Introduction
Heart development is a complex morphological process
requiring precise spatiotemporal coordination of diverse cell
types. Cardiac defects result from abnormal heart formation and are
characterized by various phenotypes, including atrial septum,
ventricular septum, and vessel and valve defects. Some categories
of severe cardiac defects can cause perinatal morbidity and
mortality with a prevalence of 2.5-3 per 1,000 live births
(1). Numerous studies have
investigated the etiology of cardiac defects. Mutations and
differential expression of genes have been identified in several
cardiac defect phenotypes (2).
Chromosomal and structural DNA abnormalities could be the cause of
certain cardiac defects (2). Some
environmental risk factors have also been linked to cardiac defects
(3). However, for the majority of
cases, the etiology has not been fully explored. Extensive studies
support the role of the epigenetic mechanisms, as an interplay of
genetic and environmental factors in cardiac defects. Therefore,
the present study focused on the epigenetic process of cardiac
defect formation.
Accumulating evidence suggests that abnormal
epigenetic regulation could have an important role in the
development of cardiac defects (4).
DNA methylation, defined as methylation of cytosine residues at CpG
dinucleotides, is one of the most important epigenetic
modifications and is involved in a variety of biological process.
DNA methylation patterns are continuously changing during early
mammalian development, from germlines through to the postnatal
stage. Disturbance of these methylation patterns could lead to
abnormal development and diseases, including congenital syndromes
of immunodeficiency, growth phenotypes, neurodegeneration, and
cancer (5). Recently, DNA
methylation abnormalities were shown to be a biomarker for the
prediction of congenital heart disease and have been explored in
neonatal blood spots (6) and
placental tissues (7,8). Additionally, in heart tissue samples
from patients 1-48 months in age, decreased whole-genome
methylation levels, implied by analyzing long interspersed
nucleotide element (LINE)1 methylation status, was associated with
increased risk of tetralogy of fallot (TOF) (9). Changes in DNA methylation at the
promoter and intron regions of several heart development-related
genes, including NK2 homeobox 5, GATA binding protein 4, heart and
neural crest derivatives expressed 1, and SWI/SNF-related matrix
associated actin-dependent regulator of chromatin subfamily a
member 4, and their association with the expression of these genes,
have also been reported in the myocardium of children with
ventricular septal defect (VSD), TOF, and double-chambered right
ventricle (10,11). However, limited studies have
examined DNA methylation during the fetal period, a key stage when
cardiac structures are forming. Furthermore, most current studies
have focused on a few simple forms of cardiac defects that are not
life-threatening after birth. Such studies are insufficient to
provide cues for more severe and complex fetal cardiac defects with
or without extracardiac malformations in prenatal life.
The present study was designed to explore the
genome-wide DNA methylation landscapes in fetal heart tissues with
isolated and non-isolated cardiac defects to identify and validate
the global DNA methylation status and CpG site-specific DNA
methylation changes adjacent to genes associated with heart
development. The expression levels of these heart
development-related genes were also evaluated at the mRNA and
protein levels.
Materials and methods
Tissue samples
Fetal myocardium tissue was obtained from fetuses
whose mothers underwent abortion surgery due to fetal cardiac
defects with and without the accompanying noncardiac structure
defects (non-isolated and isolated, respectively), diagnosed with
fetal echocardiography at Fudan University Affiliated Obstetrics
and Gynecology Hospital (Shanghai, China). Normal fetal myocardium
tissue samples, as controls, were obtained from fetuses without
obvious anatomical abnormalities aborted due to severe maternal
complication or trauma. Fetal ventricle myocardium tissue was
carefully dissected at the outflow tract area of the fetal heart.
Fetal age was calculated based on last menstruation. Gestational
age was matched between samples with cardiac defects and controls.
The tissue was kept in RNAlater® (Ambion; Thermo Fisher
Scientific, Inc.) for at least 24 h at room temperature, then
frozen at -80˚C until further use. Thirty-one fetuses with cardiac
defects, including 17 isolated cardiac defects and 14 non-isolated
cardiac defects, ranging in age from 23 to 27 weeks (mean, 24.5
weeks) and 22 controls with no evidence of cardiac defects ranging
in age from 22 to 27.2 weeks (mean, 23.5 weeks) were recruited
between January 2011 and December 2012. The sample characteristics
and the cardiac defect phenotypes are listed in Table SI, Table SII, Table SIII and Table SIV.
All parents signed written informed consent prior to
tissue harvesting for scientific research. The present study was
approved by the Ethics Committee of Fudan University Affiliated
Obstetrics and Gynecology Hospital (Shanghai, China).
DNA extraction and sodium bisulfite
conversion
Genomic DNA was extracted from heart tissue using
the QIAamp DNA mini kit (Qiagen GmbH) following the manufacturer's
instructions. The DNA concentration and purity were determined by
absorbance of A260/A280≥1.8 and A260/A230≥1.9, respectively, using
a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Inc.).
Sodium bisulfite treatment of extracted DNA was performed using an
EZ DNA Methylation kit, strictly according to the manufacturer's
instructions (Zymo Research, Corp.). The sodium bisulfite-converted
DNA was resuspended in 10 µl elution buffer and stored at -20˚C
until required for experiments.
Methylated DNA immunoprecipitation
microarray (MeDIP-chip) processing and human CpG island
microarray
MeDIP-chip analysis was performed using the Roche
NimbleGen's DNA methylation assay at CapitalBio Technology, Inc. In
brief, 6 µg genomic DNA was digested into 200-1,000 bp fragments
with MseI restriction endonuclease. Some of the digested DNA
fragments were stored at -20˚C for use as control (input DNA). Some
of the remaining digested DNA fragments were immunoprecipitated
(IP) with monoclonal anti-5-methyl cytidine antibody (IP DNA), as
instructed in the NimbleGen's DNA methylation assay protocol (Roche
Diagnostics). Subsequently, IP DNA and input DNA were amplified
using the Whole Genome Amplification kit (Sigma-Aldrich; Merck
KGaA), following the manufacturer's instructions. Amplified IP and
input DNA were labeled with Cy5 and Cy3 dyes, respectively, and
co-hybridized to human CGI oligonucleotide microarrays (Roche
Diagnostics). The arrays were designed based on the University of
California at Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu) CpG island list and contained
27,728 CpG islands plus RefSeq promoters covering 2,440 bp to +610
bp regions from 22,532 potential transcription start sites.
Following hybridization and washing, the arrays were scanned using
the MS200 scanner (NimbleGen; Roche Diagnostics), then NimbleScan
software (version 2.5; NimbleGen) was used to extract the raw
fluorescence intensity data from the scanned images. Along with
visual inspection, the MS200 scanner and NimbleScan software were
applied to show no apparent chip defects and/or that defected areas
covered <1% of the array. Further microarray quality assessment
was achieved using Sample Tracking Controls features and
experimental metrics reports following NimbleGen's instructions on
the manufacturer's user guide.
Microarray data analysis
For each probe, after the IP/Input log2
ratio was computed, scaling was performed by subtracting the
bi-weight mean from each log2 ratio value on the array.
Regions of significant positive enrichment in CHIP-based
methylation microarray data were identified using a modified ACME
algorithm for peak identification (12). In detail, from the scaled
log2 ratio data, a 750 bp fixed-length window was placed
around each consecutive probe and the one-side Kolmogorov-Smirnov
(KS) test was applied to determine whether the probes had a
significantly positive log2 ratio intensity distribution
compared with those in the rest of the array. The resulting score
for each probe was the -log10 P-value from the windowed
KS test around that probe. Regions with more than two probes
scoring >2 were defined as MeDIP peaks, and peaks were mapped to
genes and CpG islands on the UCSC website (http://genome.ucsc.edu; version NCBI36/hg18).
Bioinformatic analysis
DNA methylation levels of each MeDIP peak CpG
region, including several CpG sites, were compared between fetal
heart tissues with isolated and/or non-isolated cardiac defects and
controls to identify differentially methylated CpG regions (DMRs),
using limma, an R package based on linear regression (13). Filtering criteria for statistically
significant DMRs were set at P<0.01 and log2 fold
change (FC)>0.5. Bedtools (14)
was used to identify the LINEs and short-interspersed nuclear
elements (SINEs) that overlap with DMRs. Genomic distribution
characteristics of these DMRs were analyzed using ChIPpeakAnno
workflow in R package (15). Then,
DMRs of annotated genes were used for Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) enrichment analyses using
gene set information obtained from MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/). The R
script was from NetBID2 (https://github.com/jyyulab/NetBID). In addition to the
mean methylation level across all CpG sites per DMR, the
methylation levels of single CpG sites between cardiac defects
cases and normal controls were analyzed using the Mann-Whitney
nonparametric U test.
Validation of methylation levels by
MassARRAY EpiTYPER assay
The Sequenom MassARRAY EpiTYPER platform was used to
validate the methylation levels of the targeted regions identified
from microarray results (the phenotypes of samples with cardiac
defects used for MassARRAY analysis are listed in Table SII). The EpiTYPER assay quantifies
CpG dinucleotide methylation based on matrix-assisted laser
desorption ionization time-of-flight (MALDI-TOF) mass spectrometry.
This is an accurate, sensitive and high-throughput method for the
quantitative analysis of DNA methylation at CpG sites (16). The primers used in the present study
were designed using Methprimer (http://epidesigner.com; Table SV). Approximately 500 ng of
fragmented DNA from each sample was modified by bisulfite
treatment. Following PCR with specific primers, which added T7
promoter tags (reverse primer) and 10-mer tags (forward primer),
amplicons were treated with shrimp alkaline phosphatase. Fragments
were ligated to a T7 promoter segment and transcribed into RNA. The
synthesized RNA was cleaved with Rnase A and all cleavage products
were analyzed using EpiTYPER software (version 1.0; Sequenom). The
detailed protocol was described previously (9).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from 100-200 mg of frozen
fetal myocardium tissue with TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). Approximately 500 ng of extracted RNA was
used as a template for the reverse transcriptase reaction according
to the manufacturers' protocol. Transcribed cDNA was amplified in a
qPCR reaction using SYBR Premix Ex Taq system (Takara Biotechnology
Co., Ltd.) on the Applied Biosystems 7900 Real-Time PCR system
(Thermo Fisher Scientific, Inc.). The amplification reaction
conditions were set as follows: An initial 5 min at 94˚C, then 35
cycles comprising 30 sec at 94˚C, 30 sec at 58˚C, and 30 sec at
72˚C, followed by 72˚C for 3 min. Gene expression comparisons were
conducted for epidermal growth factor receptor (EGFR), solute
carrier family 19 member 1 (SLC19A1), and NOTCH1. All assays were
performed in triplicate. GAPDH was used as a housekeeping gene for
normalization and the results were analyzed using the
2-ΔΔCq method (17)
after averaging the triplicates of each assay. The primers used are
listed in Table SVI.
Western blot analysis
Heart tissues from fetuses with and without cardiac
defects were homogenized in RIPA lysis buffer (Beyotime Institute
of Biotechnology), using an electric homogenizer to break tissue
until there was no visible tissue block. Samples were kept on ice
for 20 min to fully lyse the tissues. The lysate was
ultracentrifuged at 12,000 x g (20 min, 4˚C) to obtain the
supernatant for determination of protein concentration using the
BCA method. The protein from each sample was treated with 5X
loading buffer, and the concentration was then adjusted to 1 µg/µl
by supplementing the remaining volume with protein lysate as
diluent. Finally, the protein sample was denatured in a 100˚C water
bath for 5 min and loaded onto a gel or stored at -80˚C for later
use. For each sample, the proteins in 20 µl were separated by 10%
SDS-PAGE and transferred to a nitrocellulose membrane. The
membranes were blocked with 5% non-fat milk powder for 2 h at room
temperature in TBS with 0.05% Tween-20 (TBST) and incubated with
primary antibodies overnight at 4˚C. Primary antibodies used were
rabbit polyclonal EGFR antibody (1:1,000; cat. no. sc-03; Santa
Cruz Biotechnology, Inc.), rabbit polyclonal SLC19A1 antibody
(1:1,000; cat. no. ab62302; Abcam), rabbit polyclonal NOTCH1
antibody (1:1,000; cat. no. ab27526; Abcam), and rabbit polyclonal
GAPDH antibody (1:2,500; cat. no. ab263962 Abcam) as a control. The
blots were rinsed in TBST three times and incubated in
HRP-conjugated secondary antibody (1:5,000; cat. no. ab6721; Abcam)
for 1 h at room temperature. Immunoreactive proteins were developed
in ECL Plus and were exposed using the LAS-3000 system (Fujifilm
Wako Pure Chemical Corporation). The densitometry of the bands was
calculated using ImageJ (version 1.8.0, National Institutes of
Health).
Statistical analysis
The methylation levels of single CpG sites between
31 cases of cardiac defects and 22 cases of normal controls were
analyzed using the Mann-Whitney nonparametric U test and presented
as median with the interquartile ranges. Differences in gene and
protein expression levels were assessed by a two-tailed Student's
t-test and presented as the means ± standard deviation. All
experiments were repeated three times. P<0.05 was considered as
statistically significant. All data were analyzed with GraphPad
Prims (version 6.0c; GraphPad Software, Inc.).
Results
Sample allocation
The DNA methylation patterns in fetal cardiac tissue
of isolated cardiac defects, non-isolated cardiac defects, and
control samples were determined using NimbleGen's whole genomic DNA
methylation microarray. Overall, 17 sampled from isolated cardiac
defects, 14 samples from non-isolated cardiac defects, and 22
samples with normal histology were included in the present study.
The methylation analysis was divided into two parts. In the first
part, using the methylation microarray, two isolated cardiac
defects samples and three non-isolated cardiac defects samples were
compared to four normal controls to obtain a list of DMRs (for
detailed information, see Tables
SI and SII). In the second
part, another 15 isolated cardiac defect samples, 11 non-isolated
cardiac defect samples and 18 normal controls were used to validate
the regional and CpG site-specific methylation level changes in
DMRs and expression changes in annotated genes within the DMR (for
detailed information, see Tables
SIII and SIV).
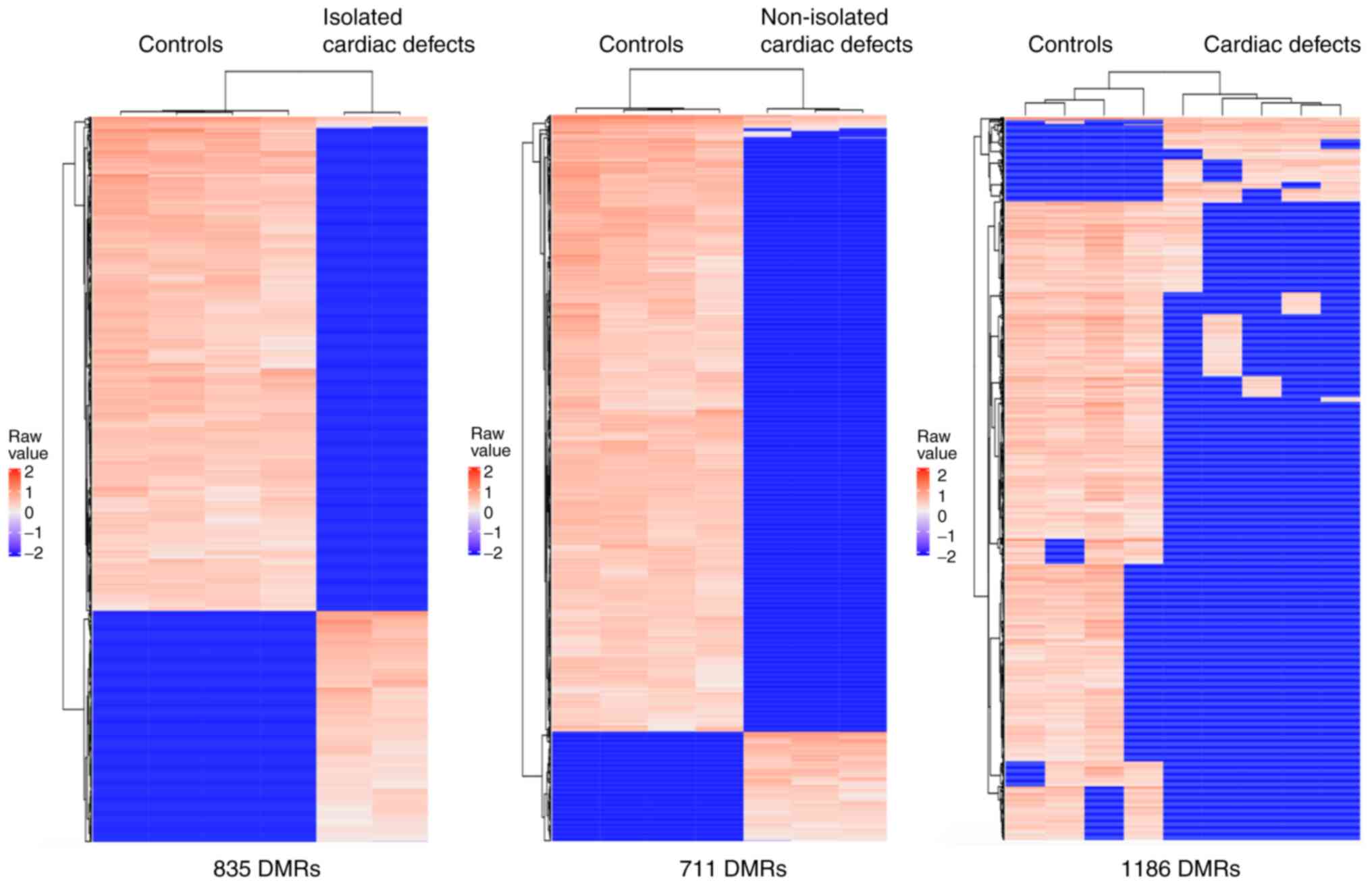
Global DNA methylation is
downregulated in fetal hearts with cardiac defects
A total of 17,386 CpG sites located in 1,546
methylated regions were captured through the present methylation
microarray data analysis. The microarray data have been deposited
in the figshare database (DOI: https://doi.org/10.6084/m9.figshare.14130173). The
results revealed more hypomethylated regions than hypermethylated
regions in both isolated and non-isolated cardiac defect cases
compared with the normal controls. In isolated cardiac defect
cases, 835 DMRs were tested (Table
SVII), of which over 68% (569 DMRs) were hypomethylated
(Fig. 1). In non-isolated cardiac
defects, 711 DMRs were tested (Table
SVIII), of which up to 84% (604 DMRs) were hypomethylated
(Fig. 1). After combining isolated
and non-isolated cardiac defect cases, the results identified
global hypomethylation alteration, at a genome-wide level, with
1,186 DMRs (Table SIX), of which
1,052 (89%) were hypomethylated compared with those in control
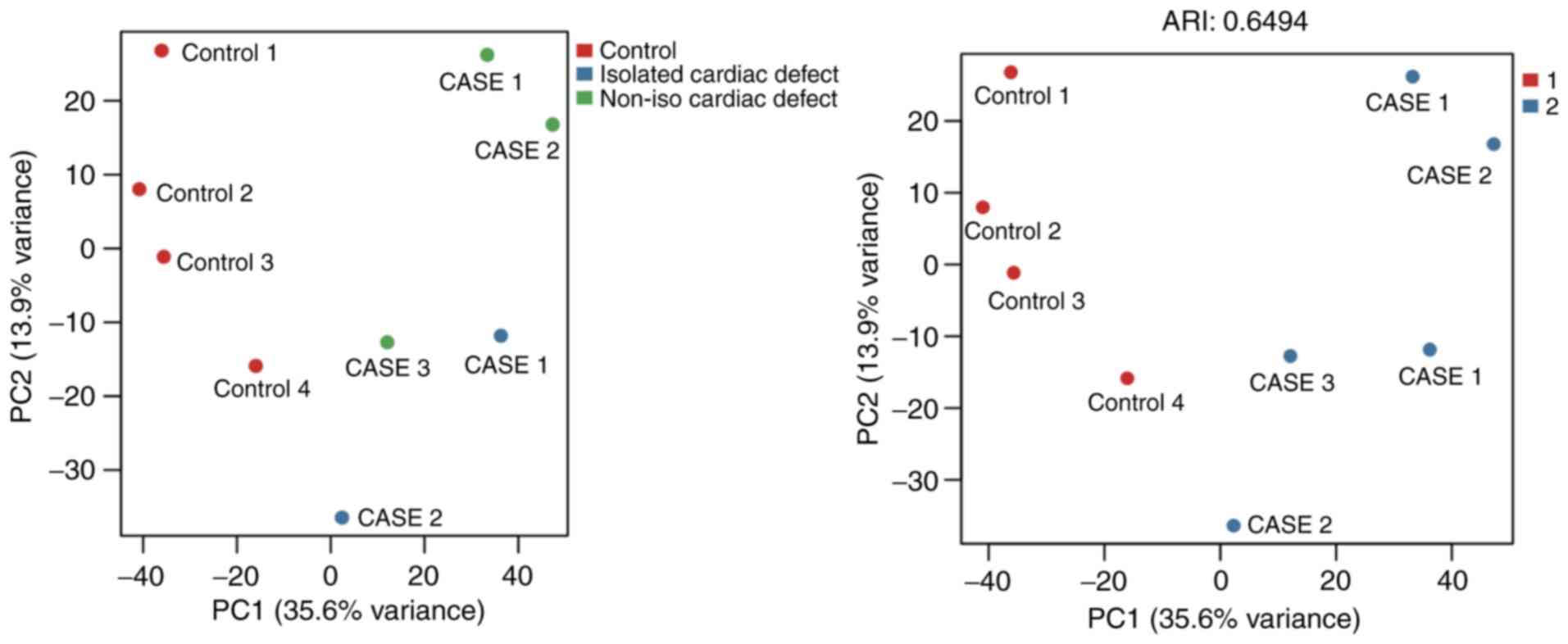
samples (Fig. 1). Additionally, the
present study conducted a principal component analysis based on the
most variable levels of 1,737 CpG methylation, which showed that
cardiac defect cases were clearly clustered from the normal
controls, but the subgroups of cardiac defects with and without
extracardiac defects did not appear to differentiate from each
other very well (Fig. 2).
Therefore, the isolated cardiac defect and the non-isolated cases
were combined to form the full set of cardiac defects in subsequent
analyses.
To further determine the global alterations of
hypomethylation in fetal heart tissue with cardiac defects, the
methylation levels of LINEs and SINEs were assessed, because
methylation levels of these elements are highly correlated with
global DNA methylation (18). In
heart tissue from cardiac defect cases, a total of 267 LINEs and
165 SINEs were present in differential methylation profiles, and
75% (200/267) of LINEs and 82% (136/165) of SINEs were
hypomethylated.
To validate the reliability of the present DNA
methylation alteration patterns of fetal heart tissues with cardiac
defects, data from a previous study using the Illumina Infinium
HumanMethylation450 BeadChips (450 K arrays) platform, analyzing 49
pediatric human cardiac tissue samples from patients with different
CHDs (19), were compared with the
MeDIP-chip data from the present study. Of the 1,168 DMRs, two CpG
islands, located on chr10:123356616-123358285 and
chr13:113772727-113773012, displayed an overlap to the 450 K assay
target ID of Hoff et al (19).
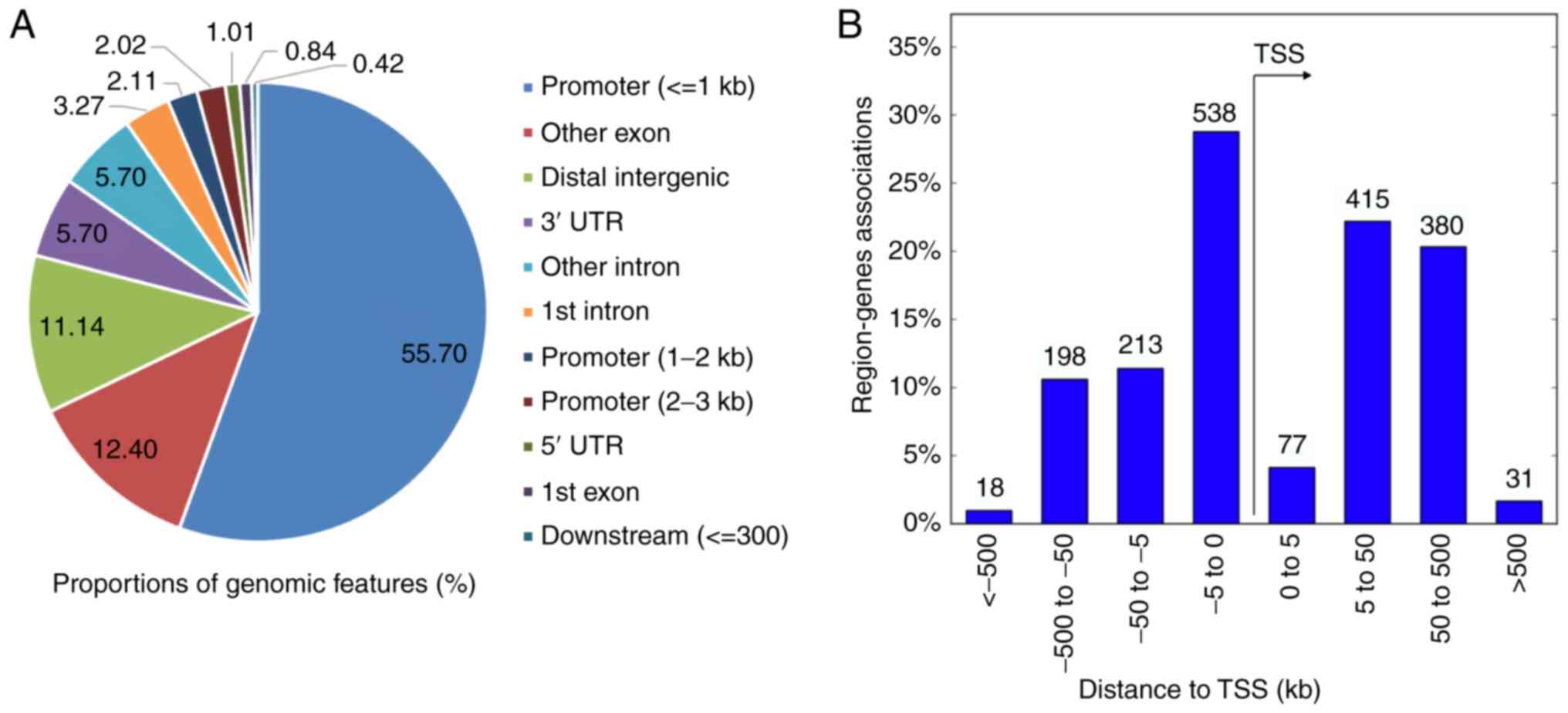
Genomic features of DMRs and
functional enrichment analysis of genes adjacent to DMRs
The genomic location of the 1,168 DMRs was examined,
the distribution of which is shown in Fig. 3. As the present microarray platform
explored CpG islands, the majority (59.83%) of the DMRs were
observed in promoters. A considerable number of DMRs were
associated with exons (13.24%), introns (8.97%), and distal
intergenic areas (11.14%). Regions up to 300 bp downstream of
transcriptional start sites, the 5' untranslated region (UTR), and
the 3'UTR overlapped with 7.13% of DMRs.
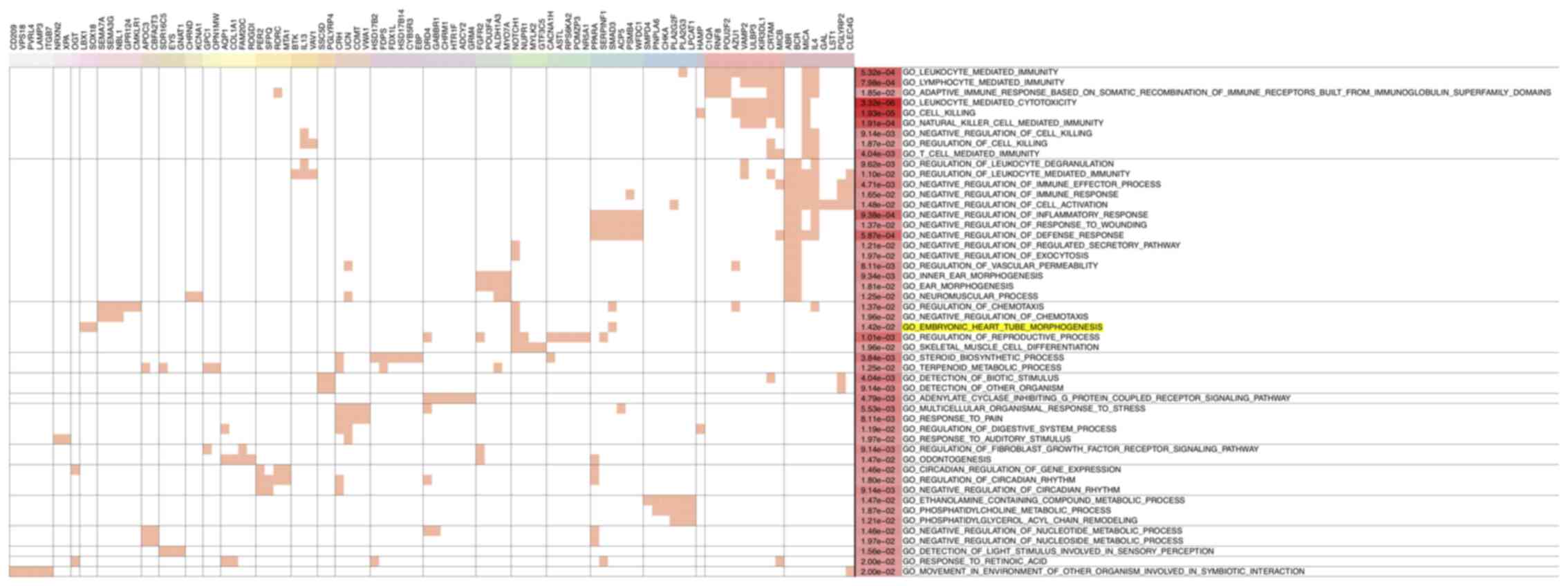
Next, GO analysis was performed for genes contiguous
to the 1,168 DMRs to identify significant functional enrichment.
The results revealed that most hypomethylated region-related genes
were enriched for immune-related functions, suggesting that the
immune regulation system may have a role during heart development
(Fig. 4). The GO term ‘embryonic
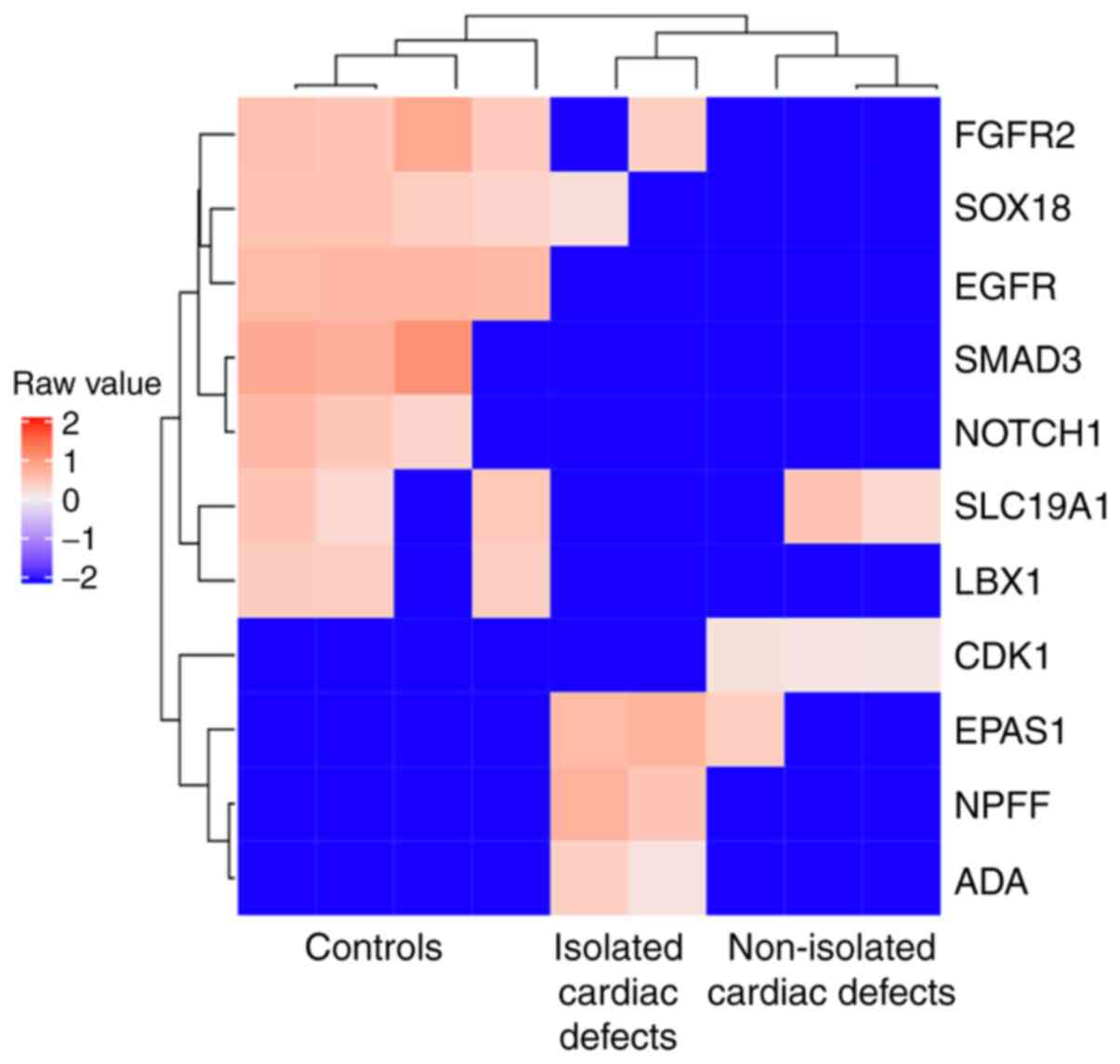
heart tube morphogenesis’ was also found to be enriched (Fig. 4). A total of 11 genes, including
fibroblast growth factor receptor 2, SRY-box transcription factor
18, epidermal growth factor receptor (EGFR), SMAD3, NOTCH1, solute
carrier family 19 member 1 (SLC19A1), ladybird homeobox 1,
cyclin-dependent kinase 1, endothelial PAS domain protein 1,
neuropeptide FF-amide peptide precursor, and adenosine deaminase,
were enriched in GO term ‘embryonic heart tube morphogenesis’ and
the associated DMRs were used to generate a corresponding heatmap
(Fig. 5). However, no significant
heart development-associated enrichment was observed for
hypermethylated region-related genes (data not shown).
Validation of regional and
site-specific DNA methylation levels in cardiac defects
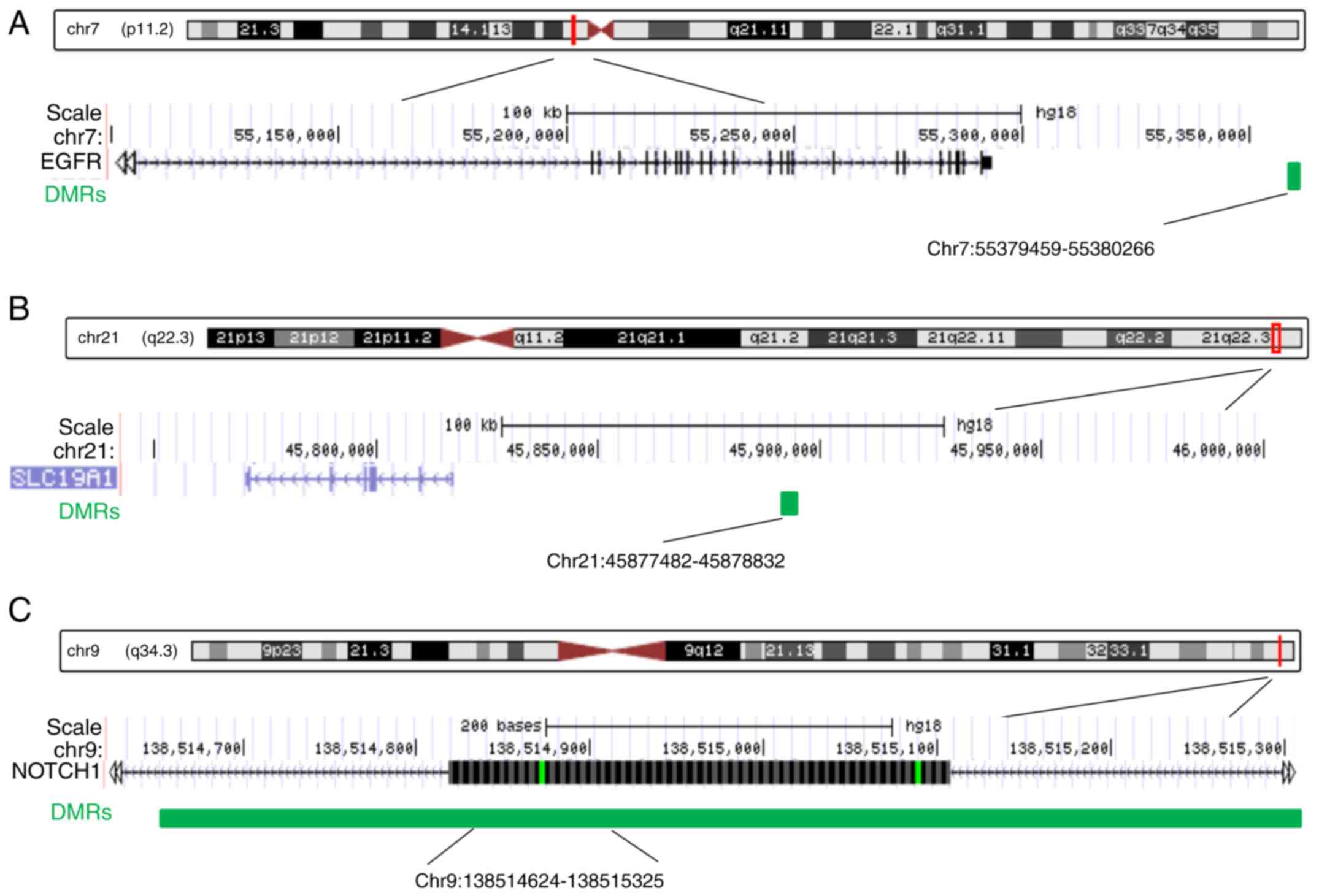
Among the 11 DMRs annotated to heart
development-related genes, the three most significant DMRs were
selected for further study. Two of the three DMRs, including the
EGFR-related and the SLC19A1-related DMRs, were located in
intergenic regions, while the NOTCH1-related DMR was intragenic,
located in the gene body (Fig. 6).
Next, the average methylation levels across all CpG sites of these
three DMRs and the methylation levels at single CpG sites were
measured. To do this, quantitative methylation analysis was used
through the MassARRAY platform in fetal heart tissue samples from
an additional 26 cases with cardiac defects, containing 15 isolated
cardiac defects and 11 non-isolated cardiac defects, and 18 normal
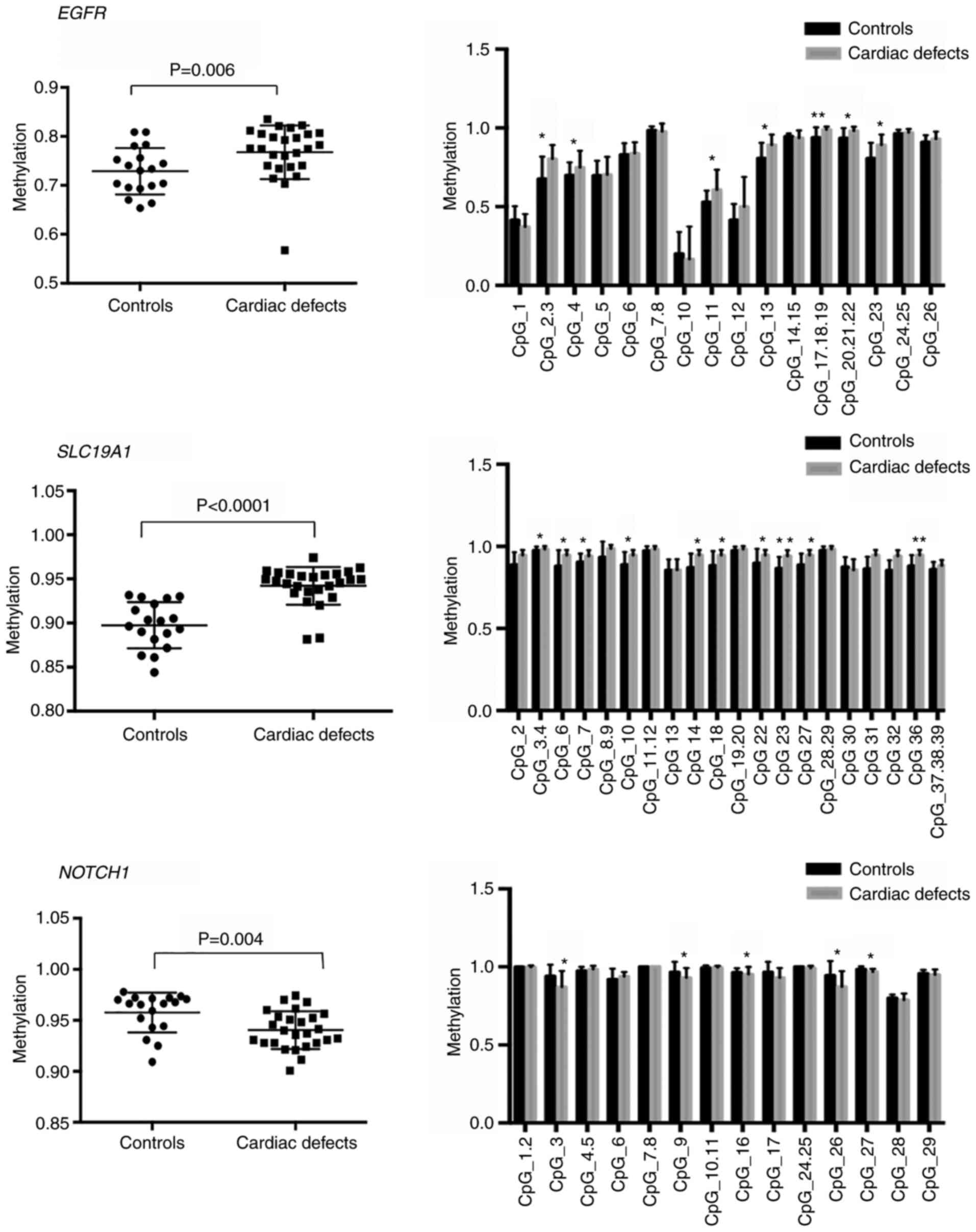
controls. For EGFR-associated DMRs and SLC19A1-associated DMRs, the
results demonstrated significantly higher methylation levels in
cardiac defect cases compared with those observed in controls
[median 0.78 vs. 0.75, interquartile range (IQR) 0.76-0.81 vs.
0.7-0.78, P=0.006 for EGFR DMRs; and median 0.95 vs. 0.91, IQR
0.94-0.96 vs. 0.88-0.93, P<0.0001 for SLC19A1 DMRs) (Fig. 7). The mean CpG region methylation
level in the NOTCH1 gene body was significantly lower in cardiac
defects cases than in controls (median 0.94 vs. 0.97, IQR 0.93-0.96
vs. 0.94-0.97, P=0.0043; Fig. 7).
Specifically, hypermethylation of seven CpG sites, including
CpG2.3, CpG4, CpG11, CpG13, CpG17.18.19, CpG20.21.22, and CpG23, at
EGFR intergenic regions, and 10 CpG sites, including CpG2, CpG6,
CpG7, CpG10, CpG14, CpG18, CpG22, CpG23, CpG27, and CpG36, at
SLC19A1 intergenic regions contributed to the increased methylation
level in EGFR and SLC19A1 DMRs (Fig.
7). Hypomethylation in the NOTCH1 gene body was mainly
attributed to decreased methylation levels of multiple CpGs,
including CpG3, CpG9, CpG16, CpG26, and CpG27 (Fig. 7).
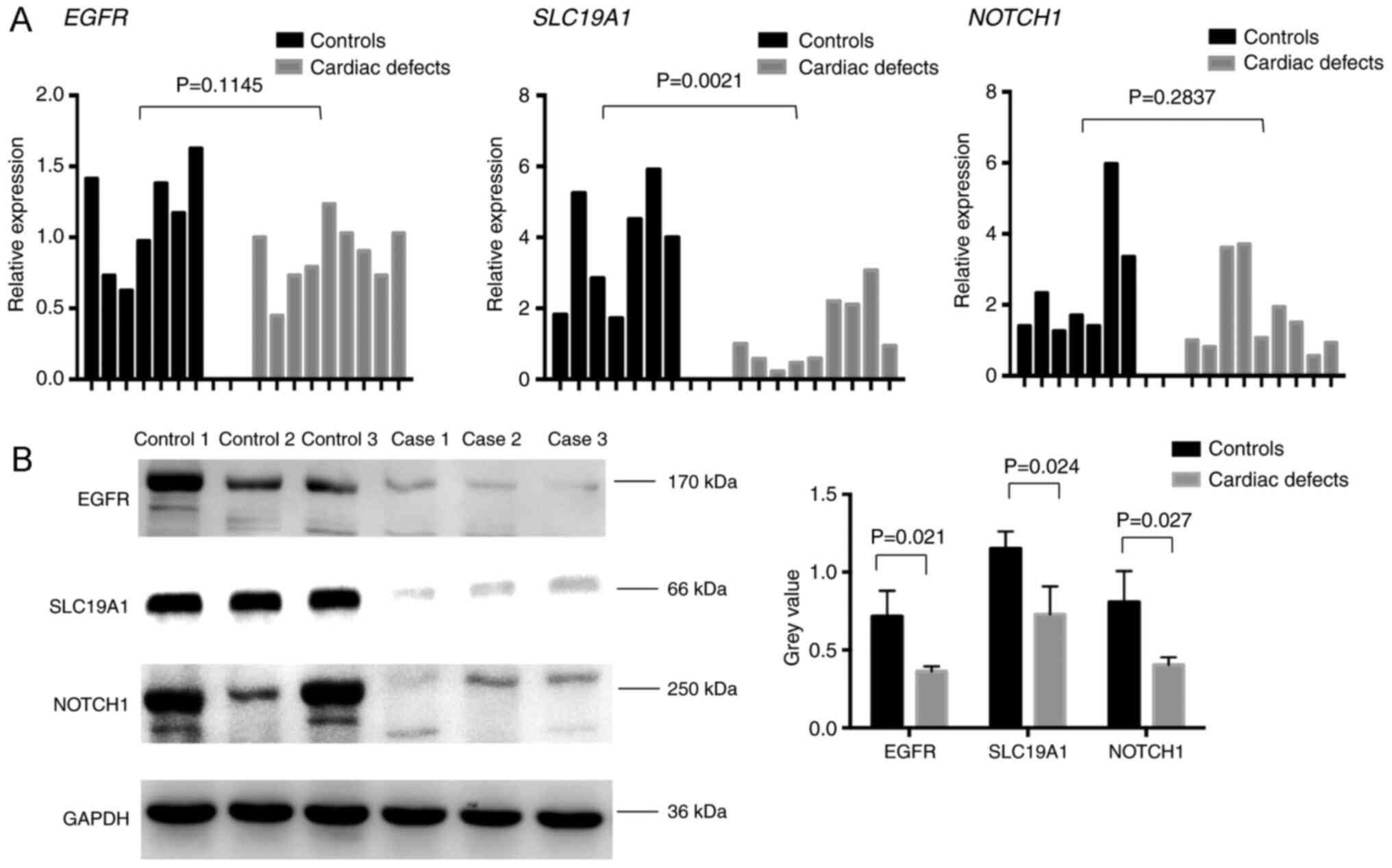
EGFR, NOTCH1, and SLC19A1 expression
changes at mRNA and protein levels in isolated cardiac defects
Since changes in global and CpG site-specific
methylation levels were observed, the present study analyzed the
corresponding gene expression levels. Given the low residual amount
of fetal heart tissue, the mRNA expression changes in EGFR, NOTCH1,
and SLC19A1 were examined in seven normal controls and nine
isolated cardiac defect cases from the previously analyzed sample
cohort. Of these, three normal controls and three isolated cardiac
defects samples with the most variable mRNA expression levels were
used to detect EGFR, NOTCH1, and SLC19A1 protein expression levels.
RT-qPCR results showed a statistically significant reduction in
SLC19A1 mRNA expression levels in cardiac defects samples compared
with normal controls, which was consistent with protein expression
results (Fig. 8). Similarly, both
EGFR and NOTCH1 displayed a tendency for decreased mRNA expression
levels and a significant decline in protein expression in isolated
cardiac defects compared with controls (Fig. 8). Notably, changes in the
methylation levels observed at EGFR and SLC19A1 intergenic regions
showed a trend opposite to that of the expression of the
corresponding gene. A consistent trend in the methylation level
changes in the NOTCH1 intragenic region and corresponding gene
expression levels was identified.
Discussion
Cardiac defects are one of the most prevalent
congenital abnormalities of complicated etiology, and their
phenotypes vary greatly. Different types of cardiac defects arise
from distinct disruptions in heart development at particular stages
of heart formation (20). Given
that epigenetic regulation is time-specific, it is necessary to
understand the underlying mechanisms using samples of the
developing heart that are approaching developmental time points
when cardiac defects occur. To the best of our knowledge, this
genome-wide methylation analysis in heart tissues from a cohort of
fetuses is the first of its kind. The present results revealed a
lower global methylation status in fetal heart tissue with isolated
and with non-isolated cardiac defects compared with normal control
tissues. In addition, in heart tissues with fetal cardiac defects,
three sets of differentially methylated CpG sites were identified,
including DMRs from EGFR and SLC19A1 intergenic regions and a DMR
from the NOTCH1 gene body. DNA methylation changes in these DMRs
may affect corresponding gene expression and contribute to cardiac
defects.
The current study represents a genome-wide
methylation analysis of relatively complicated cardiac defects.
Previous DNA methylation studies were limited to analyses of
methylation status in some congenital heart defect candidate genes
(10,11,21-23),
and were insufficient to reveal information for DNA methylation on
a global scale. One previous study conducted a genome-wide DNA
methylation study (24) and found
more hypermethylated than hypomethylated DMRs in myocardial samples
from pediatric patients with CHDs compared with controls. This is
contrary to the present results that revealed a global
hypomethylation status in heart tissue with cardiac defects.
However, the previous study selected only two CHDs, namely TOF and
VSD, obtained heart samples from pediatric patients during surgery,
and lacked appropriate age-matched normal hearts. Apart from the
above two subtypes of CHD, several complicated and severe CHD
phenotypes, possibly exerting a greater impact on pediatric
morbidity and mortality, require more in-depth research. In another
study (25), whole genome
methylation profiles were obtained using heart tissue DNA from
fetuses presenting a variety of cardiac defect phenotypes including
double outlet right ventricle, hypoplasia of the ascending aorta,
right heart hypoplasia, left heart hypoplasia, mitral valve
atresia, aorta valve atresia, tricuspid valve stenosis,
transposition of the great arteries, and absent ductus arteriosus,
but no significant differences in whole genome methylation pattern
were identified between fetuses with isolated cardiac defects and
fetuses with normal development. Nevertheless, they identified
several CpG sites that were differentially methylated in single CHD
cases, but these results were not validated in an independent
cohort (25).
The global hypomethylation status of fetuses with
cardiac defects reported in the present study is consistent with
the results of three previously published studies assessing LINE-1
methylation in venous blood samples from very young children with
complex CHD (26), mothers whose
pregnancies were affected by non-syndromic CHD (27), and in right ventricular tissue
samples from pediatric patients (9). The current observation of a global
hypomethylation change in cardiac defects is supported by an
assessment of increased hypomethylated LINE-1 in fetal heart tissue
with cardiac defects and by the detection of a larger proportion of
hypomethylated DMRs through whole-genome methylation analysis.
Furthermore, detailed comparison of methylation levels in two
intergenic DMRs and one intragenic DMR revealed several CpG sites
within these DMRs that significantly differed between cases with
cardiac defects and controls. The role of CpG site methylation in
regions outside of promoters has recently been well-studied.
Irrespective of where it is located in the genomic context, DNA
methylation serves a crucial role in the transcriptional and
splicing regulation of genes (28).
To date, several studies have confirmed that intragenic methylated
CpG islands are largely tissue-specific and that they are strongly
associated with transcription initiation and elongation (28-30).
Additionally, discrete hypomethylated regions in intergenic spaces
are more predictive of nearby gene activity than are the promoter
regions themselves (31).
Intergenic DNA hypomethylation, as a downstream target of some
signaling pathways, could explain neoplastic tissue overgrowth and
developmental disorders (32).
Therefore, it is reasonable to speculate that there might be a link
between intergenic CpG site methylation levels of EGFR and SLC19A1
DMRs, intragenic CpG sites of NOTCH1 DMRs, and gene expression in
the current study. On the other hand, changes in the opposite or
same direction between methylation levels of intergenic EGFR and
SLC19A1 DMRs and intragenic NOTCH1 DMRs and related gene expression
varied based on the genome location and nearby regulatory elements,
which was also supported by a previous report (29). This emphasizes the importance of
methylation in non-promoter regions. However, the exact mechanisms
underlying the intergenic and intragenic methylation-based
regulation of EGFR, SLC19A1, and NOTCH1 expression in the present
study remain unclear and need to be further explored.
In the present study, RT-qPCR and western blot
analyses revealed that EGFR, SLC19A1, and NOTCH1 expression were
decreased in fetal heart tissue compared with control normal
tissues. These three genes are crucial for heart development. For
instance, in mouse studies, Egfr regulates embryonic formation of
the aortic valve (33). Slc19a1
knock-out mice were shown to die post-implantation at E6.5, even
with folic acid supplementation, and the mice presented with
cardiac malformations, including VSDs, thin myocardial wall and VSD
with overriding aorta (34).
Additionally, Notch signaling is required for cardiac fate
determination, patterning of the primitive heart (3), and cardiac valve morphogenesis
(35). Mutations in NOTCH1 have
been associated with left ventricular outflow tract malformations,
including aortic valve stenosis, coarctation of the aorta, and
hypoplastic left heart syndrome (36). Based on these previous studies, it
can be inferred that hypermethylation of EGFR and SLC19A1
intergenic regions and hypomethylation of NOTCH1 intragenic regions
might be associated with downregulation of gene expression,
resulting in the occurrence of fetal cardiac defects.
Of note, immune regulation was one of the most
highly enriched biological functions identified in the present GO
enrichment analysis. Previously, the function of an endogenous
complement inhibitor, which had an impact on cardiac neural crest
cell migration in the zebrafish model, was described, suggesting
that immune system molecules may be involved in cardiac tissue
development (37). Single-cell
transcriptome analysis in the human fetal heart by Cui et al
(38) confirmed the role of immune
cells in heart development and found that the proportion of these
cells greatly increased with the development of the heart.
There are several limitations in the present study.
Firstly, the number of cases for some cardiac defect subtypes was
small. Secondly, the present study did not use multiple methods to
validate the methylation microarray data, because of undetectable
methylation level of some CpG sites by EpiTYPER analysis and
certain methodological limitations. Thirdly, some interfering
factors, including fetal chromosomal abnormalities, the overlap
between CpG sites and single-nucleotide polymorphisms and adverse
maternal exposure, should also be considered and excluded.
Fourthly, subsequent studies are required to clarify the
relationship between gene expression and CpG site-specific
methylation levels at specific areas and at CpG sites other than
those near the EGFR, SLC19A1, and NOTCH1 promoters.
The present study is the first to explore
methylation level changes in heart tissue from fetuses with
isolated and non-isolated cardiac defects. The results revealed
both a global hypomethylation status and absolute changes in DNA
methylation at particular CpG sites for cardiac defect samples. The
mRNA and protein expression analyses of heart
development-associated genes adjacent to DMRs may provide new
insights into the possible epigenetic mechanisms of heart
development during the fetal period. In the future, it would be
worth examining the causality of the three identified DMRs,
especially the CpG sites and related gene expression.
Supplementary Material
Characteristics of samples used for
genome-wide methylation microarray analysis to obtain a list of
differentially methylated regions.
Phenotypes of samples with cardiac
defects used for genome-wide methylation microarray analysis.
Characteristics of samples used for
genome-wide methylation microarray analysis to validate
differentially methylated regions.
Phenotypes of samples with cardiac
defects used for MassARRAY analysis.
Primers used for validation of
methylation level of CpG sites adjacent to heart
development-related genes by MassARRAY.
Primers used for reverse
transcription-quantitative PCR.
DMRs in isolated cardiac defects
DMRs in non-isolated cardiac
defects
DMRs in cardiac defects irrespective
of whether they are isolated
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by grants from the Shanghai Key
Program of Clinical Science and Technology Innovation (grant nos.
17411950500, 17411950501 and 18511105602), the National Natural
Science Foundation of China (grant nos. 81901500, 81741047,
81971411 and 81801468), and the Shanghai Medical Center of Key
Programs for Female Reproductive Diseases (grant no.
2017ZZ01016).
Availability of data and materials
The datasets generated during the current study are
available in the figshare database (https://doi.org/10.6084/m9.figshare.14130173).
Authors' contributions
DM and XL designed the study and provided funding
for the study. JZ participated in the experiments and writing. YX
participated in sample acquisition and helped to draft the
manuscript. XD was responsible for bioinformatics analysis and for
generating some of the charts. HW participated in the study concept
and design. YQ was responsible for part of the experiments. XL and
JZ confirmed the authenticity of all the raw data. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Fudan University Affiliated Obstetrics and Gynecology
Hospital (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hoffman JI and Kaplan S: The incidence of
congenital heart disease. J Am Coll Cardiol. 39:1890–1900.
2002.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yuan S, Zaidi S and Brueckner M:
Congenital heart disease: Emerging themes linking genetics and
development. Curr Opin Genet Dev. 23:352–359. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Vecoli C, Pulignani S, Foffa I and
Andreassi MG: Congenital heart disease: The crossroads of genetics,
epigenetics and environment. Curr Genomics. 15:390–399.
2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jarrell DK, Lennon ML and Jacot JG:
Epigenetics and mechanobiology in heart development and congenital
heart disease. Diseases. 7(52)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Greenberg MVC and Bourc'his D: The diverse
roles of DNA methylation in mammalian development and disease. Nat
Rev Mol Cell Biol. 20:590–607. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bahado-Singh RO, Zaffra R, Albayarak S,
Chelliah A, Bolinjkar R, Turkoglu O and Radhakrishna U: Epigenetic
markers for newborn congenital heart defect (CHD). J Matern Fetal
Neonatal Med. 29:1881–1887. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Radhakrishna U, Albayrak S, Zafra R, Baraa
A, Vishweswaraiah S, Veerappa AM, Mahishi D, Saiyed N, Mishra NK,
Guda C, et al: Placental epigenetics for evaluation of fetal
congenital heart defects: Ventricular septal defect (VSD). PLoS
One. 14(e0200229)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bahado-Singh R, Vishweswaraiah S, Mishra
NK, Guda C and Radhakrishna U: Placental DNA methylation changes
for the detection of tetralogy of Fallot. Ultrasound Obstet
Gynecol. 55:768–775. 2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Sheng W, Wang H, Ma X, Qian Y, Zhang P, Wu
Y, Zheng F, Chen L, Huang G and Ma D: LINE-1 methylation status and
its association with tetralogy of fallot in infants. BMC Med
Genomics. 5(20)2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sheng W, Qian Y, Wang H, Ma X, Zhang P,
Diao L, An Q, Chen L, Ma D and Huang G: DNA methylation status of
NKX2-5, GATA4 and HAND1in patients with tetralogy of fallot. BMC
Med Genomics. 6(46)2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Qian Y, Xiao D, Guo X, Chen H, Hao L, Ma
X, Huang G, Ma D and Wang H: Hypomethylation and decreased
expression of BRG1 in the myocardium of patients with congenital
heart disease. Birth Defects Res. 109:1183–1195. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Scacheri PC, Crawford GE and Davis S:
Statistics for ChIP-chip and DNase hypersensitivity experiments on
NimbleGen arrays. Methods Enzymol. 411:270–282. 2006.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43(e47)2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Quinlan AR and Hall IM: BEDTools: A
flexible suite of utilities for comparing genomic features.
Bioinformatics. 26:841–842. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin
SM, Lapointe DS and Green MR: ChIPpeakAnno: A Bioconductor package
to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics.
11(237)2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ehrich M, Nelson MR, Stanssens P, Zabeau
M, Liloglou T, Xinarianos G, Cantor CR, Field JK and van den Boom
D: Quantitative high-throughput analysis of DNA methylation
patterns by base-specific cleavage and mass spectrometry. Proc Natl
Acad Sci USA. 102:15785–15790. 2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Weisenberger DJ, Campan M, Long TI, Kim M,
Woods C, Fiala E, Ehrlich M and Laird PW: Analysis of repetitive
element DNA methylation by MethyLight. Nucleic Acids Res.
33:6823–6836. 2005.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hoff K, Lemme M, Kahlert AK, Runde K,
Audain E, Schuster D, Scheewe J, Attmann T, Pickardt T, Caliebe A,
et al: DNA methylation profiling allows for characterization of
atrial and ventricular cardiac tissues and hiPSC-CMs. Clin
Epigenetics. 11(89)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Collins-Nakai R and McLaughlin P: How
congenital heart disease originates in fetal life. Cardiol Clin.
20:367–383, v-vi. 2002.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sheng W, Qian Y, Zhang P, Wu Y, Wang H, Ma
X, Chen L, Ma D and Huang G: Association of promoter methylation
statuses of congenital heart defect candidate genes with Tetralogy
of Fallot. J Transl Med. 12(31)2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sheng W, Chen L, Wang H, Ma X, Ma D and
Huang G: CpG island shore methylation of ZFPM2 is identified in
tetralogy of fallot samples. Pediatr Res. 80:151–158.
2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang J, Ma X, Wang H, Ma D and Huang G:
Elevated methylation of the RXRA promoter region may be responsible
for its downregulated expression in the myocardium of patients with
TOF. Pediatr Res. 75:588–594. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Grunert M, Dorn C, Cui H, Dunkel I, Schulz
K, Schoenhals S, Sun W, Berger F, Chen W and Sperling SR:
Comparative DNA methylation and gene expression analysis identifies
novel genes for structural congenital heart diseases. Cardiovasc
Res. 112:464–477. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Serra-Juhé C, Cuscó I, Homs A, Flores R,
Torán N and Pérez-Jurado LA: DNA methylation abnormalities in
congenital heart disease. Epigenetics. 10:167–177. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Obermann-Borst SA, Van Driel LM, Helbing
WA, de Jonge R, Wildhagen MF, Steegers EA and Steegers-Theunissen
RP: Congenital heart defects and biomarkers of methylation in
children: A case-control study. Eur J Clin Invest. 41:143–150.
2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chowdhury S, Cleves MA, Macleod SL, James
SJ, Zhao W and Hobbs CA: Maternal DNA hypomethylation and
congenital heart defects. Birth Defects Res A Clin Mol Teratol.
91:69–76. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Schultz MD, He Y, Whitaker JW, Hariharan
M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, et
al: Human body epigenome maps reveal noncanonical DNA methylation
variation. Nature. 523:212–216. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Neri F, Rapelli S, Krepelova A, Incarnato
D, Parlato C, Basile G, Maldotti M, Anselmi F and Oliviero S:
Intragenic DNA methylation prevents spurious transcription
initiation. Nature. 543:72–77. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Schlesinger F, Smith AD, Gingeras TR,
Hannon GJ and Hodges E: De novo DNA demethylation and noncoding
transcription define active intergenic regulatory elements. Genome
Res. 23:1601–1614. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Weinberg DN, Papillon-Cavanagh S, Chen H,
Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht
H, et al: The histone mark H3K36me2 recruits DNMT3A and shapes the
intergenic DNA methylation landscape. Nature. 573:281–286.
2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chen B, Bronson RT, Klaman LD, Hampton TG,
Wang JF, Green PJ, Magnuson T, Douglas PS, Morgan JP and Neel BG:
Mice mutant for Egfr and Shp2 have defective cardiac semilunar
valvulogenesis. Nat Genet. 24:296–299. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Taparia S, Gelineau-van Waes J, Rosenquist
TH and Finnell RH: Importance of folate-homocysteine homeostasis
during early embryonic development. Clin Chem Lab Med.
45:1717–1727. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
MacGrogan D, D'Amato G, Travisano S,
Martinez-Poveda B, Luxán G, Del Monte-Nieto G, Papoutsi T, Sbroggio
M, Bou V, Gomez-Del Arco P, et al: Sequential ligand-dependent
notch signaling activation regulates valve primordium formation and
morphogenesis. Circ Res. 118:1480–1497. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
McBride KL, Riley MF, Zender GA,
Fitzgerald-Butt SM, Towbin JA, Belmont JW and Cole SE: NOTCH1
mutations in individuals with left ventricular outflow tract
malformations reduce ligand-induced signaling. Hum Mol Genet.
17:2886–2893. 2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Mortensen SA, Skov LL, Kjaer-Sorensen K,
Hansen AG, Hansen S, Dagnæs-Hansen F, Jensenius JC, Oxvig C, Thiel
S and Degn SE: Endogenous natural complement inhibitor regulates
cardiac development. J Immunol. 198:3118–3126. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong
J, Hu Y, Dong J, Li Q, Wu X, et al: Single-cell transcriptome
analysis maps the developmental track of the human heart. Cell Rep.
26:1934–1950.e5. 2019.PubMed/NCBI View Article : Google Scholar
|