Introduction
Diabetes is a metabolic disease during which the
pancreas loses the ability to secrete insulin, resulting in high
blood sugar levels (1). Severe
diabetes may lead to seizures, heart attack, stroke and coma
(2). Most patients with type 1 or
type 2 diabetes die from complications rather than from diabetes
itself (3,4). Heart disease is the most common and
deadliest complication among patients with diabetes (5). Among the types of heart disease in
patients with diabetes, ischemic heart disease is the most common,
followed by cardiac failure (6).
Reperfusion injury is a common prognostic problem for percutaneous
transluminal coronary intervention therapy (7). A previous study has demonstrated that
myocardial ischemia-reperfusion (IR) injury (IRI) is induced in
patients with diabetes more easily compared with that in healthy
populations, which may be caused by the alteration of mitochondrial
function and reactive oxygen species production (8).
Peroxisome proliferator-activated receptor α (PPARα)
activation has been reported to improve cardiac function and
increase ATP production in myocardial IRI (9,10).
PPARα serves a protective role against cardiac IRI by regulating
the expression of uncoupling protein-3(11). A previous study has demonstrated
that PPARα activation depends on the decrease of glucose uptake and
susceptibility to ischemic injury (12). NF-κB is a transcription factor
involved in diverse cellular activities, including the cardiac IRI
(13). A previous study has
reported that NF-κB activation is induced by diabetic metabolic
disorders with myocardial IRI, whereas inhibition of NF-κB reverses
diabetic-IRI (D-IRI) (14).
Emerging evidence demonstrated that PPARα overexpression can
inactivate NF-κB signaling (15).
Pemafibrate, a selective PPARα modulator, is
effective at regulating lipid and glucose metabolism in patients
with type 2 diabetes mellitus (16). A growing body of literature has
shown that pemafibrate protects against multiple diabetic
complications, for instance diabetic retinopathy, endothelial
dysfunction and cardiovascular disease (17,18).
However, the exact effects of pemafibrate on D-IRI and the
potential mechanisms remains to be elucidated. The present study
aimed to investigate whether pemafibrate may protect against
diabetic IRI in vivo and in vitro, and to examine the
molecular mechanism of action of pemafibrate.
Materials and methods
Animals
Male Sprague-Dawley rats (age, 7 weeks; weight,
220-300 g) were provided by the Tianjin Experimental Animal Center.
Rats were housed in a 12 h light/dark cycle, and food and water was
available ad libitum during the experimental period. A
constant temperature and humidity of 25˚C and 50%, respectively,
was maintained. Rats were subjected to experiments following 2
weeks of acclimatization. All animal care and experimental
procedures were in compliance with the guidelines of the
International Guiding Principles for Biomedical Research Involving
Animals of the Council for International Organizations of Medical
Sciences. All experimental procedures were approved by the Ethical
Committee of Tianjin Teda Hospital (approval no.
EC-20190722-1039).
Diabetic-myocardial IRI (D-IRI)
model
The D-IRI experimental model was established as
previously described (19). Rats
were administered a single dose of 65 mg/kg streptozotocin (STZ;
Sigma-Aldrich; Merck KGaA) by tail vein injection. Following
induction for 8 weeks, the fasting blood (50 µl) were collected by
caudal vein blood once a week and the concentration of fasting
blood glucose was measured, with ≥16.7 mmol/l indicating successful
establishment of the type 1 diabetes mellitus (T1DM) model.
Subsequently, rats were anesthetized with an intraperitoneal
injection of 50 mg/kg body weight pentobarbital. The rats were
mechanically ventilated with air through a rodent ventilator
followed by oral endotracheal intubation. The body temperature of
the rats was maintained at 37±0.5˚C. The thoracic cavity was opened
between the fourth and fifth intercostal space of the left edge of
the thoracotomy, and the heart of the rat was exposed after
pericardial incision. A 7-0 silk thread was sutured around the left
anterior descending coronary artery of the rat, 2 mm from the tip
of the left auricle; 10 min later, when the thread was clamped and
tightened, a snare that blocked the artery was formed. Following
ligation, regional epicardial cyanosis appearance in the heart was
observed as a sign of successful ischemia. At 30 min after the
ischemia, coronary artery reperfusion was performed by releasing
the clamp for 2 h. The behavior of the rats was observed every 20
min. The rats were randomly divided into two groups (n=6 in each
group): D-IRI and D-IRI + pemafibrate (intragastric administration;
1 mg/kg). Pemafibrate was administered immediately after IRI. If
the rats went into shock, decreased activity, lethargy or dyspnea,
the animals were euthanized prior to the experimental endpoint. The
rats were euthanized by an overdose of sodium pentobarbital (200
mg/kg intraperitoneal injection), followed by exsanguination from
the abdominal aorta; death was confirmed by respiratory and cardiac
arrest.
Detection of infarction size
To observe the infarct area,
2,3,5-triphenyltetrazolium chloride (TTC) staining was performed.
The fresh hearts were cut into transverse slices (1-2 mm-thick) and
stained with 1.5% TTC (Sigma-Aldrich; Merck KGaA) for 20 min at
37˚C. Subsequently, the stained slices were then fixed with 4%
formaldehyde for 24 h. The unstained pale area (infarct area) and
TTC-stained red area (ischemic but viable myocardium) were
evaluated through planimetry with the Image-Pro Plus 4.5 software
(Media Cybernetics, Inc.).
Immunohistochemistry
Cardiac tissues were embedded in paraffin as
previously described (20).
Paraffin-embedded specimens (4-µm-thick) were deparaffinized and
rehydrated with a graded ethanol and xylene series at room
temperature. Antigen retrieval was performed by heating the slides
in 1 mmol/l EDTA (pH 8.0) for 30 min. After blocking with 10%
normal goat serum (Wuhan Servicebio Technology Co., Ltd.) for 10
min at 37˚C, slides were probed with primary antibodies targeting
PPARα (cat. no. ab215270; 1:200; Abcam) overnight at 4˚C.
Subsequently, a horseradish peroxidase (HRP)-conjugated goat
anti-rabbit IgG (cat. no. ab6721; 1:1,000; Abcam) antibody was
added, incubated for 1 h at room temperature and visualized using
the eBioscience DAB Advanced Chromogenic kit (Thermo Fisher
Scientific, Inc.). The immunohistochemical images were acquired
using a light microscope (DMI3000; Leica, Microsystems, Inc.) at
x200 magnification from five random fields.
Transmission electron microscopy
(TEM)
The mitochondrial morphology in the rat hearts was
assessed using TEM. Following the fixation of heart sections (1x1x1
mm) with 2.5% glutaraldehyde, an ethanol gradient dehydration (30,
50, 70, 80, 85, 90 and 100% ethanol gradient for 15-20 min) was
performed. The tissues were subsequently embedded in Epon 812 resin
(Sinopharm Chemical Reagent Co., Ltd.). The heart slices (0.1-µm)
were stained with 2% uranylacetate and 1% lead citrate. Tissue
sections were observed and photographed using a Hitachi TEM system
(Hitachi, Ltd.) at x6000 magnification from three random
fields.
Cell culture and treatment
The embryonic rat cardiomyocyte-derived cell line
H9c2 was obtained from the Cell Resource Center, Institute of Basic
Medicine, Chinese Academy of Medical Sciences and cultured in
high-glucose DMEM (cat. no. 11965092; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin at 37˚C in a
humidified incubator with 5% CO2. The cells
(1x106 cells/well) were seeded into 6-well plates. Prior
to the experiments, the cells were starved in 1% FBS-supplemented
low glucose DMEM (cat. no. 11885076; Thermo Fisher Scientific,
Inc.) for 24 h and divided into the following groups: i) Low
glucose (control; final concentration, 5.5 mmol/l); ii) high
glucose (HG; final concentration, 33 mmol/l); iii) HG +
hypoxia/reoxygenation (HG + H/R); and iv) HG + H/R + 50 nmol/l
pemafibrate (MedChemExpress). Briefly, when the cells reached 60%
confluence, they were pre-treated with control or HG media for 48
h. Subsequently, the H/R model was induced by culturing the cells
for 6 h in hypoxic conditions (95% N2 and 5%
CO2) with 1% FBS-DMEM, followed by 4 h of reoxygenation
in normal culture conditions. Pemafibrate was dissolved in DMSO
(203.85 mmol/l) before being added to media.
Cell transfection
An NF-κB overexpression plasmid (NF-κB) and its
empty negative control vector (NC) were purchased from Shanghai
GenePharma Co., Ltd. H9c2 cells were seeded into 6-well plates at a
density of 1x106 cells/well 24 h prior to transfection
and incubated overnight. Transfection (100 nmol/l) was performed
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Transfection efficiency was assessed using reverse
transcription-quantitative (RT-q)PCR 24 h post-transfection.
Detection of mitochondrial
function
Mitochondrial dysfunction was determined by
detecting the mitochondrial ROS (mtROS) levels, ATP content,
malondialdehyde (MDA) release and superoxide dismutase (SOD)
activity. mtROS levels were detected by mitochondrial superoxide
(MitoSOX; Invitrogen; Thermo Fisher Scientific, Inc.) staining.
Briefly, H9c2 cells (1x106 cells) were cultured with 5
mmol/l MitoSOX in serum-free DMEM for 15 min at 37˚C. The
mitochondrial ROS production was measured using flow cytometry (BD
Biosciences) and quantified using FlowJo software (BD Biosciences).
ATP content was evaluated using an ATP Determination kit
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions using a multifunctional plate reader
(BD Biosciences) at a wavelength of 650 nm. MDA release and SOD
activity were determined using commercial kits (cat. nos. A003-1-2
and A001-3-2; Nanjing Jiancheng Bioengineering Institute) according
to the manufacturer's instructions at a wavelength of 532 nm or 450
nm, respectively, using a multifunctional plate reader (BD
Biosciences).
Apoptosis analysis
H9c2 cells (1x106 cells) were collected
and resuspended in 500 µl Annexin binding buffer to determine cell
apoptosis using a Dead Cell Apoptosis kit with Annexin V Alexa
Fluor™ 488 and propidium iodide (PI) (Thermo Fisher Scientific,
Inc.). The cells were incubated with FITC-conjugated Annexin V and
PI each for 10 min at room temperature. A FACSCalibur instrument
(BD Biosciences) was used to measure the levels of cell
fluorescence. The data were analyzed using CellQuest™ v5.1 software
(BD Biosciences).
RT-qPCR
Total RNA was isolated from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Subsequently, cDNA was synthesized using a Reverse
Transcription kit (Takara Biotechnology Co., Ltd.) according to the
manufacturer's instructions. qPCR was performed using
SYBR® Premix Ex Taq (Takara Biotechnology Co., Ltd.) and
an ABI 7500 system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The following thermocycling conditions for PCR was used:
95˚C for 5 min and then 40 cycles of 94˚C for 15 sec, 55˚C for 20
sec, 72˚C for 20 sec, followed by 72˚C for 7 min. The primers
sequences used in the present study were as follows: NF-κB,
forward, 5'-CTGAGTCCCGCCCCTTCTAA-3' and reverse,
5'-CTCCACCAGCTCTTTGATGGT-3'; GAPDH, forward,
5'-ATGTGTCCGTCGTGGATCTGA-3' and reverse,
5'-GATGCCTGCTTCACCACCTT-3'. The 2-ΔΔCt method was
carried out to compare relative expressions (21). GAPDH was considered as an internal
reference gene.
Western blotting
Total proteins in cardiac tissues and H9c2 cells
were isolated using RIPA reagent buffer (Beyotime Institute of
Biotechnology) at 4˚C and a BCA protein assay kit (Beyotime
Institute of Biotechnology) was used to determine protein
concentration. Equal amounts of protein (40 µg per lane) were
separated using SDS-PAGE (8-10% gel) and transferred to
polyvinylidene difluoride (PVDF) membranes. Subsequently, 5%
non-fat milk was used to block the membrane for 1 h at room
temperature. The membrane was incubated with primary antibodies
against PPARα (cat. no. ab3484; 1:1,000; Abcam), cleaved-caspase-3
(cat. no. 9664; 1:1,000), caspase-3 (cat. no. 14220; 1:1,000),
cleaved-caspase-9 (cat. no. 9057; 1:1,000), caspase-9 (cat. no.
9508; 1:1,000), cytochrome c (Cyt-c; cat. no. 11940T;
1:1,000), NF-κB (cat. no. 8242; 1:1,000) and GAPDH (cat. no. 5174;
1:1,000) (all from Cell Signaling Technology, Inc.) overnight at
4˚C. The membranes were incubated with goat-anti-rabbit or
goat-anti-mouse HRP-conjugated IgG (cat. nos. 7074 and 7076,
respectively; 1:10,000; Cell Signaling Technology, Inc.) for 2 h at
room temperature. The protein bands were visualized using an
enhanced chemiluminescence detection system (Applygen Technologies,
Inc.) and subsequently quantified using ImageJ software (version
1.52r; National Institutes of Health). GAPDH was used for
normalization. Data are presented as relative levels compared with
those in the control rats.
Statistical analysis
Data are presented as the mean ± SD and were
analyzed using GraphPad Prism 6.0 (GraphPad Software, Inc.). The
statistical differences were calculated using one-way ANOVA
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Pemafibrate alleviates diabetic
cardiac IRI
STZ injections were used to induce the T1DM model in
rats. Subsequently, the hearts were subjected to
ischemia-reperfusion surgery to mimic cardiac IRI. TTC staining was
performed to evaluate the extent of the infarction induced by
diabetic IRI. Fig. 1A and B demonstrate that the infarct volume was
significantly alleviated in rats treated with pemafibrate compared
with that in rats in the D-IRI group. Immunohistochemical staining
revealed that pemafibrate increased the protein expression levels
of PPARα compared with those in the D-IRI group (Fig. 1C). According to the TEM results, the
D-IRI group displayed marked destruction of the intracellular
structures including the myofibrils and mitochondria; the
mitochondria were rounded, with a large space between cristae, and
a number of mitochondria presented with membrane ruptures (Fig. 1D). The pemafibrate-treated group
presented with intact mitochondria, which were irregularly arranged
between the myofibrils, with normal striation of the cardiac muscle
(Fig. 1D). These results suggest
that pemafibrate increases PPARα expression and may alleviate
diabetic IRI.
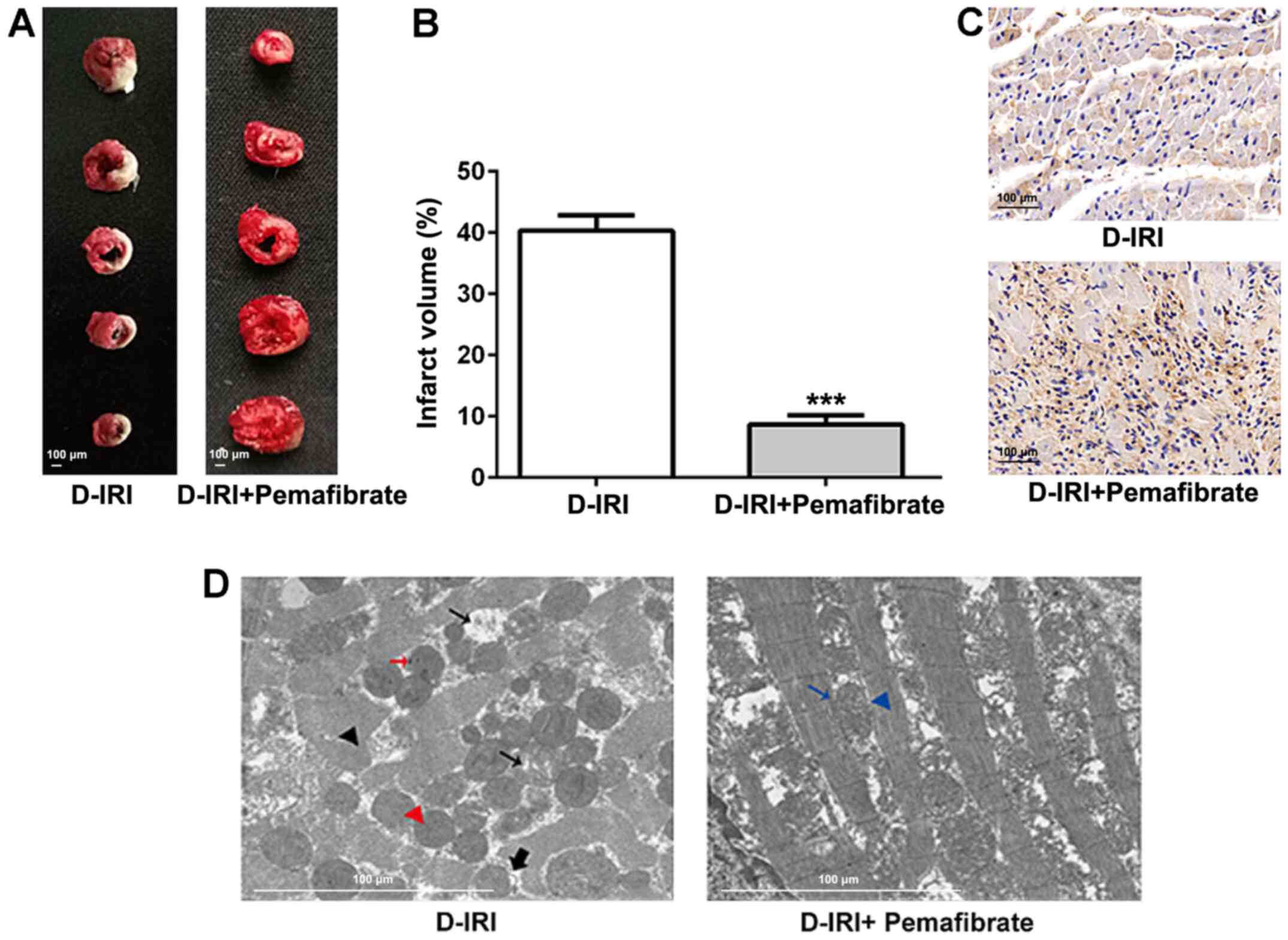 | Figure 1Effects of pemafibrate on the extent
of myocardial infarction and myocardial damage. (A and B) TTC
staining of D-IRI induced infarction. (C) Immunohistochemistry for
PPARα expression in cardiac tissues. Magnification, x200. (D) The
ultrastructure of myocardial tissue from the D-IRI and D-IRI +
pemafibrate groups revealed disrupted myofibrils (black arrowhead),
rounded mitochondria with increased distance between cristae (small
black arrow), electron-dense particles (red arrow), rupturing of
the inner membranes resulting in protrusions from the mitochondria
(thick black arrow), rupturing of the inner and outer membranes
(red arrow head), normal mitochondria (blue arrow) and normal
myofibrils (blue arrowhead). Scale bar, 100 µm.
***P<0.001 vs. D-IRI. D-IRI, diabetic-myocardial
ischemia reperfusion injury; PPARα, peroxisome
proliferator-activated receptor α; TTC, 2,3,5-triphenyltetrazolium
chloride. |
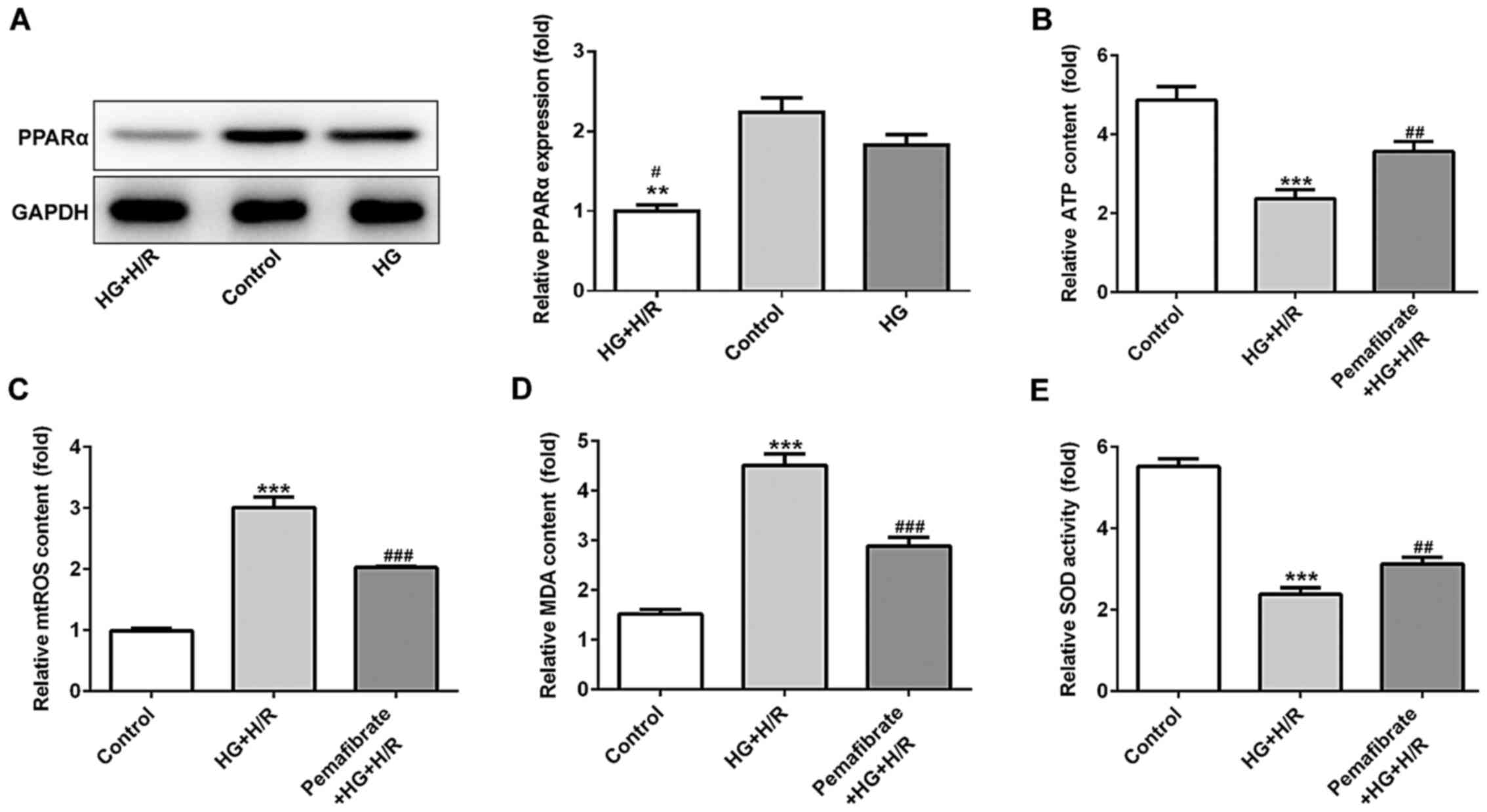
Pemafibrate inhibits mitochondrial
dysfunction by increasing PPARα expression
H9c2 cells were treated with HG and subsequently
with H/R treatment to mimic diabetic IRI in vitro. The
expression levels of PPARα were evaluated using western blotting in
the control, HG and HG + H/R groups. The results demonstrated that
PPARα protein expression levels were downregulated under HG
conditions compared with those in the control group, although there
was no significant difference found, while H/R treatment
significantly exacerbated this reduction (P<0.05; Fig. 2A). As the mitochondria are the major
energy producers (through the production of ATP) that use oxygen in
cells, the ATP activity in treated and untreated H9c2 cells was
investigated. The results demonstrated that compared with those in
the control group, the relative ATP levels were reduced under HG +
H/R treatment, whereas pemafibrate significantly increased the ATP
levels in HG and H/R conditions (P<0.01; Fig. 2B). The relative mtROS levels were
enhanced by HG + H/R treatment compared with those in the control
group (P<0.001; Fig. 2C), while
pemafibrate intervention notably reversed the level of mtTOS
relative to the HG + H/R group (P<0.001; Fig. 2C). To determine the potential
protective effects of pemafibrate on oxidative stress, the levels
of MDA and SOD activity were examined; compared with those in cells
under HG + H/R conditions, treatment with pemafibrate significantly
decreased the MDA levels and increased the SOD activity (P<0.001
and P<0.01, respectively; Fig.
2D and E).
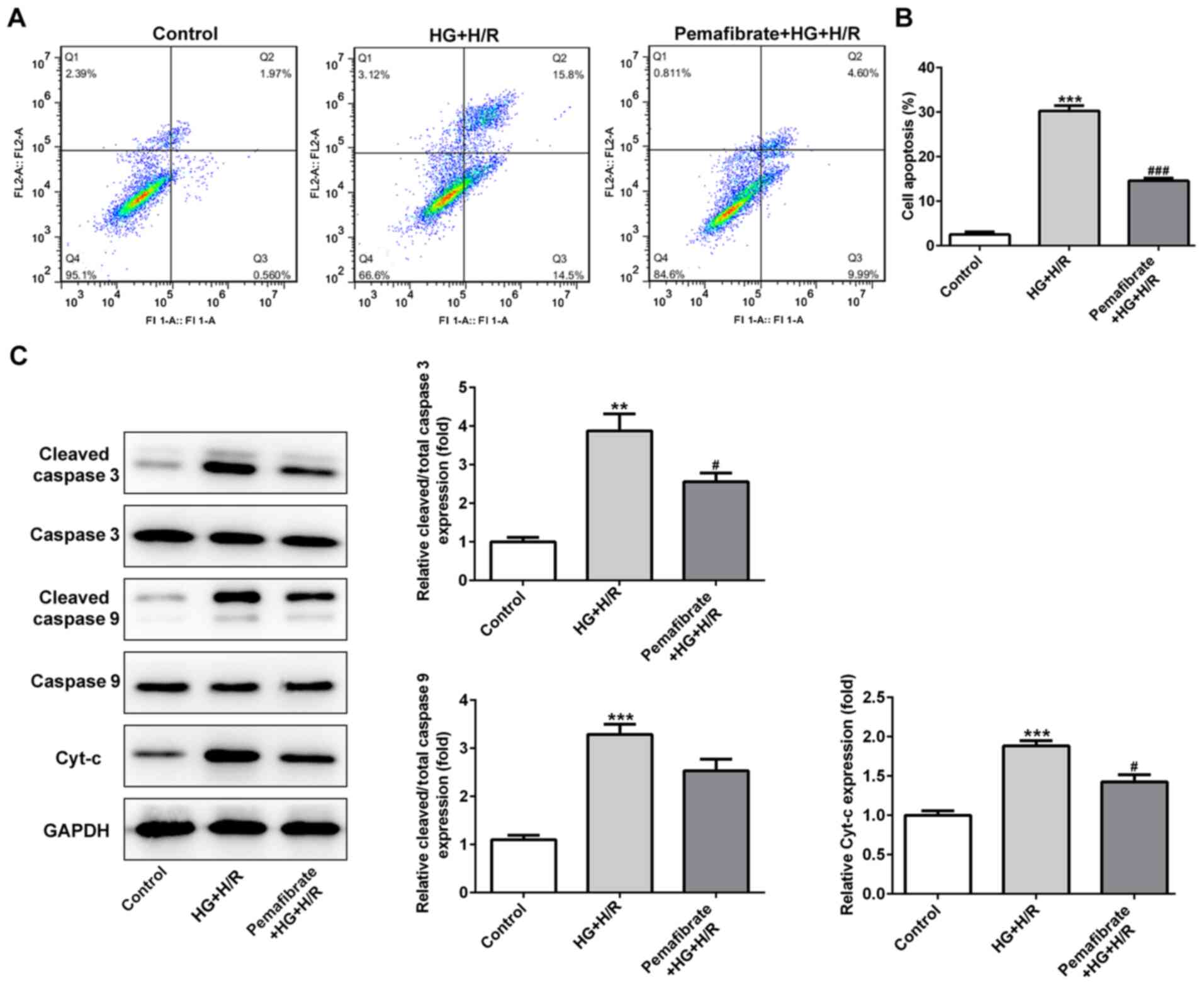
Pemafibrate suppresses
mitochondria-induced apoptosis
To assess whether pemafibrate affected cell death
under HG and H/R conditions, apoptotic rates were determined in the
HG + H/R-treated H9c2 cells using flow cytometry. Compared with
that in the control group, the apoptotic rate was significantly
enhanced in the HG + H/R group, whereas treatment with pemafibrate
decreased the number of apoptotic cells (P<0.001; Fig. 3A and B). Hyperglycemia and H/R treatment also
increased the protein levels of cleaved-caspase-3 and Cyt-c in H9c2
cells compared with those in the control cells (P<0.01; Fig. 3C), which was significantly reversed
by pemafibrate treatment (P<0.05; Fig. 3C). The levels of cleaved-caspase-9
in the HG + H/R group were increased compared with those in the
control group, and they also appeared to be suppressed by
pemafibrate, although the difference was not significant (Fig. 3C).
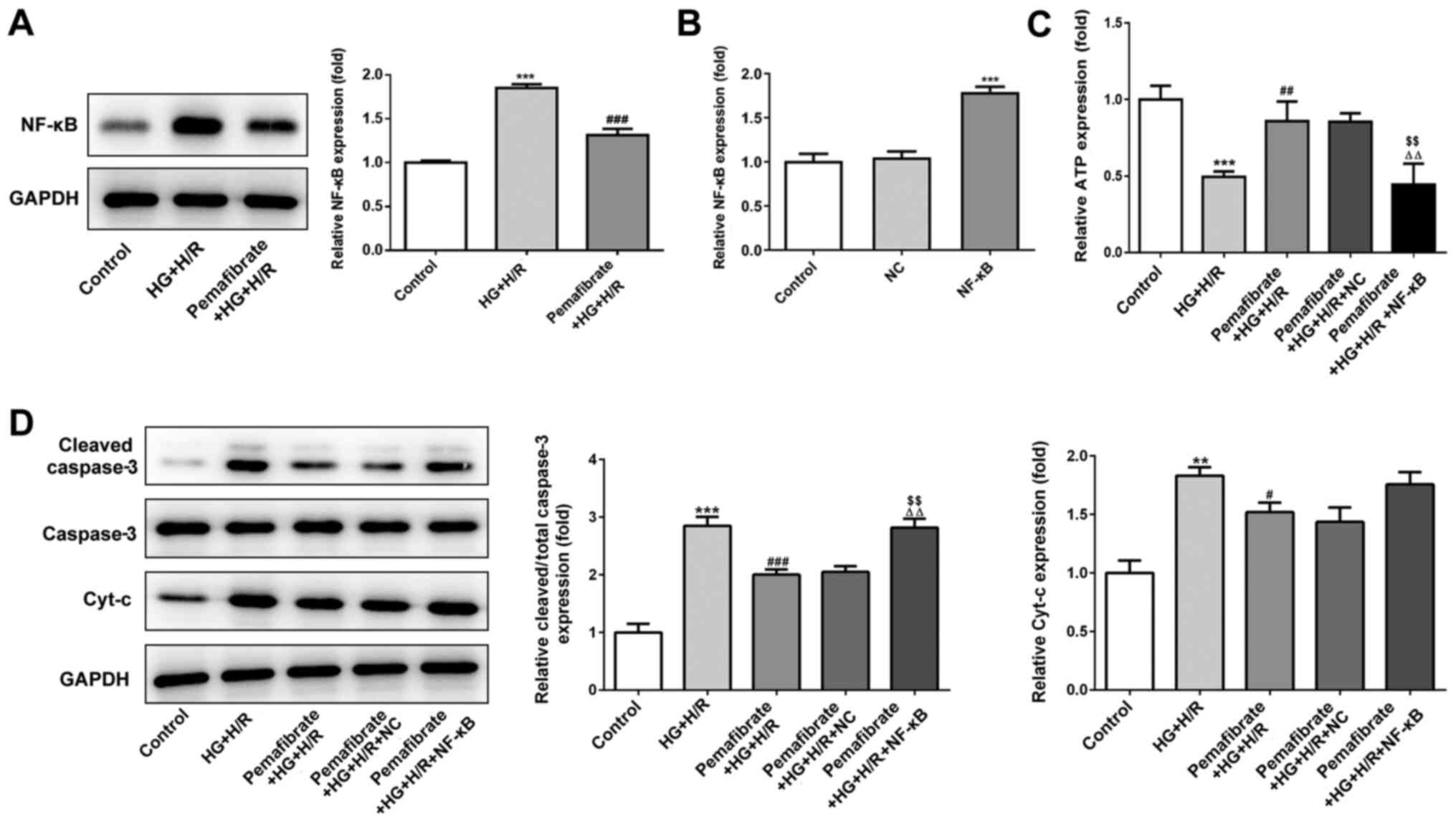
Pemafibrate prevents mitochondrial
dysfunction via the NF-κB signaling pathway
Under HG + H/R conditions, NF-κB protein expression
levels were significantly enhanced in H9c2 cells compared with
those in the control group (P<0.001; Fig. 4A). Pemafibrate significantly
suppressed the protein expression levels of NF-κB compared with
those in the HG + H/R group (P<0.001; Fig. 4A). To evaluate whether pemafibrate
may regulate mitochondrial dysfunction and apoptosis through the
NF-κB signaling pathway, NF-κB was overexpressed in H9c2 cells
(P<0.001; Fig. 4B). The results
demonstrated that overexpression of NF-κB reversed the
pemafibrate-induced increase in ATP levels in the HG + H/R
treatment group (P<0.01 vs. pemafibrate + HG + H/R and
pemafibrate + HG + H/R + NC; Fig.
4C). Pemafibrate decreased the expression levels of
cleaved-caspase-3 compared with those in the HG + H/R group;
however, overexpression of NF-κB reversed this effect (P<0.01
vs. pemafibrate + HG + H/R and pemafibrate + HG + H/R + NC;
Fig. 4D), indicating that
pemafibrate inhibited the HG + H/R-induced apoptosis by regulating
the NF-κB signaling pathway. However, overexpression of NF-κB
expression failed to significantly reverse the pemafibrate-induced
reduction in Cyt-c expression levels (Fig. 4D). These results suggest that
pemafibrate may prevent mitochondrial dysfunction by interacting
with the NF-κB signaling pathway.
Discussion
In the present study, pemafibrate was demonstrated
to protect H9c2 cells against IRI and exposure to HG in
vitro, as well as to limit the extent of the myocardial infarct
in vivo in T1DM model rats. TEM and functional analysis
(including ROS, ATP, SOD and MDA analyses) indicated mitochondrial
dysfunction following diabetic IRI in vivo. Additionally,
immunohistochemistry analysis and western blotting results revealed
that pemafibrate exerted a protective effect against diabetic IRI
through the activation of PPARα in vivo. Therefore, the
protective effects of pemafibrate were likely due to PPARα
activation, which exerted antioxidative and antiapoptotic effects,
thus, preventing further cardiac injury.
The expression levels of PPARα have been reported to
be decreased under high glucose conditions in an animal model of
diabetes (22). Additionally, PPARα
is downregulated in the hearts of offspring from
Ins2Akita mothers with elevated blood glucose
concentration (23). In the present
study, pemafibrate was notably reduced the extent of cardiac
infarction and enhanced the expression of PPARα in the T1DM IR rat
model, which was in agreement with a previous study that
demonstrated that PPARα activation reduced cardiac IRI (24). To determine how pemafibrate may
affect the myocardial IRI under diabetic conditions, the
ultrastructure of the myocardium was evaluated in the present study
and marked destruction of the intracellular structures were
observed in the D-IRI group. Similar alterations about myocardial
injury and mitochondrial dysfunction in the diabetic IRI have been
observed in other reports (25,26).
In vivo analysis in the present study demonstrated that
pemafibrate partially restored the mitochondrial structure in the
myocardium. Furthermore, pemafibrate enhanced the ATP and SOD
activity levels, and decreased ROS and MDA levels in the HG + H/R
treated H9c2 cells.
Both diabetes and IRI induce oxidative stress and
apoptosis in cardiomyocytes (27,28).
Hyperglycemic conditions and IR damage the mitochondrial function
to induce oxidative stress and apoptosis (29,30).
In the present study, pemafibrate reduced apoptosis induced by high
glucose and IR treatment. Cyt-c is an electron carrier in the
mitochondrial electron transport chain (31). Under cellular stress such as high
glucose or IR, Cyt-c release from mitochondria is a key step that
leads to apoptosis, resulting in the formation of the apoptosome
and caspase activation (32).
Pemafibrate markedly reduced Cyt-c protein expression levels and
the extent of apoptosis in the cells under HG + H/R conditions in
the present study. Furthermore, the generation of ROS induces
apoptosis through mitochondria-dependent pathways (33). Therefore, pemafibrate may reduce
mitochondria-dependent apoptosis in H9c2 cells under HG + H/R.
Treatment with pemafibrate in the doses used in the
present study did not significantly decrease the blood glucose
levels in the D-IRI model rats (data not shown). Diabetes enhances
the susceptibility of cardiomyocytes to IRI and disrupts the
cellular signaling pathway responsible for conditioning-induced
enhancement of resistance to cell death to remodel cardiac
responses to IRI (34). The
combination of pemafibrate and hypoglycemic agents may be an
effective strategy for the treatment of D-IRI; however, this
requires further study.
NF-κB is a transcription factor involved in diverse
cellular activities, both in normal and pathological conditions,
including inflammation and proliferation (35). A recent study has demonstrated that
NF-κB activation is induced by diabetic metabolic disorders with
myocardial IRI, whereas inhibition of NF-κB reverses D-IRI-induced
apoptosis (36). The results of the
present study suggested that pemafibrate inhibited the expression
of NF-κB, whereas overexpression of NF-κB reversed the upregulation
of ATP and the downregulation of cleaved-caspase-3 levels after
pemafibrate treatment. However, Cyt-c expression levels were not
significantly enhanced by overexpression of NF-κB. Activation of
PPARα reverses the HG-induced increases in the expression levels of
NF-κB and subsequently inhibits apoptosis in diabetic
cardiomyopathy (37), indicating
that NF-κB may mediate the effects of pemafibrate on mitochondrial
dysfunction and apoptosis.
In conclusion, pemafibrate may be a potential
therapeutic agent for the treatment of myocardial IRI under
diabetic conditions and may alleviate the diabetic-myocardial IRI
by preventing mitochondrial dysfunction and inhibiting
cardiomyocyte apoptosis via inhibition of the NF-κB signaling
pathway. The results of the present study further enriched the
theoretical basis for clinical research into the use of pemafibrate
for the treatment of myocardial IRI in patients with T1DM. However,
the lack of a sham control group is a limitation of the present
study and whether the present findings are applicable to type 2
diabetes remains to be further determined.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WL and JX designed the research, interpreted the
data and performed the experiments. XG and XX collected the data
and searched the literature. WL wrote the manuscript. YS analyzed
and interpreted the data and revised the manuscript. All authors
read and approval the final manuscript.
Ethics approval and consent to
participate
This experiment was approved by the Ethics Committee
of Tianjin Teda Hospital, China (EC-20190722-1039).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bhawal UK, Yoshida K, Kurita T, Suzuki M,
Okada Y, Tewari N, Oka S, Kuboyama N and Hiratsuka K: Effects of
830 nm low-power laser irradiation on body weight gain and
inflammatory cytokines in experimental diabetes in different animal
models. Laser Ther. 28:257–265. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Odak M, Douedi S, Upadhyaya V, Fadhel M
and Cosentino J: Focal neurological seizure due to hyperglycemic
hyperosmolar non-ketotic syndrome in undiagnosed diabetes mellitus.
Cureus. 12(e9909)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hegazy GA, Awan Z, Hashem E, Al-Ama N and
Abunaji AB: Levels of soluble cell adhesion molecules in type 2
diabetes mellitus patients with macrovascular complications. J Int
Med Res. 48(300060519893858)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Šimonienė D, Platūkiene A, Prakapienė E,
Radzevičienė L and Veličkiene D: Insulin resistance in type 1
diabetes mellitus and its association with patient's micro- and
macrovascular complications, sex hormones, and other clinical data.
Diabetes Ther. 11:161–174. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Guan Y, Zhou L, Zhang Y, Tian H, Li A and
Han X: Effects of PP2A/Nrf2 on experimental diabetes
mellitus-related cardiomyopathy by regulation of autophagy and
apoptosis through ROS dependent pathway. Cell Signal.
62(109339)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Low Wang CC, Hess CN, Hiatt WR and
Goldfine AB: Clinical update: Cardiovascular disease in diabetes
mellitus: Atherosclerotic cardiovascular disease and heart failure
in type 2 diabetes mellitus-mechanisms, management, and clinical
considerations. Circulation. 133:2459–2502. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dehaini H, Awada H, El-Yazbi A, Zouein FA,
Issa K, Eid AA, Ibrahim M, Badran A, Baydoun E, Pintus G and Eid
AH: MicroRNAs as potential pharmaco-targets in ischemia-reperfusion
injury compounded by diabetes. Cells. 8(152)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Russo I, Penna C, Musso T, Popara J,
Alloatti G, Cavalot F and Pagliaro P: Platelets, diabetes and
myocardial ischemia/reperfusion injury. Cardiovasc Diabetol.
16(71)2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lam VH, Zhang L, Huqi A, Fukushima A,
Tanner BA, Onay-Besikci A, Keung W, Kantor PF, Jaswal JS, Rebeyka
IM and Lopaschuk GD: Activating PPARα prevents post-ischemic
contractile dysfunction in hypertrophied neonatal hearts. Circ Res.
117:41–51. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yue TL, Bao W, Jucker BM, Gu JL, Romanic
AM, Brown PJ, Cui J, Thudium DT, Boyce R, Burns-Kurtis CL, et al:
Activation of peroxisome proliferator-activated receptor-alpha
protects the heart from ischemia/reperfusion injury. Circulation.
108:2393–2399. 2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Song JW, Kim HJ, Lee H, Kim JW and Kwak
YL: Protective effect of peroxisome proliferator-activated receptor
α activation against cardiac ischemia-reperfusion injury is related
to upregulation of uncoupling protein-3. Oxid Med Cell Longev.
2016(3539649)2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Panagia M, Gibbons GF, Radda GK and Clarke
K: PPAR-alpha activation required for decreased glucose uptake and
increased susceptibility to injury during ischemia. Am J Physiol
Heart Circ Physiol. 288:H2677–H2683. 2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Fu ZH, Mui D, Zhu H and Zhang Y: Exenatide
inhibits NF-κB and attenuates ER stress in diabetic cardiomyocyte
models. Aging (Albany NY). 12:8640–8651. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li L, Luo W, Qian YY, Zhu W, Qian J, Li J,
Jin Y, Xu X and Liang G: Luteolin protects against diabetic
cardiomyopathy by inhibiting NF-κB-mediated inflammation and
activating the Nrf2-mediated antioxidant responses. Phytomedicine.
59(152774)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jiang W, Cai X, Xu T, Liu K, Yang D, Fan
L, Li G and Yu X: Tripartite motif-containing 46 promotes viability
and inhibits apoptosis of osteosarcoma cells by activating NF-B
signaling through ubiquitination of PPAR. Oncol Res. 28:409–421.
2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Araki E, Yamashita S, Arai H, Yokote K,
Satoh J, Inoguchi T, Nakamura J, Maegawa H, Yoshioka N, Tanizawa Y,
et al: Effects of pemafibrate, a novel selective PPARα modulator,
on lipid and glucose metabolism in patients with type 2 diabetes
and hypertriglyceridemia: A randomized, double-blind,
placebo-controlled, phase 3 trial. Diabetes Care. 41:538–546.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tomita Y, Lee D, Miwa Y, Jiang X, Ohta M,
Tsubota K and Kurihara T: Pemafibrate protects against retinal
dysfunction in a murine model of diabetic retinopathy. Int J Mol
Sci. 21(6243)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Pradhan AD, Paynter NP, Everett BM, Glynn
RJ, Amarenco P, Elam M, Ginsberg H, Hiatt WR, Ishibashi S, Koenig
W, et al: Rationale and design of the pemafibrate to reduce
cardiovascular outcomes by reducing triglycerides in patients with
diabetes (PROMINENT) study. Am Heart J. 206:80–93. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang D, He Y, Ye X, Cai Y, Xu J, Zhang L,
Li M, Liu H, Wang S and Xia Z: Activation of autophagy inhibits
nucleotide-binding oligomerization domain-like receptor α protein 3
inflammasome activation and attenuates myocardial
ischemia-reperfusion injury in diabetic rats. J Diabetes Investig.
11:1126–1136. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Srisowanna N, Choijookhuu N, Yano K,
Batmunkh B, Ikenoue M, Nhat Huynh Mai N, Yamaguchi Y and Hishikawa
Y: The effect of estrogen on hepatic fat accumulation during early
phase of liver regeneration after partial hepatectomy in rats. Acta
Histochem Cytochem. 52:67–75. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Hu Y, Chen Y, Ding L, He X, Takahashi Y,
Gao Y, Shen W, Cheng R, Chen Q, Qi X, et al: Pathogenic role of
diabetes-induced PPARα- down-regulation in microvascular
dysfunction. Proc Natl Acad Sci USA. 110:15401–15406.
2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Lindegaard ML and Nielsen LB: Maternal
diabetes causes coordinated down-regulation of genes involved with
lipid metabolism in the murine fetal heart. Metabolism. 57:766–773.
2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Qi K, Li X, Geng Y, Cui H, Jin C, Wang P,
Li Y and Yang Y: Tongxinluo attenuates reperfusion injury in
diabetic hearts by angiopoietin-like 4-mediated protection of
endothelial barrier integrity via PPARα- pathway. PLoS One.
13(e0198403)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Muráriková M, Ferko M, Waczulíková I,
Jašová M, Kancirová I, Murínová J and Ravingerová T: Changes in
mitochondrial properties may contribute to enhanced resistance to
ischemia-reperfusion injury in the diabetic rat heart. Can J
Physiol Pharmacol. 95:969–976. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yu L, Gong B, Duan W, Fan C, Zhang J, Li
Z, Xue X, Xu Y, Meng D, Li B, et al: Melatonin ameliorates
myocardial ischemia/reperfusion injury in type 1 diabetic rats by
preserving mitochondrial function: Role of AMPK-PGC-1α-SIRT3
signaling. Sci Rep. 7(41337)2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wilson AJ, Gill EK, Abudalo RA, Edgar KS,
Watson CJ and Grieve DJ: Reactive oxygen species signalling in the
diabetic heart: Emerging prospect for therapeutic targeting. Heart.
104:293–299. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhang M, Zhu J, Qin X, Zhou M, Zhang X,
Gao Y, Zhang T, Xiao D, Cui W and Cai X: Cardioprotection of
tetrahedral DNA nanostructures in myocardial ischemia-reperfusion
injury. ACS Appl Mater Interfaces. 11:30631–30639. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Hou J, Zheng D, Xiao W, Li D, Ma J and Hu
Y: Mangiferin enhanced autophagy via inhibiting mTORC1 pathway to
prevent high glucose-induced cardiomyocyte injury. Front Pharmacol.
9(383)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhou H, Toan S, Zhu P, Wang J, Ren J and
Zhang Y: DNA-PKcs promotes cardiac ischemia reperfusion injury
through mitigating BI-1-governed mitochondrial homeostasis. Basic
Res Cardiol. 115(11)2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang X, Tang D, Zou Y, Wu X, Chen Y, Li H,
Chen S, Shi Y and Niu H: A mitochondrial-targeted peptide
ameliorated podocyte apoptosis through a HOCl-alb-enhanced and
mitochondria-dependent signalling pathway in diabetic rats and in
vitro. J Enzym Inhib Med Chem. 34:394–404. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kalpage HA, Wan J, Morse PT, Zurek MP,
Turner AA, Khobeir A, Yazdi N, Hakim L, Liu J, Vaishnav A, et al:
Cytochrome c phosphorylation: Control of mitochondrial electron
transport chain flux and apoptosis. Int J Biochem Cell Biol.
121(105704)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lejay A, Fang F, John R, Van JA, Barr M,
Thaveau F, Chakfe N, Geny B and Scholey JW: Ischemia reperfusion
injury, ischemic conditioning and diabetes mellitus. J Mol Cell
Cardiol. 91:11–22. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wen Y, Geng L, Zhou L, Pei X, Yang Z and
Ding Z: Betulin alleviates on myocardial inflammation in diabetes
mice via regulating Siti1/NLRP3/NF-κB pathway. Int Immunopharmacol.
85(106653)2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Liu Y, Wang T, Zhang M, Chen P and Yu Y:
Down-regulation of myocardial infarction associated transcript 1
improves myocardial ischemia-reperfusion injury in aged diabetic
rats by inhibition of activation of NF-κB signaling pathway. Chem
Biol Interact. 300:111–122. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Nan WQ, Shan TQ, Qian X, Ping W, Bing GA
and Ying LL: PPARα agonist prevented the apoptosis induced by
glucose and fatty acid in neonatal cardiomyocytes. J Endocrinol
Invest. 34:271–275. 2011.PubMed/NCBI View Article : Google Scholar
|