Introduction
Hypertrophic cardiomyopathy (HCM) is a primary
autosomal-dominant inherited myocardial disorder characterized by
unexplained left ventricular hypertrophy (1). Studies over the last 50 years have
indicated that HCM is a familial heart disease with a risk of
sudden cardiac death (SCD). SCD, heart failure and thromboembolism
are the three major causes of HCM-related death (2). SCD is mostly associated with fatal
arrhythmias, which mainly include ventricular tachycardia
(continuous or non-sustained), ventricular fibrillation and
atrioventricular blockage. Furthermore, ~1 million individuals in
China suffer from this disease, with an annual mortality rate of
3-6%. HCM is one of the leading causes of sudden death in
adolescents and athletes (3,4).
HCM is the most common monogenic heart disease, and
its causative genes are relatively well defined (5). It is mainly caused by a mutation in a
gene that encodes a sarcomeric structural protein. The most common
disease-causing genes encode myosin heavy chain 7 (MYH7), myosin
binding protein C3 (MYBPC3), tropomyosin 1 (TPM1), troponin T2,
cardiac type (TNNT2), TNNI3, actin alpha cardiac muscle 1 (ACTC1),
myosin regulatory light chain 2 (MYL2) and MYL3 proteins (6-11).
FH2 domain-containing protein 3 is a novel pathogenic gene of HCM
(12). Among these genes, HCM
caused by MYH7 gene mutations accounts for 30-50% of the total
number of cases (13). Although the
MYH7 gene is currently considered to be the major causative gene of
HCM, the mechanisms by which its mutations cause HCM have remained
to be fully elucidated. Currently recognized mechanisms include
Ca2+ homeostasis, myocardial fibrosis and energy
imbalance (14,15). The present study performed
whole-exome sequencing (WES) combined with Sanger sequencing to
identify genetic abnormalities in a pedigree with familial HCM.
Patients and methods
Subjects
The subjects of the present study were a Han Chinese
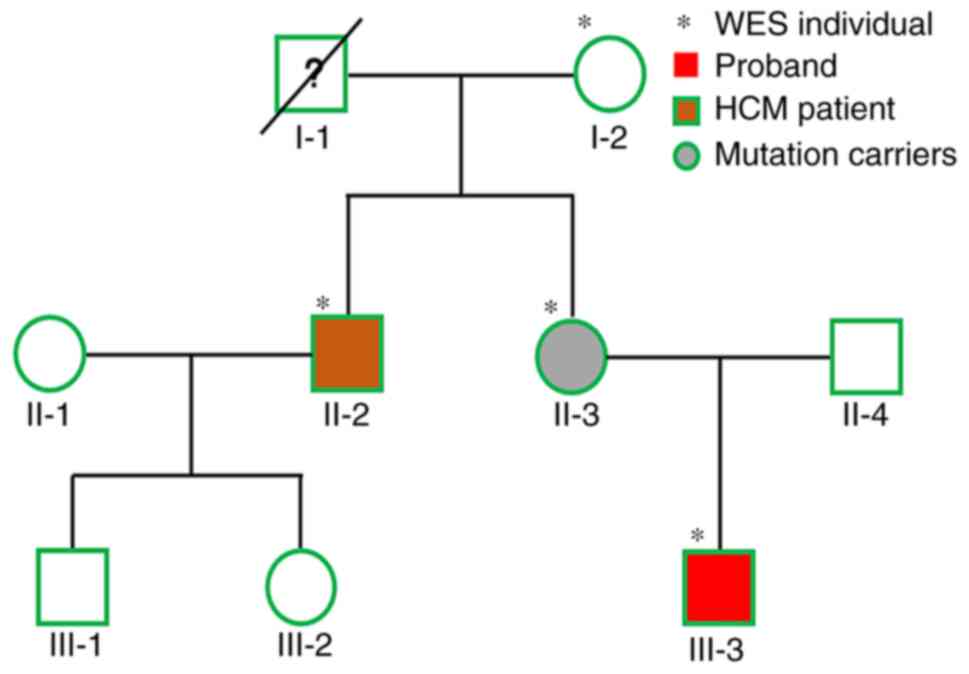
family with HCM from Guangdong (China). A pedigree chart is
provided in Fig. 1. The proband
(patient III-3) was a 17-year-old male (born in November 2000) who
was admitted to the Second Affiliated Hospital of Shantou
University Medical College (Shantou, China) with sudden cardiac
arrest in September 2017. Based on the 2014 European Society of
Cardiology (ESC) guidelines (16),
the diagnostic criteria for HCM in adults are as follows: Arbitrary
imaging (echocardiography, cardiac magnetic resonance imaging or
computed tomography) indicated that a segment or segments of the
left ventricular myocardium are not entirely due to abnormal
cardiac load and segment wall thickness ≥15 mm (in adults) or left
ventricular wall thickness ≥ predicted mean +2X the standard
deviation (in children aged <14 years). If for the first-degree
relatives of the patient, after exclusion of other causes, cardiac
imaging suggests that a segment of the left ventricular wall or a
multi-stage thickness of ≥13 mm is present, a diagnosis of familial
HCM may be made (16). According to
the aforementioned information, the proband's uncle (patient II-2)
met the diagnostic criteria for HCM but had no clinical symptoms.
In addition, the proband's mother (patient II-3) also exhibited
structural changes on echocardiography but no clinical symptoms.
The proband's grandfather (I-1) suffered a sudden cardiac death at
40 years of age. The echocardiography result was normal in the
grandmother (65-year-old) of the proband (I-2), who was set as the
control in the present study. Hence, in the present study, four
participants from three generations were investigated (I-2, II-2,
II-3 and III-3; Fig. 1). Clinical
data included results of physical examinations, laboratory tests
(liver and kidney function, blood routine, rheumatoid immune index
and sex hormone levels), electrocardiography and two-dimensional
and Doppler echocardiographic examinations. All subjects signed
informed consent forms. The present study was implemented strictly
in accordance with the Declaration of Helsinki and approved by the
Ethics Committee of the Second Affiliated Hospital of Shantou
University Medical College (Shantou, China).
WES and mutation screening
To extract genomic DNA, peripheral venous blood was
collected from the participants and EDTA was used as the
anticoagulant. A blood extraction kit (cat. no. 51304; Invitrogen;
Thermo Fisher Scientific, Inc.) was used to extract genomic DNA
from leukocytes according to the manufacturer's protocol. Extracted
DNA samples were sent to Beijing Nuohe Zhiyuan Technology Co., Ltd.
for WES. Genomic DNA was sheared into fragments with a length of
150-200 bp through sonication (17). End-repairing, A-tailing and adaptor
ligation, a four-cycle pre-capture PCR amplification and xGen Exome
Research Panel (Integrated DNA Technologies, Inc.) enrichment were
then performed (17). The
sequencing was performed on an Illumina Hiseq Xten platform
(Illumina, Inc.) with a mean sequence coverage of ≥90X and a
coverage of ≥20X in >95% of the target bases. Cutadapt
(https://cutadapt.readthedocs.io/en/stable/) and FastQC
(http://darlinglab.org/tutorials/fastqc/) were used to
perform quality control. Clean reads were mapped to the human
reference genome (University of California Santa Cruz hg19) via
Sentieon BWA (version 0.7.15) (18). Duplicate sequence reads were removed
using Picard (version 1.85) and variants were detected by GATK
(version 3.1) (19). Variants were
annotated using ANNOVAR software (version from 14 December 2015;
http://annovar.openbioinformatics.org/en/latest/).
All exome variants were filtered for allele frequencies <0.001
in the ExAC database (20). The
nomenclature of variants was based on the guidelines of the Human
Genome Variation Society (21).
Sanger sequencing
Sanger sequencing was performed to verify the
variants in MYH7 identified by WES. PCR was performed to amplify
the exon of MYH7. The upstream sequence of the primer was
5'-CATCTCCTGGCCTCTTCACTTA-3' and the downstream sequence was
5'-ATGTCCATCAGAGTGCCTTACA-3'. PCR was done using SYBR Premix EX
Taq™ II (Takara Bio, Inc.). The following thermocycling
conditions were used for the PCR: Initial denaturation at 94˚C for
3 min; 33 cycles of denaturation at 94˚C for 30 sec, annealing at
57˚C for 45 sec and extension at 72˚C for 45 sec, followed by a
final extension at 72˚C for 10 min. PCR products were then examined
for sequence variations by Sanger sequencing.
Bioinformatics analysis
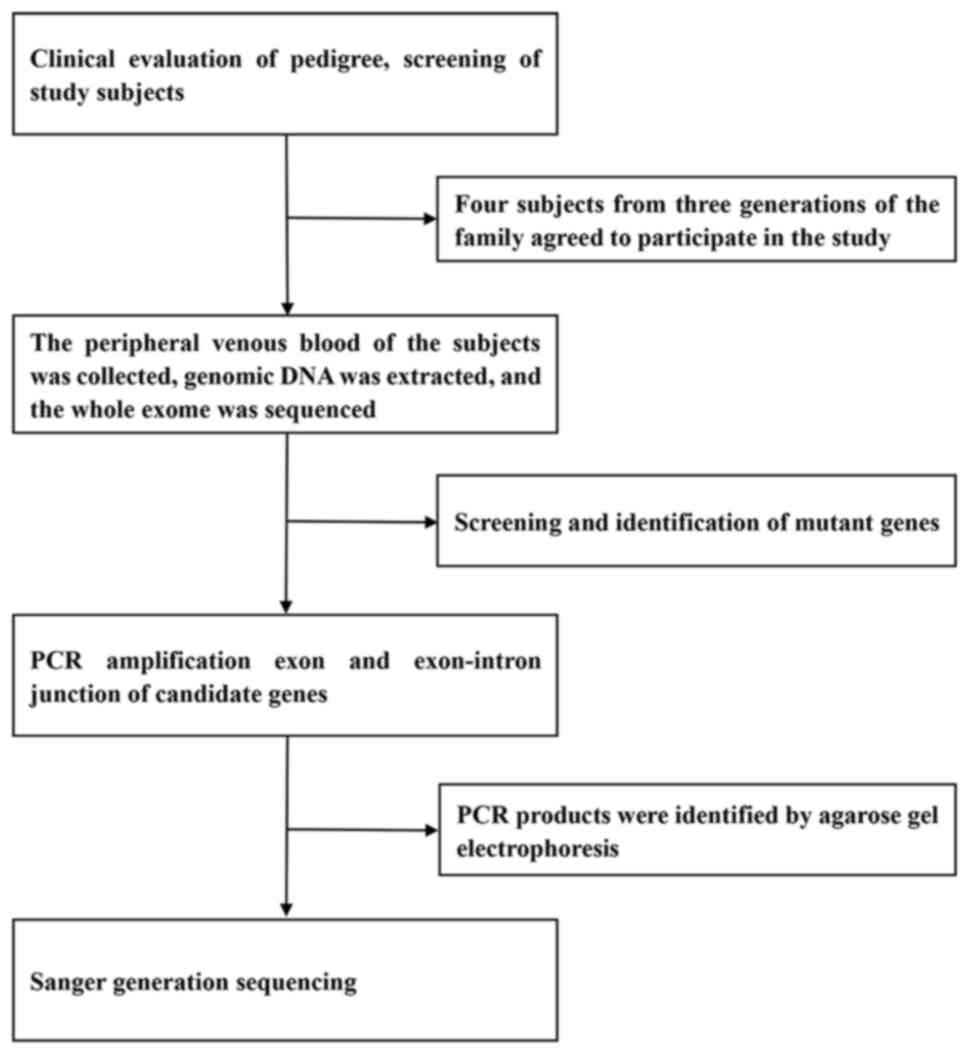
The gene detection strategies adopted in the present
study are presented in Fig. 2. The
frequencies of mutations in the general population were confirmed
using the ExAC_EAS database (https://exac.hms.harvard.edu), gnomAD database
(version r2.0; http://gnomad.brodinstitute.org) and 1000 Genomes
project (http://www.1000genomes.org). The
pathogenicity of the mutations was predicted using the single
nucleotide polymorphism database (dbSNP; http://www.ncbi.nlm.nih.gov/SNP/) and Polyphen-HCM
(http://www.genetics.bwh.harvard.edu/hcm) and they were
annotated according to the recommendations of the American College
of Medical Genetics and Genomics (22).
Results
Clinical phenotype
The proband (III-3) was a 17-year-old male who was
admitted to The Second Affiliated Hospital of Shantou University
Medical College (Shantou, China) after successful cardio-pulmonary
resuscitation (CPR) due to cardiac arrest in September 2017. The
patient had no chest pain, no history of syncope and no known
family history of hereditary diseases. The patient's grandfather
had died of a heart attack at the age of 40 years, but it was not
confirmed whether this was due to HCM. On admission, the proband
had a grade III systolic murmur at the apex of the heart.
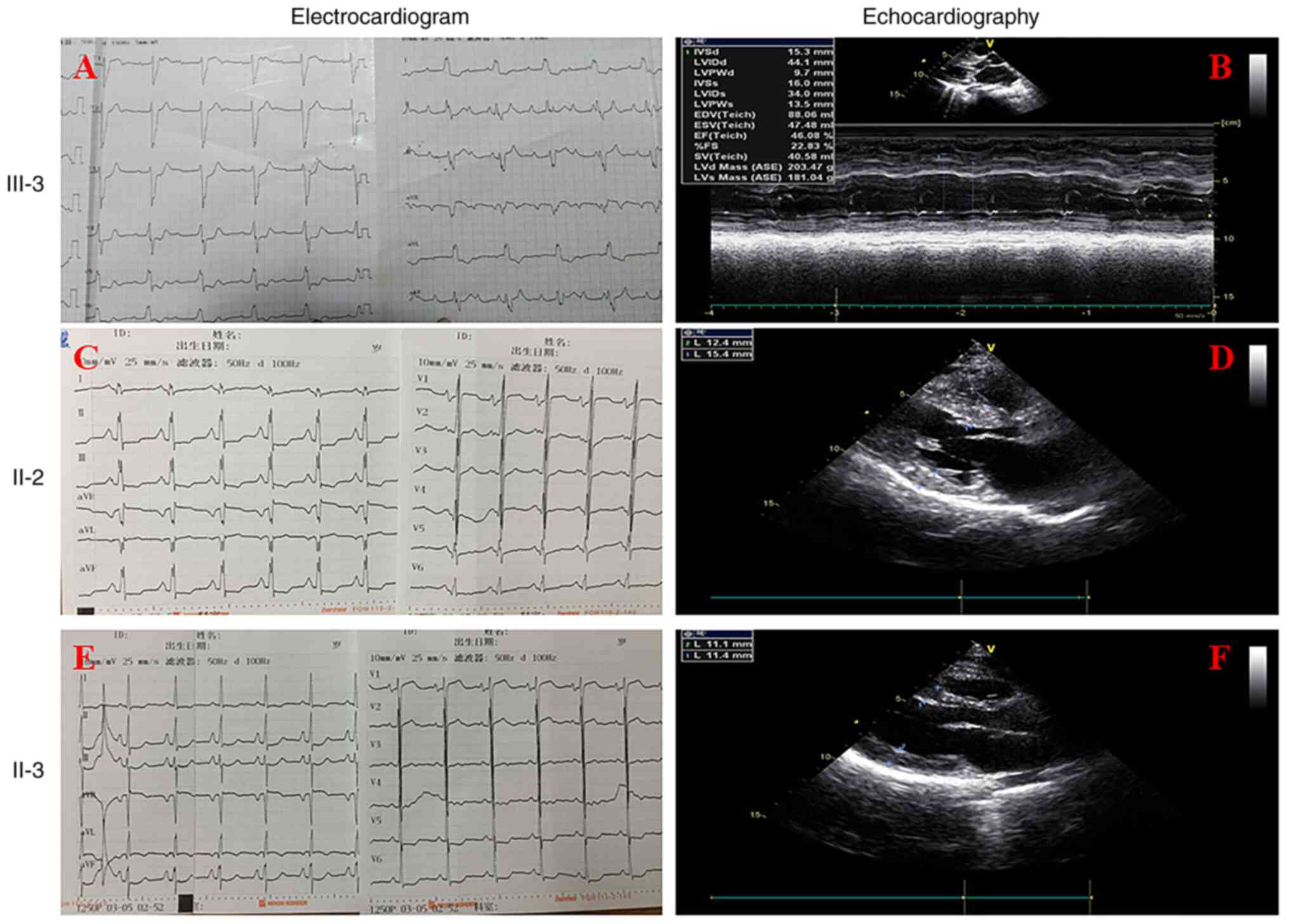
Electrocardiographic examination indicated abnormal Q waves, ST
segment depression, T wave inversion and third-degree
atrioventricular block (Fig. 3A).
Echocardiography displayed the ventricular septum (IVS; 15.3 mm)
and the posterior wall of the left ventricle (LVPW; 13.5 mm;
Table I; Fig. 3B). Examination of the thickness of
the left ventricular short-axis papillary muscle suggested that the
thickness of the hypertrophic myocardium was 20 mm (normal value,
<12 mm), the anterior wall was 19 mm (normal value, <12 mm)
and the lower wall was 19 mm (normal value, <12 mm). All of
these data were above the diagnostic lower limit of 15 mm for HCM.
Therefore, the proband was diagnosed with HCM according to the ESC
criteria aforementioned (16).
 | Table IClinical data of patients and
asymptomatic carriers in the family. |
Table I
Clinical data of patients and
asymptomatic carriers in the family.
| |
Electrocardiogram | Echocardiography |
|---|
| Patient | Sex | Age (years) | Clinical
manifestation | ST-segment
depression | Abnormal T wave | Abnormal Q wave | Other
abnormalities | IVS (mm) | LVPW (mm) | Left ventricular
dysfunction |
|---|
| III-3 (the
proband) | Male | 17 | HCM, sudden cardiac
arrest | Yes | Yes | Yes | III˚ AVB | 15.3 | 13.5 | Systolic |
| II-2 (the proband's
uncle; affected) | Male | 47 | HCM, cardiac
dysfunction | Yes | Yes | Yes | Biatrial
enlargement | 15.4 | 12.4 | Diastolic |
| II-3 (the proband's
mother; carrier without clinical symptoms) | Female | 45 | Slight structural
abnormalities of the heart, but without cardiac dysfunction | Yes | Yes | Yes | None | 11.4 | 11.1 | None |
Detailed clinical data of the family are presented
in Table I and Fig. 3. The proband's uncle (II-2) was also
confirmed to have HCM by echocardiography (IVS, 15.4 mm; LVPW, 12.4
mm) and presenting with cardiac dysfunction (Table I; Fig.
3D). Although this was not clinically confirmed according to
the HCM diagnostic criteria, echocardiology indicated that the
proband's mother (II-3) had an enlarged left atrium, slightly
thicker right anterior wall and anterior septum and an expanded
atrial septum (Table I; Fig. 3F). The function of the heart was
normal in the proband's grandmother. Laboratory tests (liver and
kidney function, blood routine testing, rheumatoid immune index and
sex hormone levels) of all subjects in the present study provided
normal results. In addition, none of the subjects in this study had
living habits that are considered to be risk factors, including
smoking and drinking.
Genetic screening
DNA samples from four members (I-2, II-2, II-3 and
III-3) of the family were subjected to WES analysis (Fig. 1). Previous studies have reported
that mutations in 57 genes may cause or be associated with HCM,
including eight definitive genes (MYBPC3, MYH7, TNNT2, TNNI3, TPM1,
ACTC1, MYL2 and MYL3), three moderately evidenced genes [cysteine
and glycine-rich protein 3 (CSRP3), troponin C and junctophilin-2]
and other genes with limited or no evidence (titin, Krueppel-like
factor 10, myopalladin, ankyrin repeat domain-containing protein 1,
myosin light chain kinase 2, myozenin-2, nexilin, vinculin, E3
ubiquitin-protein ligase TRIM63, ryanodine receptor 2, MYH6,
obscurin, PDZ and LIM domain protein 3, telethonin, myomesin-1 and
calreticulin-3) (23-27).
Therefore, the present study focused on the genetic variations
(SNPs and indels) occurring in any of the aforementioned genes.
Furthermore, the present study considered only the genetic
variations that existed in both affected patients. The left
non-synonymous single nucleotide variants (SNVs) were prioritized
by the SNV Prioritization via the Integration of Genomic data
(SPRING) (http://bioinfo.au.tsinghua.edu.cn/spring).
Based on the aforementioned strategy for variation
analysis, 21 variants were obtained in the present study (19
missense SNVs and two stopgain SNVs) in 21 genes, of which only the
MYH7 mutation exhibited a close association with HCM (Table II). The mutation is located on
chromosome 14 and the SNP site (ID: rs727503263) is a heterozygous
missense mutation in the 18th exon. This mutation corresponds to a
one-base substitution at 2011 (C to T), at position 671 of the
amino acid sequence. The mutation resulted in an amino acid change
(arginine to cysteine) with a frequency of 0 in gnomAD and 1,000
Genomes and <0.001 in the ExAC_EAS exome or genome sequencing
databases (Table III). The
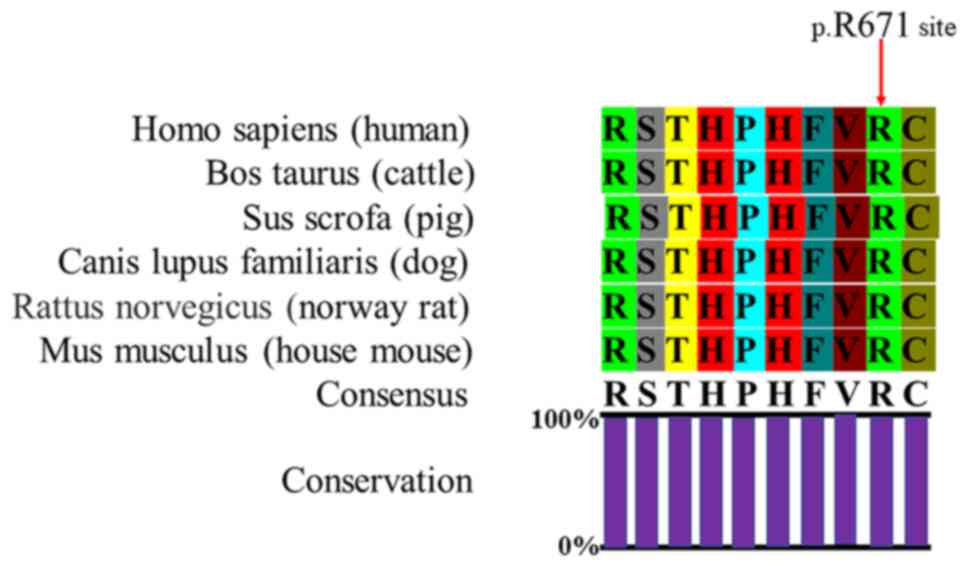
p.Arg671 variant is highly conserved in mammals, from humans to
house mouse (Fig. 4). In
silico analysis by Polyphen-HCM and SIFT suggested that the
p.R671C variant is a disease-causing variant (Table III). This variant was also
interpreted as being a likely pathogenic variant by ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/variation/164350/).
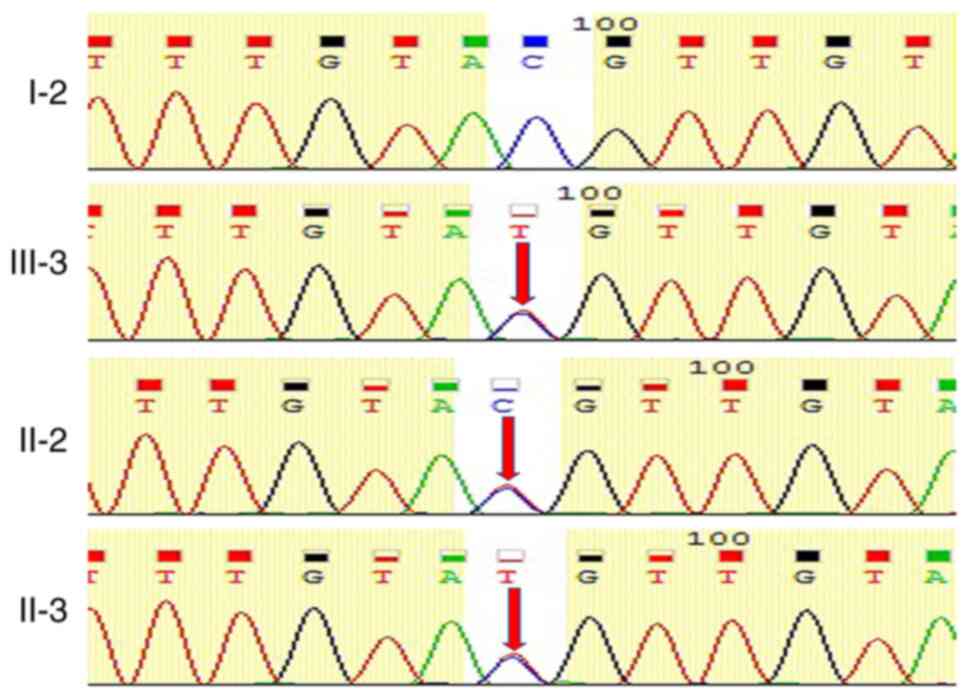
In order to confirm the mutation, the Sanger generation of the
family members were sequenced and verified for the mutation. Family
members with echocardiology changes (II-2, II-3 and III-3) harbored
the same combination of MYH7 mutations, while the healthy member
(I-2) exhibited no mutations at this position (Fig. 5). These results are consistent with
the electrocardiograms, echocardiography and clinical symptoms.
| Table IISNVs with P<0.05 analyzed by SNV
Prioritization via the Integration of Genomic data. |
Table II
SNVs with P<0.05 analyzed by SNV
Prioritization via the Integration of Genomic data.
| Gene | Amino acid
change | Type of
variant | dbSNP (rsID) | SIFT | PolyPhen2 | Likelihood ratio
test | MutationTaster |
|---|
| MYH7 | R671C | Missense | rs727503263 | D | D | D | D |
| MIB2 | R385W | Missense | rs376615315 | D | D | D | D |
| SIGLEC1 | R400C | Missense | rs76254218 | D | D | N | D |
| FNDC1 | L1839P | Missense | rs201022798 | D | D | D | D |
| HTR1B | I368T | Missense | rs761871198 | D | D | D | D |
| PCDHB10 | R289X | Stopgain | rs782513627 | Absent | Absent | Absent | D |
| OR7A5 | M59V | Missense | rs756036560 | D | D | U | N |
| DNAH14 | R2911C | Missense | rs770222940 | D | D | N | N |
| YEATS2 | T739A | Missense | rs201274382 | D | D | D | D |
| TRAPPC4 | D160H | Missense | Absent | D | D | D | D |
| DONSON | P328L | Missense | Absent | D | D | D | D |
| DBX2 | G11S | Missense | rs746897983 | D | D | U | N |
| RIMKLA | P326Q | Missense | rs199674761 | D | D | D | D |
| ALDH1L1 | D741H | Missense | rs746372949 | D | D | D | D |
| SEMA3G | D64N | Missense | rs775338504 | D | D | D | D |
| TMEM108 | S293C | Missense | Absent | D | D | N | N |
| WRAP53 | R250P | Missense | rs780547823 | D | D | D | D |
| SGCA | R110Q | Missense | rs145697858 | D | D | D | D |
| EXOSC9 | K67N | Missense | Absent | D | D | D | D |
| DNAH7 | R2105X | Stopgain | rs186849698 | Absent | Absent | D | Absent |
| THAP6 | F207S | Missense | rs180792819 | D | D | Absent | D |
| Table IIIIn silico analysis of MYH7
variants. |
Table III
In silico analysis of MYH7
variants.
| Variant | Amino acid
change |
Polyphen-HCMa | SIFTb |
ExAC_EASc | gnomADd | 1000
Genomese |
|---|
| c.2011C>T | p.R671C | Probable damage
(1.0) | Damaging (0) | <0.001 | 0 | 0 |
Family genotype-phenotype
analysis
After screening by exome sequencing, it was
determined that three subjects in the family (II-2, II-3 and
III-3), including the proband, carried a gene mutation of MYH7.
However, these three subjects exhibited differences in their
electrocardiograms, i.e. the degree of cardiac hypertrophy was not
similar. The proband and the proband's uncle were diagnosed with
HCM, but the proband's mother was not. In addition, the proband is
a survivor of sudden cardiac arrest, while the proband's uncle
presented with cardiac dysfunction. The proband's mother had no
obvious clinical symptoms and was without cardiac dysfunction. All
three family members had the same mutated gene, but had three
different clinical phenotypes, which was particularly exhibited in
the degree of cardiac hypertrophy and severity of clinical
symptoms. To improve the prognosis, the family members were
recommended to undergo clinical evaluations every 12-24 months,
including 24 h ambulatory electrocardiogram and
echocardiography.
Discussion
The present study was the first, to the best of our
knowledge, to reveal a MYH7 p.R671C mutation in three members of
the same family with HCM based on WES combined with Sanger
sequencing. Previous studies have confirmed that HCM is a
single-gene disease with an autosomal-dominant inheritance pattern
and nearly 60% of patients have familial aggregation (28,29).
The discovery of the p.Arg403Glu mutation in the MYH7 gene encoding
the β-myosin heavy chain 7 protein in a French-Canadian family
described by Perryman et al (30) was the foundation of subsequent
important discoveries. Multiple independent mutations were
identified from all genes encoding sarcomeric proteins and HCM is
currently considered a genetically heterogeneous disease. Among the
pathogenic genes known at this stage, MYH7 and MYBPC3 are the two
most common and approximately half of HCM cases were caused by
these two genes (7,31-33).
Mutations in the TNNT2, TNNI3 and TPM1 genes, which account for
HCM, are relatively rare and ~10% of cases are caused by the
aforementioned genes (33). Mutant
genes such as ACTC1, MYL2, MYL3 and CSRP3 were also detected, these
occur more rarely in patients with HCM (13). So far, although multitudinous
disease-causing genes aforementioned have been found, their causal
effects have remained to be fully elucidated. In the present study,
an affected pedigree was subjected to gene screening through WES,
which differs from multi-gene targeted sequencing used in most
previous studies. The results suggested that the family carried a
mutation in the MYH7 gene. This mutation corresponds to a one-base
substitution at base 2,011 (C to T), resulting in an amino acid
change at position 671 (arginine to cysteine). Although the
mutation of MYH7 p.R671C has been previously reported in HCM, it
was only in sporadic single cases (10,34).
To the best of our knowledge, the present study was the first to
identify the mutation in three members of the same family with
HCM.
In the present study, the family members carrying
the same gene mutation displayed differences in sex and clinical
phenotype. The proband and the proband's uncle were diagnosed with
HCM, and while the proband's mother was classified as a mutation
carrier. Although the proband's mother had some structural
abnormalities in her heart, they did not meet the diagnostic
criteria for HCM. The proband was a 17-year-old survivor of sudden
cardiac arrest with systolic dysfunction. The proband's uncle
presented with diastolic dysfunction. However, his mother has no
obvious clinical symptoms, and without cardiac dysfunction. WES
revealed that the three members of the family carried the mutant
gene MYH7, which was the likely pathogenic mutation. The functional
significance of this mutation, however, is not well understood in
familial HCM. Among the pathogenic genes known at this stage, MYH7
and MYBPC3 are the two most common, and about half of all HCM cases
are caused by these two genes. Studies have reported that MYH7
mutation at different sites may result in different prognoses of
patients, which also includes the influence of environmental
differences, such as smoking and alcohol abuse. In view of
phenotypic heterogeneity and gene polymorphisms in the family
examined in the present study, an HCM model of MYH7 p.R671c
mutation may be established by inducing pluripotent stem cells and
cardiomyocytes to further study the potential molecular mechanism
of cardiac hypertrophy (35).
In the present study, the results of WES and Sanger
generation sequencing indicated that the proband, the proband's
uncle and the proband's mother all carried a heterozygous mutation
in the MYH7 gene. In addition, members of the family with the same
mutation had different phenotypes. The degree of cardiac
hypertrophy and cardiac dysfunction were different among family
members of different sexes and age, reflecting genetic
polymorphisms and phenotypic heterogeneity in HCM.
In conclusion, the present study first reported the
p.Arg671Cys mutation of MYH7 as a likely pathogenic variant in
familial HCM. In addition, the family members carrying the same
mutated gene exhibited three distinct clinical phenotypes.
Furthermore, the present study demonstrated that WES is a powerful
tool for identifying genetic variants in HCM and may contribute to
precise diagnosis and treatment.
Acknowledgements
Not applicable.
Funding
Funding: The study was supported by grants from the Medical
Scientific Research Foundation of Guangdong Province (grant nos.
A2019130 and A2019568).
Availability of data and materials
The sequencing data used and/or analyzed during the
current study are not deposited in a public database to protect
patient privacy but are available from the corresponding author on
reasonable request.
Authors' contributions
WY, MMH, CJC and JDC were involved in the analysis
of the WES data. WY, MMH, GHZ and WW were involved in clinical data
collection, performed data analysis and interpreted the results.
CJC and JDC checked and confirmed the authenticity of the raw data.
WY, CJC and JDC were involved in manuscript preparation. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments were approved by the Ethics
Committee of the Second Affiliated Hospital of Shantou University
Medical College (Shantou, China).
Patient consent for publication
Written informed consents were obtained from all the
patients for the publication of all accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Maron BJ, Ommen SR, Semsarian C, Spirito
P, Olivotto I and Maron MS: Hypertrophic cardiomyopathy: Present
and future, with translation into contemporary cardiovascular
medicine. J Am Coll Cardiol. 64:83–99. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ho CY, Day SM, Ashley EA, Michels M,
Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, et
al: Genotype and lifetime burden of disease in hypertrophic
cardiomyopathy: Insights from the sarcomeric human cardiomyopathy
registry (SHaRe). Circulation. 138:1387–1398. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Spirito P and Autore C: Management of
hypertrophic cardiomyopathy. BMJ. 332:1251–1255. 2006.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Maron BJ: Clinical course and management
of hypertrophic cardiomyopathy. N Engl J Med. 379:655–668.
2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Coppini R, Santini L, Olivotto I, Ackerman
MJ and Cerbai E: Abnormalities in sodium current and calcium
homoeostasis as drivers of arrhythmogenesis in hypertrophic
cardiomyopathy. Cardiovasc Res. 116:1585–1599. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jääskeläinen P, Vangipurapu J, Raivo J,
Kuulasmaa T, Heliö T, Aalto-Setälä K, Kaartinen M, Ilveskoski E,
Vanninen S, Hämäläinen L, et al: Genetic basis and outcome in a
nationwide study of Finnish patients with hypertrophic
cardiomyopathy. ESC Heart Fail. 6:436–445. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Seeger T, Shrestha R, Lam CK, Chen C,
McKeithan WL, Lau E, Wnorowski A, McMullen G, Greenhaw M, Lee J, et
al: A premature termination codon mutation in MYBPC3 causes
hypertrophic cardiomyopathy via chronic activation of
nonsense-mediated decay. Circulation. 139:799–811. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lopes LR, Zekavati A, Syrris P, Hubank M,
Giambartolomei C, Dalageorgou C, Jenkins S and McKenna W: Uk10k
Consortium. Plagnol V and Elliott PM: Genetic complexity in
hypertrophic cardiomyopathy revealed by high-throughput sequencing.
J Med Genet. 50:228–239. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Maron BJ, Maron MS and Semsarian C:
Genetics of hypertrophic cardiomyopathy after 20 years: Clinical
perspectives. J Am Coll Cardiol. 60:705–715. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Richard P, Charron P, Carrier L, Ledeuil
C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M,
et al: Hypertrophic cardiomyopathy: distribution of disease genes,
spectrum of mutations, and implications for a molecular diagnosis
strategy. Circulation. 107:2227–2232. 2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Toepfer CN, Wakimoto H, Garfinkel AC,
McDonough B, Liao D, Jiang J, Tai AC, Gorham JM, Lunde IG, Lun M,
et al: Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate
myosin. Sci Transl Med. 11(eaat1199)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ochoa JP, Sabater-Molina M, Garcia-Pinilla
JM, Mogensen J, Restrepo-Córdoba A, Palomino-Doza J, Villacorta E,
Martinez-Moreno M, Ramos-Maqueda J, Zorio E, et al: Formin homology
2 domain containing 3 (FHOD3) is a genetic basis for hypertrophic
cardiomyopathy. J Am Coll Cardiol. 72:2457–2467. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kaski JP, Syrris P, Esteban MT, Jenkins S,
Pantazis A, Deanfield JE, McKenna WJ and Elliott PM: Prevalence of
sarcomere protein gene mutations in preadolescent children with
hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2:436–441.
2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Frey N, Luedde M and Katus HA: Mechanisms
of disease: Hypertrophic cardiomyopathy. Nat Rev Cardiol. 9:91–100.
2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sequeira V, Bertero E and Maack C:
Energetic drain driving hypertrophic cardiomyopathy. FEBS Lett.
593:1616–1626. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Authors/Task Force members. Elliott PM,
Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege
AA, Lafont A, Limongelli G, et al: 2014 ESC guidelines on diagnosis
and management of hypertrophic cardiomyopathy: The task force for
the diagnosis and management of hypertrophic cardiomyopathy of the
European society of cardiology (ESC). Eur Heart J. 35:2733–2779.
2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Guo D, Shi Y, Jian W, Fu Y, Yang H, Guo M,
Yong W, Chen G, Deng H, Qin Y, et al: A novel nonsense mutation in
the L1CAM gene responsible for X-linked congenital hydrocephalus. J
Gene Med. 22(e3180)2020.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Li H and Durbin R: Fast and accurate
long-read alignment with burrows-wheeler transform. Bioinformatics.
26:589–595. 2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Lek M, Karczewski KJ, Minikel EV, Samocha
KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ,
Cummings BB, et al: Analysis of protein-coding genetic variation in
60,706 humans. Nature. 536:285–291. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
den Dunnen JT, Dalgleish R, Maglott DR,
Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T,
Antonarakis SE and Taschner PE: HGVS recommendations for the
description of sequence variants: 2016 update. Hum Mutat.
37:564–569. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ren MB, Chai XR, Li L, Wang X and Yin C:
Potential digenic inheritance of familial hypertrophic
cardiomyopathy identified by whole-exome sequencing. Mol Genet
Genomic Med. 8(e1150)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Das K J, Ingles J, Bagnall RD and
Semsarian C: Determining pathogenicity of genetic variants in
hypertrophic cardiomyopathy: Importance of periodic reassessment.
Genet Med. 16:286–293. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Green RC, Berg JS, Grody WW, Kalia SS,
Korf BR, Martin CL, McGuire AL, Nussbaum RL, O'Daniel JM, Ormond
KE, et al: ACMG recommendations for reporting of incidental
findings in clinical exome and genome sequencing. Genet Med.
15:565–574. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Liew AC, Vassiliou VS, Cooper R and
Raphael CE: Hypertrophic cardiomyopathy-past, present and future. J
Clin Med. 6(118)2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ingles J, Goldstein J, Thaxton C, Caleshu
C, Corty EW, Crowley SB, Dougherty K, Harrison SM, McGlaughon J,
Milko LV, et al: Evaluating the clinical validity of hypertrophic
cardiomyopathy genes. Circ Genom Precis Med.
12(e002460)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Valdés-Mas R, Gutiérrez-Fernández A, Gómez
J, Coto E, Astudillo A, Puente DA, Reguero JR, Álvarez V, Morís C,
León D, et al: Mutations in filamin C cause a new form of familial
hypertrophic cardiomyopathy. Nat Commun. 5(5326)2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Digilio MC, Pacileo G, Sarkozy A,
Limongelli G, Conti E, Cerrato F, Marino B, Pizzuti A, Calabrò R
and Dallapiccola B: Familial aggregation of genetically
heterogeneous hypertrophic cardiomyopathy: A boy with LEOPARD
syndrome due to PTPN11 mutation and his nonsyndromic father lacking
PTPN11 mutations. Birth Defects Res A Clin Mol Teratol. 70:95–98.
2004.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Perryman MB, Yu QT, Marian AJ, Mares A Jr,
Czernuszewicz G, Ifegwu J, Hill R and Roberts R: Expression of a
missense mutation in the messenger RNA for beta-myosin heavy chain
in myocardial tissue in hypertrophic cardiomyopathy. J Clin Invest.
90:271–277. 1992.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lafreniere-Roula M, Bolkier Y, Zahavich L,
Mathew J, George K, Wilson J, Stephenson EA, Benson LN, Manlhiot C
and Mital S: Family screening for hypertrophic cardiomyopathy: Is
it time to change practice guidelines? Eur Heart J. 40:3672–3681.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lopes LR, Brito D, Belo A and Cardim N:
Portuguese Registry of Hypertrophic Cardiomyopathy. Genetic
characterization and genotype-phenotype associations in a large
cohort of patients with hypertrophic cardiomyopathy-an ancillary
study of the Portuguese registry of hypertrophic cardiomyopathy.
Int J Cardiol. 278:173–179. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Millat G, Bouvagnet P, Chevalier P,
Dauphin C, Jouk PS, Da Costa A, Prieur F, Bresson JL, Faivre L,
Eicher JC, et al: Prevalence and spectrum of mutations in a cohort
of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J
Med Genet. 53:261–267. 2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Mohiddin SA, Begley DA, McLam E, Cardoso
JP, Winkler JB, Sellers JR and Fananapazir L: Utility of genetic
screening in hypertrophic cardiomyopathy: Prevalence and
significance of novel and double (homozygous and heterozygous)
beta-myosin mutations. Genet Test. 7:21–27. 2003.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Jaffré F, Miller CL, Schänzer A, Evans T,
Roberts AE, Hahn A and Kontaridis MI: Inducible pluripotent stem
cell-derived cardiomyocytes reveal aberrant extracellular regulated
kinase 5 and mitogen-activated protein kinase kinase 1/2 signaling
concomitantly promote hypertrophic cardiomyopathy in
RAF1-associated noonan syndrome. Circulation. 140:207–224.
2019.PubMed/NCBI View Article : Google Scholar
|