward, 5'-CCTCCTTGCCTCTGATGG-3' and reverse,
5'-AGTGCTGCCTAATGTCCC-3'; IL-6 forward, 5'-AGTTGCCTTCTTGGGACTGA-3'
and reverse, 5'-TCCACGATTTCCCAGAGAAC-3' and GAPDH forward,
5'-CTGAGCACCAGGTGGTCTC-3' and reverse, 5'-CATGACAAGGTGCGGCTCC-3'.
The thermocycling conditions were 95˚C for 30 sec, followed by 40
cycles of 95˚C for 5 sec and 60˚C and 34 sec. qPCR was performed
using an ABI 7500 Fast Real-time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Relative expression of miR-16 was
normalized to that of U6, whereas the relative mRNA expression was
normalized to GAPDH, both using the 2-ΔΔCq method
(24-26).
Luciferase reporter assay
293T cells (1x105 cells) were
co-transfected with 10 pmol/ml miR-16 mimics or miR-NC and
pmirGLO3-TLR4-3'UTR-wild-type (Wt) or pmirGLO3-TLR4-3'UTR-mutant
(MUT) (0.2 pmol/ml all from Shanghai GenePharma Co., Ltd.) by
Lipofectamine 2000. Following incubation for 48 h at 37˚C, the
luciferase activity was measured using the
Dual-Luciferase® Reporter Assay System (Promega
Corporation) according to the manufacturer's protocols. The results
of firefly luciferase activity were normalized to the
Renilla luciferase activity.
Western blot analysis
Total proteins of NHBE cells were extracted using a
protein extraction kit (NE-PER™ Nuclear and Cytoplasmic Extraction
Reagents; Pierce, Thermo Fisher Scientific, Inc.) and the protein
concentration was measured using bicinchoninic acid protein assay
reagent (Beyotime Institute of Biotechnology). A 10% SDS-PAGE gel
was prepared with 20 µg protein samples loaded in each lane. The
samples were mixed with the loading buffer and boiled at 100˚C for
5 min. The protein on the gel was then transferred to a
nitrocellulose membranes and blocked with 5% skimmed milk powder at
room temperature 1 h. GAPDH primary antibody (cat. no. ab70699;
1:5,000; Abcam) was chosen as the internal reference. The membranes
were first probed with the anti-TLR4 mouse polyclonal antibody
incubated at 4˚C overnight (cat. no. sc-293072; dilution 1:1,500;
Santa Cruz Biotechnology, Inc.) and then with a corresponding
horseradish peroxidase-conjugated secondary antibody (cat. no.
sc-525409; 1:5,000; Santa Cruz Biotechnology, Inc.) at room
temperature for 1.5 h. An enhanced chemiluminescence kit (EMD
Millipore) was used for visualization of the results and
quantification of the bands was performed using the Image J
software (v1.8.0; National Institutes of Health).
Statistical analysis
All statistical analyses were performed using
unpaired Student's t-test for comparisons between two groups.
One-way ANOVA and Tukey's post hoc test were used for multiple
group comparisons using the SPSS software (version 17.0; SPSS,
Inc.) Data are expressed as the mean ± SD from three independent
experiments performed in triplicate. P<0.05 was considered to
indicate a statistically significant difference.
Results
miR-16 expression is downregulated in
the lung tissues of ALI rats and LPS-induced NHBE cells
An ALI rat model was established by LPS, whilst
animals treated with PBS served as control. The lung tissues from
the LPS group showed lung injury, including hemorrhage,
interstitial edema and infiltration of inflammatory cells (Fig. 1A). Subsequent RT-qPCR analysis
showed that the expression of miR-16 was significantly decreased in
the lung tissues of rats treated with LPS compared with that in
control tissues treated with PBS (Fig.
1B). ELISA results revealed that the secretion levels of IL-1β,
TNF-α and IL-6 were significantly increased in the peripheral blood
of LPS-treated rats compared with those in the control PBS group
(Fig. 1C-E).
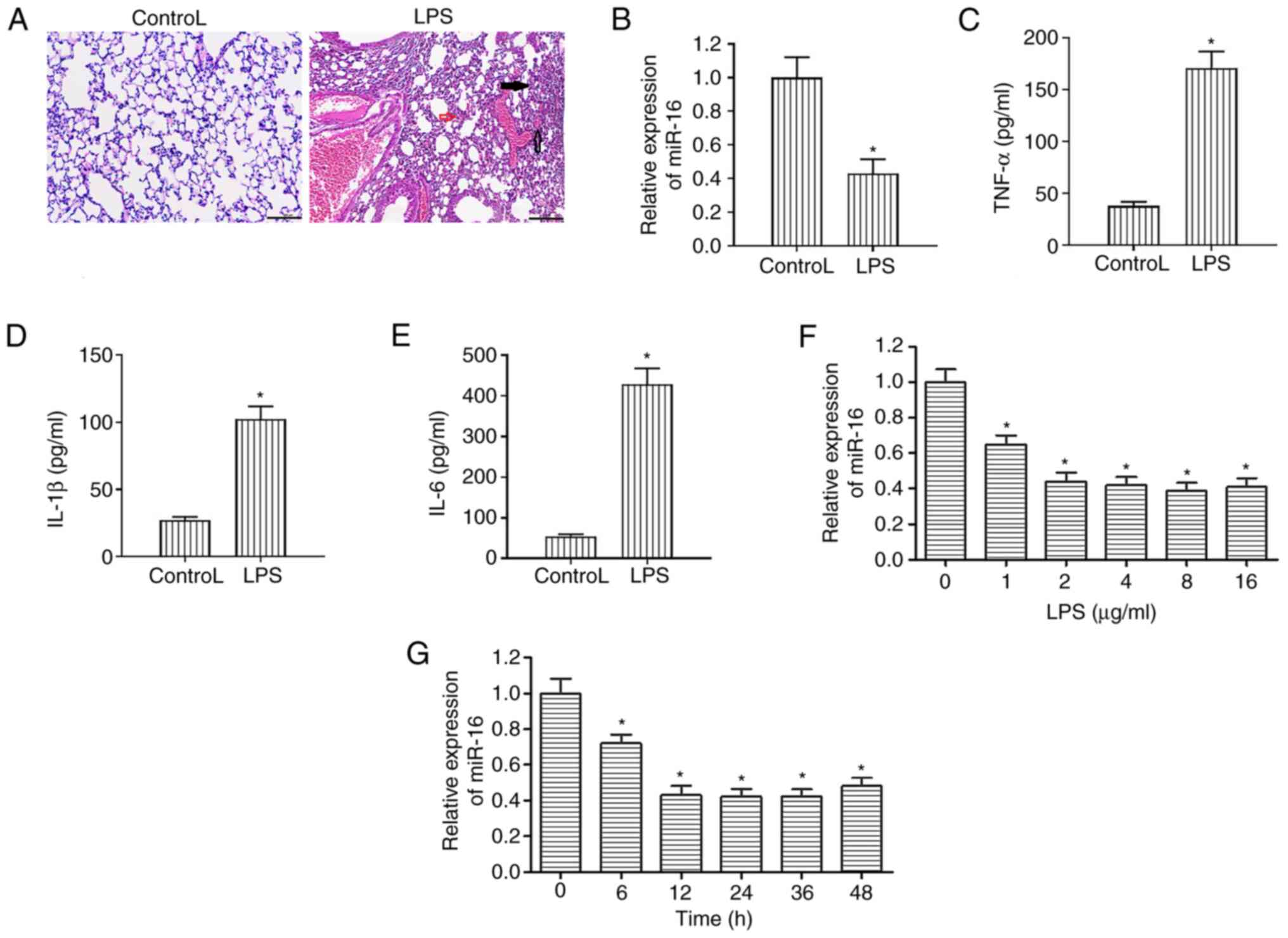 | Figure 1Treatment of rats and NHBE cells with
LPS downregulates miR-16 expression. (A) Hematoxylin and eosin
staining revealed histological differences between the lung tissues
from rats in the LPS and control groups. The lung tissues from the
LPS group showed lung injury, including hemorrhage (clear arrow),
interstitial edema (red arrow) and infiltration of inflammatory
cells (black arrow). Scale bar, 100 µm. (B) Differences in miR-16
expression between rats in the LPS and control groups were assessed
by reverse transcription-quantitative PCR. The levels of (C) TNF-α,
(D) IL-1β and (E) IL-6 were measured in rats treated with LPS or
PBS (control) using ELISA. *P<0.05 vs. Control. (F)
miR-16 expression was decreased following treatment of NHBE cells
with increasing concentrations of LPS (0, 1, 2, 3, 4, 8 and 16
µg/ml) for 12 h as assessed by reverse transcription-quantitative
PCR. *P<0.05 vs. 0 group. (G) miR-16 expression was
gradually reduced in NHBE cells incubated with 2 µg/ml LPS for a
range of indicated time durations (0, 6, 12, 24, 36 and 48 h) as
assessed by reverse transcription-quantitative PCR.
*P<0.05 vs. 0 group. *P<0.05. LPS,
lipopolysaccharide; ALI, acute lung injury; TNF-α, tumor necrosis
factor α; IL, interleukin; NHBE, normal human bronchial epithelial;
NC, negative control; miR, microRNA. |
In NHBE cells treated with different concentrations
(Fig. 1F) of LPS for different time
periods (Fig. 1G), the expression
levels of miR-16 were markedly decreased compared with those in the
0 group. These data suggested a potential association between the
expression of miR-16 and the LPS-induced inflammatory response in
the lung.
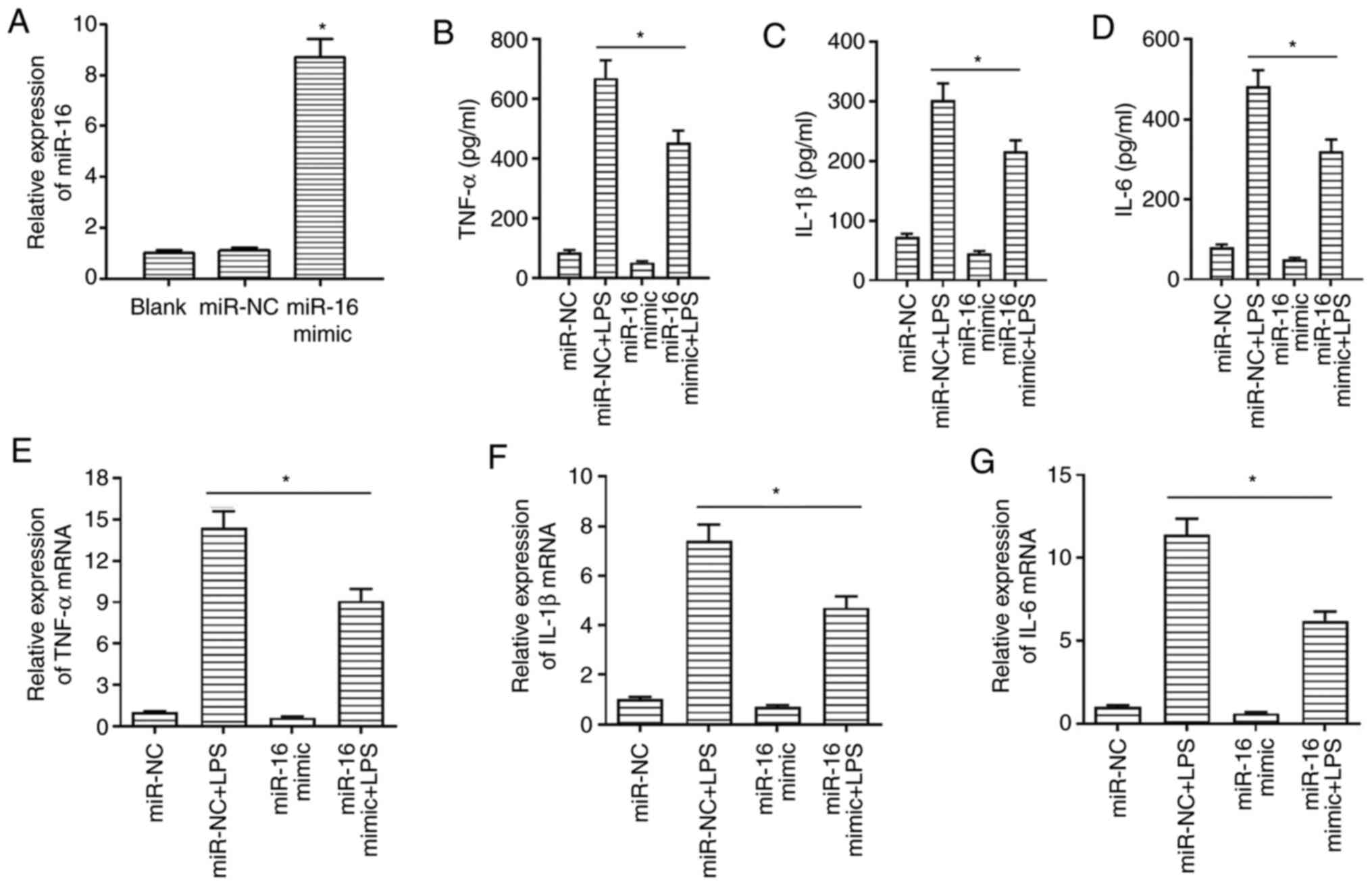
miR-16 attenuates the secretion of
proinflammatory cytokines by LPS-induced NHBE cells
To investigate the role of miR-16 in ALI, NHBE cells
were transfected with either miR-16 mimics or miR-NC, which
significantly increased miR-16 expression compared with that in the
miR-NC group (Fig. 2A). The
secretion levels of IL-1β, TNF-α and IL-6 were determined by ELISA
in cell culture supernatant treated for 12 h with LPS (2 µg/ml) and
control cells after transfection. The results demonstrated that the
secretion levels of proinflammatory cytokines were markedly reduced
in cells in the miR-16 mimic + LPS group compared with those in the
miR-NC + LPS group (Fig. 2B-D).
Additionally, mRNA expression levels of TNF-α, IL-1β and IL-6 were
also assessed by RT-qPCR in NHBE cells (Fig. 2E-G). Consistent with the ELISA data,
the expression levels of the proinflammatory cytokines were also
decreased in the miR-16 mimic + LPS group compared with those in
the miR-NC + LPS group. These findings suggest that miR-16 can
negatively regulate the expression of inflammatory factors.
However, whether miR-16 can directly target these inflammatory
cytokines requires further investigation.
TLR4 is directly targeted by
miR-16
A conserved binding region between the 3'-UTR of
TLR4 mRNA and miR-16 was identified by TargetScan analysis
(Fig. 3A). It was therefore
hypothesized that miR-16 could directly target TLR4. Western blot
analysis revealed that TLR4 was downregulated in NHBE cells
transfected with miR-16 mimics compared with that in cells
transfected with miR-NC (Fig. 3C).
To verify if TLR4 can be directly targeted by miR-16 in
vitro, a luciferase reporter assay was performed. The data
revealed that the luciferase activity of the
pmirGLO3-TLR4-3'-UTR-Wt reporter was significantly suppressed in
the miR-16 mimics group compared with the that in miR-NC group
(Fig. 3B). However, miR-16 mimic
had no effect on the luciferase activity of the
pmirGLO3-TLR4-3'-UTR-MUT construct (Fig. 3B). These findings indicated that
miR-16 could directly target TLR4 3'-UTR to suppress its expression
on the post-transcriptional level.
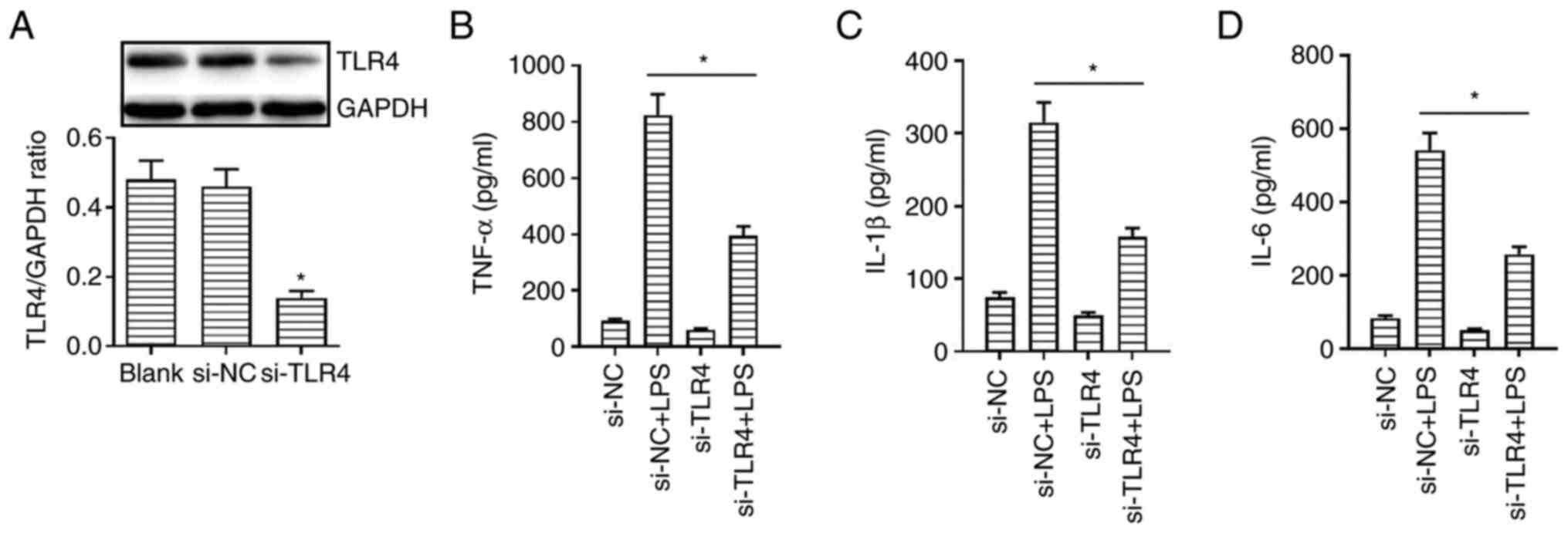
TLR4 silencing alleviates LPS-induced
inflammatory responses
To further evaluate the effect of TLR4 on
LPS-induced inflammatory responses, cells were transfected with
specific siRNAs targeting TLR4. A shown in Fig. 4A, transfection of cells with si-TLR4
significantly downregulated TLR4 protein expression compared with
that in cells transfected with si-NC. TLR4 knockdown also
significantly attenuated the LPS-induced secretion of IL-1β, TNF-α
and IL-6 by NHBE cells compared with those in LPS-treated cells
transfected with si-NC (Fig. 4B-D).
Therefore, these data suggested that TLR4 knockdown could reduce
the production of proinflammatory cytokines.
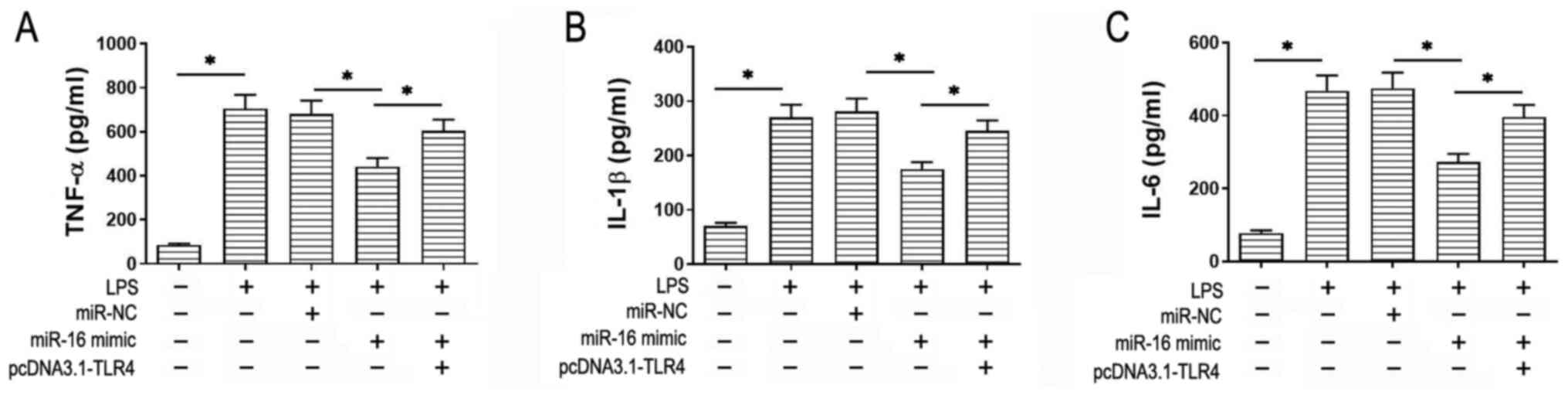
miR-16 regulates inflammatory
responses to LPS in NHBE cells by targeting TLR4
Subsequently, TLR4 was overexpressed in NHBE cells
that were also transfected with miR-16 mimics to investigate
whether miR-16 could regulate the inflammatory responses by
targeting TLR4. Following transfection with miR-16 mimics and or
pcDNA3.1-TLR4, cells were treated with LPS (2 µg/ml) before the
levels of the proinflammatory cytokines were measured by ELISA. As
shown in Fig. 5A-C, cell treatment
with LPS increased the secretion levels of IL-1β, TNF-α and IL-6
compared with those in the control, which were reversed by miR-16
overexpression compared with those in the LPS + miR-NC. However,
the protective mechanism of miR-16 against this particular
inflammatory response as reported by IL-1β, TNF-α and IL-6 levels
were significantly negated following TLR4 overexpression. Strict
transfection efficiency controls were also set up (Fig. S1). To conclude, these
aforementioned findings suggested that miR-16 may suppress the
secretion of proinflammatory cytokines by targeting TLR4.
Discussion
ALI is an inflammatory pulmonary condition that is
characterized by refractory hypoxemia and dyspnea, with substantial
risk of mortality (3). However, the
mechanism underlying ALI remains poorly understood (27). It has been previously reported that
inflammatory disorders and alveolar barrier disruption are the main
causes of ALI (28). Therefore,
inhibiting inflammatory responses can be considered to effectively
improve LPS-induced ALI (29).
Acute respiratory distress syndrome (ARDS) is considered to be
severe type of ALI (3,30). In 2003, a number of patients with
severe acute respiratory syndrome (SARS) died from ARDS (31,32).
In addition, since the end of 2019, a severe pneumonia caused
coronavirus disease 2019 (COVID-19) has become a global pandemic.
This disease is caused by the SARS-coronavirus-2 (SARS-CoV-2),
which is similar to SARS-CoV (33).
The majority of patients with COVID-19 suffer from a series of
systemic symptoms, which may progress into ARDS (34). However, a number of studies have
shown that the TLR4 signaling pathway can control the progress of
ALI (32,35).
TLR4 serves an essential role in the immne and
inflammatory response (36). TLR4
is not only involved in acquired immunity, but also in innate
immunity (37). LPS is a component
of the bacterial endotoxin that can activate TLR4 via the
TLR4/myeloid differentiation primary response 88 or
TLR4/TIR-domain-containing adapter-inducing interferon-β/tumor
necrosis factor receptor-associated factor 6 pathway to promote
inflammation in the lungs (38).
Therefore, IL-1β, TNF-α and IL-6 may be produced and aggravate ALI
(27). Additionally, suppression of
TLR4 may be associated with the alleviation of inflammatory
reactions in a mouse lung injury model induced by LPS (39). Consistent with this hypothesis, it
was previously demonstrated that dioscin (40), 25-hydroxycholesterol (41) and antagonists of the TLR4 signaling
may alleviate LPS-induced ALI (42).
miRNAs have become a popular topic of scientific
research, due to reports of their involvement in several biological
processes, including cell apoptosis, proliferation and
tumorigenesis (43). Therefore, it
has been suggested that miRNAs may act as regulatory factors that
can reduce the risk of a various diseases and improve the efficacy
of therapy by regulating the expression of target mRNAs (44). For example, a previous study
revealed that miR-302b could inhibit alveolar macrophages and
epithelial cell inflammatory responses against bacterial infections
by providing negative feedback to TLRs-mediated immunity (45). Another study reported that miR-203
inhibited ovarian tumor metastasis by targeting the transforming
growth factor-β pathway (46).
Furthermore, miRNAs may be associated with the release of
inflammatory cytokines. It has been documented that miR-182-5p
(47), miR-106a (24) and miR-302b (45) upregulate the expression levels of
inflammatory cytokines by targeting TLR4 to promote lung injury.
miR-183-5p overexpression was previously reported to downregulate
TLR4, which lead to the inhibition of inflammatory cytokine
secretion (48). It has also been
previously suggested that miR-16 may be used as a prognostic
biomarker for chronic lymphocytic leukemia (CLL), since miR-16
downregulated bcl-2 expression to induce apoptosis in B-cell CLL
(49). In addition, another study
suggested that miR-16 may improve the prognosis of malignant
mesothelioma, where after delivery of a miR-16 TargomiR, patients
assessed by CT achieved disease control (50). Although some molecular mechanisms
remain unknown, it is becoming well supported that miR-16 serves an
significant role in the progression of several diseases. However,
the role of miR-16 in LPS-induced ALI remains unclear.
A similar study has been recently published by Yang
et al (21), which reported
that upregulation of miR-16 can inhibit the expression of the
nucleotide-binding oligomerization domain, leucine rich repeat and
pyrin domain containing 3(NPRP3) inflammasome via TLR4 in A549
cells and a ALI model (21).
However, this previous study did not perform rescue experiments to
prove that TLR4 is an essential component, where miR-16 may
regulate the NLRP3 inflammasome via other pathways. The present
study used pcDNA3.1-TLR4 to confirm that TLR4 overexpression can
reverse the downregulation of inflammatory cytokines caused by
miR-16 overexpression. Also in the present study the ELISA results
demonstrated that LPS-induced NHBE cells secreted increased levels
of the inflammatory cytokines TNF-α, IL-1β and IL-6. However,
miR-16 overexpression reduced the levels of IL-1β, TNF-α and IL-6,
whilst this reduction in inflammatory cytokine secretion was
negated when miR-16 and TLR4 were simultaneously overexpressed.
RT-qPCR analysis revealed that miR-16 overexpression suppressed the
expression of TLR4. Subsequently, the present study investigated
the mechanism underlying the inhibitory effects of miR-16 on the
inflammatory response. The results revealed that TLR4
downregulation downstream of miR-16 attenuated the inflammatory
response in NHBE. Unfortunately, the protective effect of miR-16
was not verified in the animal experiments, which is a limitation
of the present study. However, data from the present study appeared
to be consistent with the data from the animal experiments
previously performed by Yang et al (21), which provide support for the present
findings.
To conclude, the present study demonstrated that
miR-16 exerted its protective effects against ALI-induced
inflammatory responses by modulating TLR4. The effective control of
ALI remains challenging. miR-16 may provide reference for the
clinical treatment of ALI, or even ARDS. Finally, although the
present study demonstrated that miR-16 may play a key role in
regulating LPS-induced ALI, further extensive scientific research
is required before these findings will be applicable in clinical
practice.
Supplementary Material
Transfection efficiency of miR-16 and
TLR4 in NHBE cells. (A) Expression levels of miR-16: i) The levels
of miR-16 was upregulated after cells were transfected with the
miR-16 mimic compared with miR-NC; ii) miR-16 expression was
downregulated in LPS-treated NHBE cells, which was reversed by
transfection with the miR-16 mimic compared with the miR-NC + LPS
group; and iii) transfection with pcDNA.3.1-TLR4 alone exerted no
significant effects on the expression levels of miR-16 regardless
of LPS presence. (B) Expression levels of TLR4: i) Without LPS
treatment, the protein expression levels of TLR4 was significantly
increased in NHBE following pcDNA-TLR4 transfection compared with
the pcDNA3.1-NC group; ii) in the absence of LPS treatment, TLR4
expression was significantly downregulated by miR-16 mimic
transfection in NHBE cells compared with the miR-NC group; iii)
There was no significant effect on TLR4 expression after
stimulation by LPS alone; iv) TLR4 protein expression was
significantly increased in LPS-treated NHBE cells following
pcDNA-TLR4 transfection compared with the pcDNA3.1-NC + LPS group;
and v) TLR4 expression was decreased in LPS-treated NHBE cells
following miR-16 mimic and pcDNA.3.1-TLR4 co-transfection compared
with the pcDNA3.1-TLR4 + LPS group miR, microRNA; TLR4, toll-like
receptor 4; NHBE, normal human bronchial epithelial cells; LPS,
lipopolysaccharide. *P<0.05. LPS, lipopolysaccharide;
miR, microRNA; NC, negative control; TLR4, toll-like receptor 4;
NHBE, normal human bronchial epithelial.
Acknowledgements
Not applicable.
Funding
Funding: The present study is supported by the project of
National Natural Science Foundation of China (grant no.
81972182).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HW conceived and designed the experiments. XL and QC
performed the experiments. XL and HW analyzed the data. XL and HW
can authenticate the raw data in this study. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Animal protocols were approved by the Ethics
Committee Of The First Affiliated Hospital Of Zhengzhou University
(Zhengzhou, China) and all animal experiments were operated
according to National Institutes of Health Guidelines for the Care
and Use of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wheeler AP and Bernard GR: Acute lung
injury and the acute respiratory distress syndrome: A clinical
review. Lancet. 369:1553–1564. 2007.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Fan E, Brodie D and Slutsky AS: Acute
respiratory distress syndrome: Advances in diagnosis and treatment.
JAMA. 319:698–710. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
He YQ, Zhou CC, Yu LY, Wang L, Deng JL,
Tao YL, Zhang F and Chen WS: Natural product derived phytochemicals
in managing acute lung injury by multiple mechanisms. Pharmacol
Res. 163(105224)2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Matthay MA and Zimmerman GA: Acute lung
injury and the acute respiratory distress syndrome: Four decades of
inquiry into pathogenesis and rational management. Am J Respir Cell
Mol Biol. 33:319–327. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sahetya SK, Goligher EC and Brower RG:
Fifty years of research in ARDS. setting positive end-expiratory
pressure in acute respiratory distress syndrome. Am J Respir Crit
Care Med. 195:1429–1438. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Deng JC and Standiford TJ: Growth factors
and cytokines in acute lung injury. Compr Physiol. 1:81–104.
2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Grommes J, Morgelin M and Soehnlein O:
Pioglitazone attenuates endotoxin-induced acute lung injury by
reducing neutrophil recruitment. Eur Respir J. 40:416–423.
2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Johnston CJ, Finkelstein JN, Gelein R and
Oberdorster G: Pulmonary cytokine and chemokine mRNA levels after
inhalation of lipopolysaccharide in C57BL/6 mice. Toxicol Sci.
46:300–307. 1998.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Martins HC and Schratt G:
MicroRNA-dependent control of neuroplasticity in affective
disorders. Transl Psychiatry. 11(263)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhao W, Sun Q, Yu Z, Mao S, Jin Y, Li J,
Jiang Z, Zhang Y, Chen M, Chen P, et al: MiR-320a-3p/ELF3 axis
regulates cell metastasis and invasion in non-small cell lung
cancer via PI3K/Akt pathway. Gene. 670:31–37. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ferruelo A, Penuelas O and Lorente JA:
MicroRNAs as biomarkers of acute lung injury. Ann Transl Med.
6(34)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Nusslein-Volhard C and Wieschaus E:
Mutations affecting segment number and polarity in
Drosophila. Nature. 287:795–801. 1980.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Ciesielska A, Matyjek M and Kwiatkowska K:
TLR4 and CD14 trafficking and its influence on LPS-induced
pro-inflammatory signaling. Cell Mol Life Sci. 78:1233–1261.
2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Plociennikowska A, Hromada-Judycka A,
Borzecka K and Kwiatkowska K: Co-operation of TLR4 and raft
proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life
Sci. 72:557–581. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sukkar MB, Xie S, Khorasani NM, Kon OM,
Stanbridge R, Issa R and Chung KF: Toll-like receptor 2, 3, and 4
expression and function in human airway smooth muscle. J Allergy
Clin Immunol. 118:641–648. 2006.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Baumgarten G, Knuefermann P, Wrigge H,
Putensen C, Stapel H, Fink K, Meyer R, Hoeft A and Grohé C: Role of
Toll-like receptor 4 for the pathogenesis of acute lung injury in
Gram-negative sepsis. Eur J Anaesthesiol. 23:1041–1048.
2006.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jiang K, Guo S, Zhang T, Yang Y, Zhao G,
Shaukat A, Wu H and Deng G: Downregulation of TLR4 by miR-181a
provides negative feedback regulation to lipopolysaccharide-induced
inflammation. Front Pharmacol. 9(142)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang J, Chen Y, Jiang K, Zhao G, Guo S,
Liu J, Yang Y and Deng G: MicroRNA-182 supplies negative feedback
regulation to ameliorate lipopolysaccharide-induced ALI in mice by
targeting TLR4. J Cell Physiol. 235:5925–5937. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Acute Respiratory Distress Syndrome
Network. Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT
and Wheeler A: Ventilation with lower tidal volumes as compared
with traditional tidal volumes for acute lung injury and the acute
respiratory distress syndrome. N Engl J Med. 342:1301–1308.
2000.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Guillamat-Prats R, Puig F,
Camprubi-Rimblas M, Herrero R, Serrano-Mollar A, Gómez MN, Tijero
J, Matthay MA, Blanch L and Artigas A: Intratracheal instillation
of alveolar type II cells enhances recovery from acute lung injury
in rats. J Heart Lung Transplant. 37:782–791. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yang Y, Yang F, Yu X, Wang B, Yang Y and
Zhou X, Cheng R, Xia S and Zhou X: MiR-16 inhibits NLRP3
inflammasome activation by directly targeting TLR4 in acute lung
injury. Biomed Pharmacother. 112(108664)2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
McCarter SD, Mei SH, Lai PF, Zhang QW,
Parker CH, Suen RS, Hood RD, Zhao YD, Deng Y, Han RN, et al:
Cell-based angiopoietin-1 gene therapy for acute lung injury. Am J
Respir Crit Care Med. 175:1014–1026. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wu CT, Huang Y, Pei ZY, Xi X and Zhu GF:
MicroRNA-326 aggravates acute lung injury in septic shock by
mediating the NF-kappaB signaling pathway. Int J Biochem Cell Biol.
101:1–11. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yang J, Chen Y, Jiang K, Yang Y, Zhao G,
Guo S and Deng G: MicroRNA-106a provides negative feedback
regulation in lipopolysaccharide-induced inflammation by targeting
TLR4. Int J Biol Sci. 15:2308–2319. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chungen Y, Dongfang Z and Guoyuan X:
MicroRNA-146a protects against ischemia/reperfusion liver injury
through inhibition of toll-like receptor 4 signaling pathway in
rats. Transplant Proc. 52:1007–1013. 2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Butt Y, Kurdowska A and Allen TC: Acute
lung injury: A clinical and molecular review. Arch Pathol Lab Med.
140:345–350. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Xu B, Chen SS, Liu MZ, Gan CX, Li JQ and
Guo GH: Stem cell derived exosomes-based therapy for acute lung
injury and acute respiratory distress syndrome: A novel therapeutic
strategy. Life Sci. 254(117766)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Matthay MA, Ware LB and Zimmerman GA: The
acute respiratory distress syndrome. J Clin Invest. 122:2731–2740.
2012.PubMed/NCBI View
Article : Google Scholar
|
|
30
|
Gao XQ, Li YF and Jiang ZL: Impact of
statins on ALI/ARDS: A meta-analysis. Pulm Pharmacol Ther.
39:85–91. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Shah D, Das P, Acharya S, Agarwal B,
Christensen DJ, Robertson SM and Bhandari V: Small immunomodulatory
molecules as potential therapeutics in experimental murine models
of acute lung injury (ALI)/acute respiratory distress syndrome
(ARDS). Int J Mol Sci. 22(2573)2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Imai Y, Kuba K, Neely GG,
Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen
R, Leung YH, Wang H, et al: Identification of oxidative stress and
Toll-like receptor 4 signaling as a key pathway of acute lung
injury. Cell. 133:235–249. 2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sun J, Zhuang Z, Zheng J, Li K, Wong RL,
Liu D, Huang J, He J, Zhu A, Zhao J, et al: Generation of a Broadly
useful model for COVID-19 pathogenesis, vaccination, and treatment.
Cell. 182:734–743.e5. 2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Pascarella G, Strumia A, Piliego C, Bruno
F, Del Buono R, Costa F, Scarlata S and Agrò FE: COVID-19 diagnosis
and management: A comprehensive review. J Intern Med. 288:192–206.
2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhou M, Zhang Y, Tang R, Liu H, Du M, Gao
Z, Ji Z and Fang H: HMGB1/TLR4 signaling affects regulatory T cells
in acute lung injury. J Inflamm Res. 14:1551–1561. 2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhang W, Zhuang N, Liu X, He L, He Y,
Mahinthichaichan P, Zhang H, Kang Y, Lu Y, Wu Q, et al: The
metabolic regulator Lamtor5 suppresses inflammatory signaling via
regulating mTOR-mediated TLR4 degradation. Cell Mol Immunol.
17:1063–1076. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Knoops B, Becker S, Poncin MA, Glibert J,
Derclaye S, Clippe A and Alsteens D: Specific interactions measured
by AFM on living cells between peroxiredoxin-5 and TLR4: Relevance
for mechanisms of innate immunity. Cell Chem Biol. 25:550–559.e3.
2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Martin TR and Wurfel MM: A TRIFfic
perspective on acute lung injury. Cell. 133:208–210.
2008.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Feng L, Yang N, Li C, Tian G, Wang J, Dong
ZB, Jia XB and Di LQ: Pudilan xiaoyan oral liquid alleviates
LPS-induced respiratory injury through decreasing nitroxidative
stress and blocking TLR4 activation along with NF-ΚB
phosphorylation in mice. J Ethnopharmacol. 214:292–300.
2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wang C, Li Q and Li T: Dioscin alleviates
lipopolysaccharide-induced acute lung injury through suppression of
TLR4 signaling pathways. Exp Lung Res. 46:11–22. 2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Ouyang W, Zhou H, Liu C, Wang S, Han Y,
Xia J and Xu F: 25-Hydroxycholesterol protects against acute lung
injury via targeting MD-2. J Cell Mol Med. 22:5494–5503.
2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Shirey KA, Lai W, Scott AJ, Lipsky M,
Mistry P, Pletneva LM, Karp CL, McAlees J, Gioannini TL, Weiss J,
et al: The TLR4 antagonist Eritoran protects mice from lethal
influenza infection. Nature. 497:498–502. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Cheng AM, Byrom MW, Shelton J and Ford LP:
Antisense inhibition of human miRNAs and indications for an
involvement of miRNA in cell growth and apoptosis. Nucleic Acids
Res. 33:1290–1297. 2005.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Liu CJ, Fu X, Xia M, Zhang Q, Gu Z and Guo
AY: MiRNASNP-v3: A comprehensive database for SNPs and
disease-related variations in miRNAs and miRNA targets. Nucleic
Acids Res. 49 (D1):D1276–D1281. 2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhou X, Li X, Ye Y, Zhao K, Zhuang Y, Li
Y, Wei Y and Wu M: MicroRNA-302b augments host defense to bacteria
by regulating inflammatory responses via feedback to TLR/IRAK4
circuits. Nat Commun. 5(3619)2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Wang B, Li X, Zhao G, Yan H, Dong P,
Watari H, Sims M, Li W, Pfeffer LM, Guo Y and Yue J: MiR-203
inhibits ovarian tumor metastasis by targeting BIRC5 and
attenuating the TGFβ pathway. J Exp Clin Cancer Res.
37(235)2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Zhu M, Li Y and Sun K: MicroRNA-182-5p
inhibits inflammation in LPS-treated RAW264.7 cells by mediating
the TLR4/NF-κB signaling pathway. Int J Clin Exp Pathol.
11:5725–5734. 2018.PubMed/NCBI
|
|
48
|
Goncalves Fernandes J, Morford LA,
Harrison PL, Kompotiati T, Huang H, Aukhil I, Wallet SM and
Macchion Shaddox L: Dysregulation of genes and microRNAs in
localized aggressive periodontitis. J Clin Periodontol.
47:1317–1325. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Braga TV, Evangelista FCG, Gomes LC,
Araujo SSDS, Carvalho MDG and Sabino AP: Evaluation of MiR-15a and
MiR-16-1 as prognostic biomarkers in chronic lymphocytic leukemia.
Biomed Pharmacother. 92:864–869. 2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Fennell D: MiR-16: Expanding the range of
molecular targets in mesothelioma. Lancet Oncol. 18:1296–1297.
2017.PubMed/NCBI View Article : Google Scholar
|