Introduction
Endothelial cells (ECs) serve to maintain vascular
integrity and homeostasis by sensing and responding to pathological
and physiological stimuli (1-4).
During the onset of vascular disease, major phenotypical
alterations occur in the ECs, resulting in increased vascular
permeability, release of large quantities of inflammatory cytokines
(IL-8, IL-6 and IL-1β) and leukocyte adhesion (5-7).
These pathological changes in the blood vessel wall structure and
function in turn increases the risk of atherosclerosis (8-10).
It has previously been reported that EC inflammation and apoptosis
serve key roles in the initiation and development of
atherosclerosis, hypertension, diabetes and other cardiovascular
diseases (11). Furthermore,
endothelial dysfunction is regarded to be one of the first stages
in the pathophysiology of atherosclerosis (12). Loss of morphological and functional
integrity in vascular ECs has been reported to be attributed to
inflammation and apoptosis (13).
Therefore, therapeutic strategies targeting inflammation, apoptosis
and vascular EC dysfunction may be important for the treatment of
atherosclerosis.
Protein tyrosine phosphatase 1B (PTP1B) is a
non-transmembrane protein tyrosine phosphatase that has been
documented to be a negative regulator in diabetes and obesity
signaling (14-16).
In addition, it has reported roles in the malignant transformation
of various cancers, including pancreatic cancer and resistance in
cancer treatments, such as dendritic cell-based cancer
immunotherapy (14-16).
Accumulating evidence indicates that PTP1B is also involved in
atherosclerosis (17). Thompson
et al (18) previously
reported that PTP1B inhibitors can prevent and reverse
atherosclerotic plaque formation in low-density lipoprotein (LDL)
receptor-/- mice with atherosclerosis, thereby reducing
the risk of cardiovascular disease. Improved glucose homeostasis,
reduced circulating lipids and atherosclerotic plaque lesions, have
also been observed in myeloid-PTP1B-knockout mice (apolipoprotein
E-/-/lysozyme M-PTP1B) with atherosclerosis (19). In addition, endothelial PTP1B has
been reported to serve as the main regulator of EC proliferation in
cardiovascular disease (20). By
contrast, PTP1B depletion was found to induce endothelium-dependent
vasorelaxation in microvessels in animal models of heart failure
and diabetes (21,22). However, to the best of our
knowledge, the potential effects of PTP1B on the inflammation,
apoptosis and dysfunction of oxidized (ox)-LDL-induced vascular ECs
and associated mechanisms remain unreported.
Therefore, the present study aimed to investigate
the possible role of PTP1B in ox-LDL-treated HUVECs. The study
reported the efficacy of PTP1B on the proliferation, inflammation,
apoptosis, oxidative stress, tubule formation, Kruppel-like factor
2 (KLF2) and 5'AMP-activated protein kinase (AMPK)/sirtuin 1
(SIRT1) signaling pathway in ox-LDL-induced HUVECs through
functional experiments and mechanism assays.
Materials and methods
Cell culture
Immortalized HUVECs were purchased from Shanghai
EK-Bioscience Biotechnology Co., Ltd. (cat. no. CC-Y1285). Cells
were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Hyclone; Cytiva) and 1%
penicillin/streptomycin in a humidified atmosphere containing 5%
CO2 at 37˚C. To establish an in vitro
atherosclerosis model, HUVECs were treated with 100 µg/ml ox-LDL
(Guangzhou Yiyuan Biological Technology Co., Ltd.) for 24 h at
37˚C. Untreated cells were regarded as the control group.
Transfection
The short hairpin (sh)RNA targeting PTP1B
(sh-PTP1B#1/2), KLF2 (sh-KLF2#1/2) and the corresponding
sh-negative control (NC; sh-NC) were synthesized and inserted into
the pRNA-U6.1 plasmid (GenScript) by GeneCopoeia, Inc. The
sequences of shRNAs were as follows: sh-PTP1B#1,
5'-GCTACAGGTTCCTGTTCAA-3'; sh-PTP1B#2, 5'-GGTTCCTGTTCAACAGCAA-3';
sh-KLF2#1, 5'-GCACCGACGACGACCTCAA-3'; sh-KLF2#2,
5'-GAGTGGTAGCTTTCTACAA-3'; sh-NC,
5'-CCGGCAACAAGATGAAGAGCACCAACTC-3'.
A pcDNA3.1 overexpression vector (GenScript)
encoding the full-length KLF2 (Ov-KLF2) and the corresponding NC
(Ov-NC) were produced by Shanghai GenePharma Co., Ltd. In total,
100 nM recombinant vectors were transfected into HUVECs for 48 h
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol at 37˚C.
The transfected HUVECs were then collected for subsequent
experiments.
Cell Counting Kit (CCK)-8 assay
HUVECs (4x104 cells/well) seeded into a
96-well plate were transfected with sh-PTP1B in the presence or
absence of sh-KLF2 before treatment with ox-LDL (100 µg/ml) for 24
h at 37C (23-25).
Subsequently, 10 µl CCK-8 solution (Beyotime Institute of
Biotechnology) was added to each well and the plates were incubated
for 2 h at 37˚C. The optical density values were analyzed using a
Thermo Multiskan FC microplate reader (Thermo Fisher Scientific,
Inc.) at 450 nm.
Lactate dehydrogenase (LDH) activity
assay
HUVECs plated at a density 1x104
cells/well in 96-well plates were transfected with sh-PTP1B in the
presence or absence of sh-KLF2 and underwent ox-LDL treatment (100
µg/ml) for 24 h at 37˚C. Cells were subsequently harvested from the
culture plate and the LDH activity levels were detected by LDH
activity kit (cat. no. A020; Nanjing Jiancheng Bioengineering
Institute) at 450 nm using a microplate reader (Thermo Fisher
Scientific, Inc.).
ELISA
Briefly, HUVECs were seeded into 96-well plates
(5x103 cells/well). Following the aforementioned
treatment, cell supernatant was collected after centrifugation at
2,000 x g for 5 min at 4˚C. IL-6 (cat. no. ab178013), IL-1β (cat.
no. ab214025) and TNF-α (cat. no. ab181421) levels in the culture
supernatant were determined using ELISA kits from Abcam according
to the manufacturer's protocols. The absorbance was determined at
450 nm using an xMark microplate absorbance spectrophotometer
(Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from HUVECs using the
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Subsequently, the quality and purity of the extracted RNA
were detected using a NanoDrop® 3000 spectrophotometer
(Thermo Fisher Scientific, Inc.). Total RNA was reverse-transcribed
into complementary DNA using the PrimeScript™ RT Master
Mix (Takara Bio, Inc.) according to the manufacturer's
instructions. qPCR was performed using the SYBR Premix Ex
Taq™ II kit (Takara Bio, Inc.). The following
thermocycling conditions were used: 95˚C for 7 min; 45 cycles of
denaturation at 95˚C for 10 sec, annealing at 60˚C for 15 sec and
extension at 72˚C for 30 sec; final extension at 60˚C for 1 min
followed by cooling at 40˚C for 5 min. The following primers were
used for qPCR: PTP1B forward (F), 5'-GCGGCCATTTACCAGTTGAC-3' and
reverse (R), 5'-ATGACGACACCCCTGCTTTT-3'; KLF2 F,
5'-TGGGCATTTTTGGGCTACCT-3' and R, 5'-GTCAGTGGGACCAGCACTTT-3'; and
GAPDH F, 5'-GGGAAACTGTGGCGTGAT-3' and R, 5'-GAGTGGGTGTCGCTGTTGA-3'.
Relative mRNA expression levels were normalized to GAPDH using the
2-ΔΔCq method (26).
TUNEL assay
Cell apoptosis was detected using an In Situ
Cell Death Detection Kit (cat. no. 11684817910; Roche Diagnostics
GmbH). Cells were fixed with 4% paraformaldehyde at room
temperature away from light for 30 min and then incubated with
proteinase K for 15 min in 37˚C. Subsequently, cells were placed in
3% H2O2 for 15 min at room temperature to
inhibit endogenous peroxide. HUVECs were then stained with the
TUNEL detection kit at 37˚C for 60 min and co-labeled with the DAPI
working solution (1 µg/ml) for 10 min at 37˚C according to the
manufacturer's protocols. Labeled HUVECs were washed with PBS
buffer. Next, cells on slips were mounted using DAPI-containing
mounting medium (Vector Laboratories, Inc.) and visualized using a
fluorescence microscope (Nikon Eclipse 80i; Nikon Corporation), and
>10 fields per section for each sample were examined. The TUNEL
positive cell rate (%) was calculated using the software of
Developer XD 1.2 (Definiens AG) according to the following formula:
(Number of positive cells/total number of cells) x100.
Malondialdehyde (MDA), superoxide
dismutase (SOD) and glutathione peroxidase (GSH-Px) assays
HUVECs were seeded into 96-well plates
(5x103 cells/well). After transfection of sh-PTP1B in
the presence or absence of sh-KLF2 and ox-LDL treatment (100 µg/ml)
for 24 h at 37˚C, oxidative stress levels were quantified by
detecting the levels of MDA (cat. no. A003-4-1) and the activity of
SOD (cat. no. A001-3-2) and GSH-Px (cat. no. A005-1-2) in the media
using the corresponding commercial kits (Nanjing Jiancheng
Bioengineering Institute) according to the manufacturer's
protocols. Absorbance at 532 nm was measured using a microplate
reader (BioTek Instruments, Inc.).
Endothelial tube formation assay
HUVECs were seeded at a density of 5x103
cells/well into 96-well plates pre-coated with 50 µl/well Matrigel
(Corning, Inc.) at 37˚C for 2 h. HUVECs were seeded into 96-well
plates and transfected with sh-PTP1B in the presence or absence of
sh-KLF2 and underwent ox-LDL treatment (100 µg/ml). Following
incubation for 24 h at 37˚C, tubules characterized by the
capillary-like structures were imaged using a light microscope in
five randomly selected fields (magnification, x40).
Dual-luciferase reporter assay
Briefly, the potential interaction between KLF2 and
the PTP1B promoter were predicted using data from the 9th release
(2022) of JASPAR database (http://jaspar.genereg.net). The wild-type and mutant
PTP1B promoter fragments, including predicted KLF2 sites, were
amplified and cloned downstream of the luciferase reporter gene in
the firefly luciferase reporter pGL3 vector (Promega Corporation).
HUVECs were transfected with 2.5 µg Ov-KLF2 or 2.5 µg Ov-NC
plasmids, with 100 ng of luciferase reporter plasmids driven by
wild-type or mutant PTP1B promoter, using Lipofectamine 2000 at
37˚C for 48 h before luciferase activity was detected using the
Dual-Luciferase Reporter Assay Kit (Promega Corporation). Firefly
luciferase activity was normalized to Renilla luciferase
activity.
Chromatin immunoprecipitation
(ChIP)
ChIP experiments were performed using the ChIP-IT
kit (cat. no. 53008; Active Motif, Inc.) according to the
manufacturer's instructions as previously described (27). Briefly, HUVECs were cross-linked
with 1% formaldehyde at 37˚C for 10 min. The cell lysates were
sonicated using a 10 sec on and 10 sec off mode for 12 cycles on
ice. Following quenching with 2.5 M glycine for 5 min at room
temperature, the supernatant was collected, added to 60 µl Protein
A Agarose beads (cat. no. 9863; Cell Signaling Technology, Inc.)
and mixed for 1 h after centrifugation. DNA was immunoprecipitated
from the 100 µl cell lysates using 2 µg KLF2 antibody (cat. no.
MBS9211982; MyBioSource) for a 2-h incubation at 4˚C. The beads
were washed using a magnetic separation rack and the bound
chromatin was eluted in ChIP Elution Buffer with Proteinase K
mixer. PCR amplification of the PTP1B binding site was then
performed using the precipitated DNA by means of SYBR Premix Ex
Taq™ II kit (cat. no. RR420A; Takara Bio, Inc.). The
following thermocycling conditions were used: 95˚C for 7 min,
followed by 45 cycles of denaturation at 95˚C for 10 sec, annealing
at 60˚C for 15 sec and extension at 72˚C for 30 sec, with a final
extension at 60˚C for 1 min followed by cooling at 40˚C for 5 min.
Next, the immunoprecipitated DNA was purified using a ChIP DNA
purification kit (cat. no. D0033; Beyotime Institute of
Biotechnology). Nonspecific antibody against IgG (2 µg; 1:40; cat.
no. sc-2025; Santa Cruz Biotechnology, Inc.) served as a negative
control.
Western blotting
Total protein was isolated from HUVECs using RIPA
buffer (Beyotime Institute of Biotechnology). The protein
concentration was determined using a BCA protein assay kit
(Beyotime Institute of Biotechnology). A total of 30 µg protein was
separated using SDS-PAGE on a 10% gel (Bio-Rad Laboratories, Inc.)
and transferred onto PVDF membranes (MilliporeSigma). After being
blocked with 5% non-fat milk for 1 h at room temperature, the
membranes were incubated with primary antibodies targeting PTP1B
(1:1,000; cat. no. ab244207; Abcam), Bcl-2 (1:1,000; cat. no.
ab196495; Abcam), Bax (1:1,000; cat. no. ab32503; Abcam), cleaved
caspase-3 (1:500; cat. no. ab32042; Abcam), caspase-3 (1:5,000;
cat. no. ab32351; Abcam), vascular endothelial growth factor A
(VEGFA; 1:1,000; cat. no. ab155944; Abcam), KLF2 (1:1,000; cat. no.
ab194486; Abcam), phosphorylated (p)-AMPK (1:1,000; cat. no.
ab92701; Abcam), AMPK (1:1,000; cat. no. ab32047; Abcam), SIRT1
(1:1,000; cat. no. ab110304; Abcam) and GAPDH (1:1,000; cat. no.
ab8245; Abcam) overnight at 4˚C. Following the primary incubation,
the membranes were incubated with the HRP-conjugated goat
anti-rabbit or mouse secondary antibodies (cat. nos. sc-2004 or
sc-2005; 1:5,000; Santa Cruz Biotechnology, Inc.) at room
temperature for 1 h. The bands were visualized using the
Pierce™ Enhanced Chemiluminescence (ECL) Western
Blotting Substrate Kit (Invitrogen; Thermo Fisher Scientific, Inc.)
and quantified by densitometry (Quantity One 4.5.0 software;
Bio-Rad Laboratories, Inc.). All data were normalized to that of
GAPDH.
Statistical analysis
All statistical analysis was performed using
GraphPad Prism software (version 5.01; GraphPad Software, Inc.).
All data are presented as the mean ± SD from at least three
independent experiments. Statistical comparisons between two groups
were performed using unpaired Student's t-test, whereas comparisons
among >2 groups was performed using one-way ANOVA followed by
Bonferroni post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
PTP1B knockdown restores cell
viability in ox-LDL-induced HUVECs
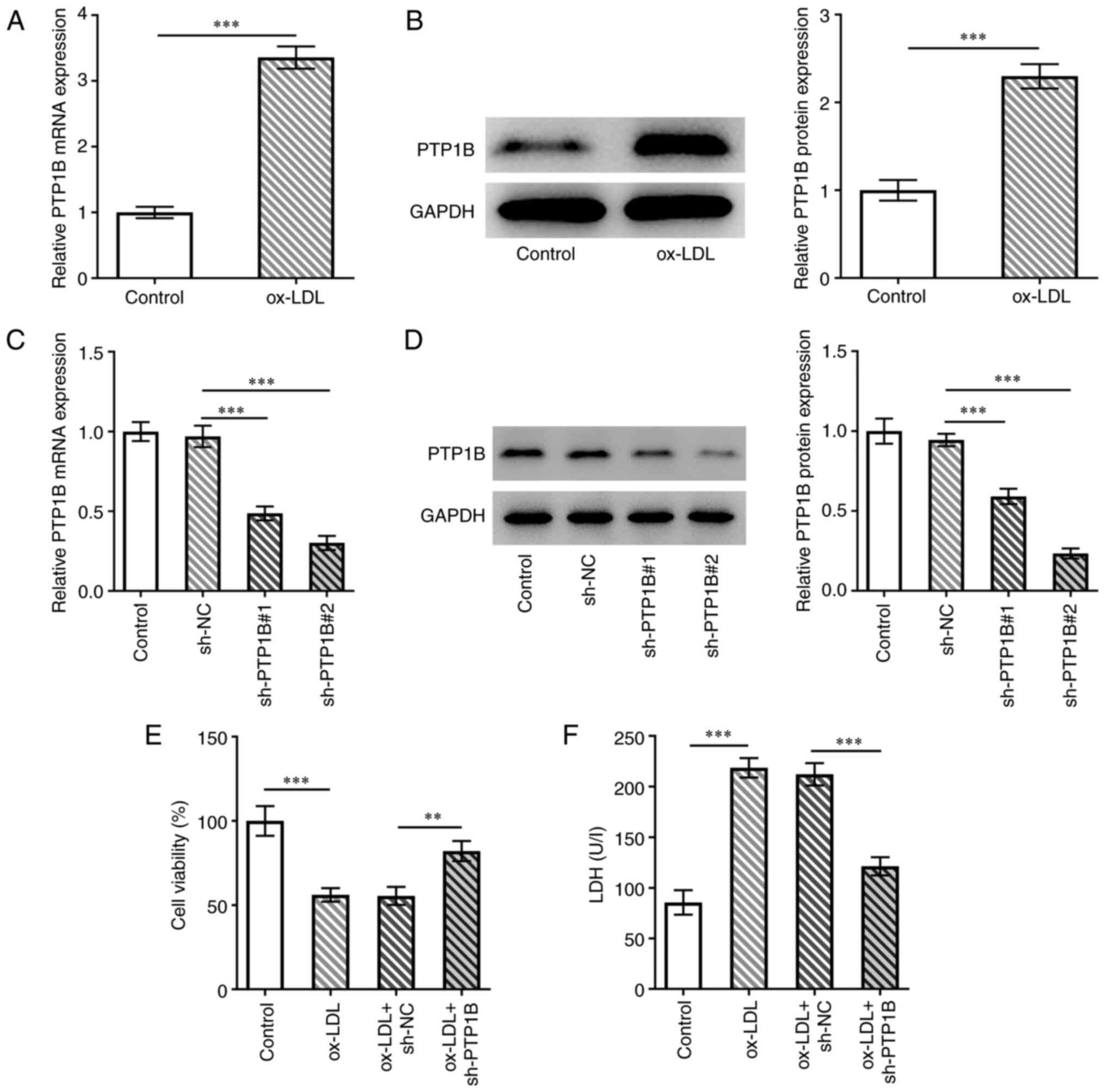
To investigate the role of PTP1B in atherosclerosis,
the expression of PTP1B in HUVECs was measured. PTP1B mRNA and
protein expression levels were significantly increased in
ox-LDL-induced cells compared with those in the control group
(Fig. 1A and B). Subsequently, sh-PTP1B constructs were
transfected into HUVECs to knock down PTP1B expression. The results
from the RT-qPCR and western blotting experiments demonstrated that
the PTP1B mRNA and protein expression levels were significantly
reduced following transfection with sh-PTP1B#1 or shPTP1B#2
(Fig. 1C and D). Since sh-PTP1B#2 demonstrated a
greater transfection efficiency, it was selected for subsequent
experiments (Fig. 1C and D). Cell viability was then assessed using
the CCK-8 assay. The results demonstrated that ox-LDL significantly
reduced the cell viability of HUVECs, which was in turn reversed by
PTP1B silencing (Fig. 1E).
Furthermore, significantly increased LDH activity was observed in
ox-LDL-treated cells compared with the control group, which was
also reversed by sh-PTP1B transfection in response to ox-LDL
(Fig. 1F).
PTP1B knockdown alleviates
ox-LDL-induced inflammatory damage in HUVECs
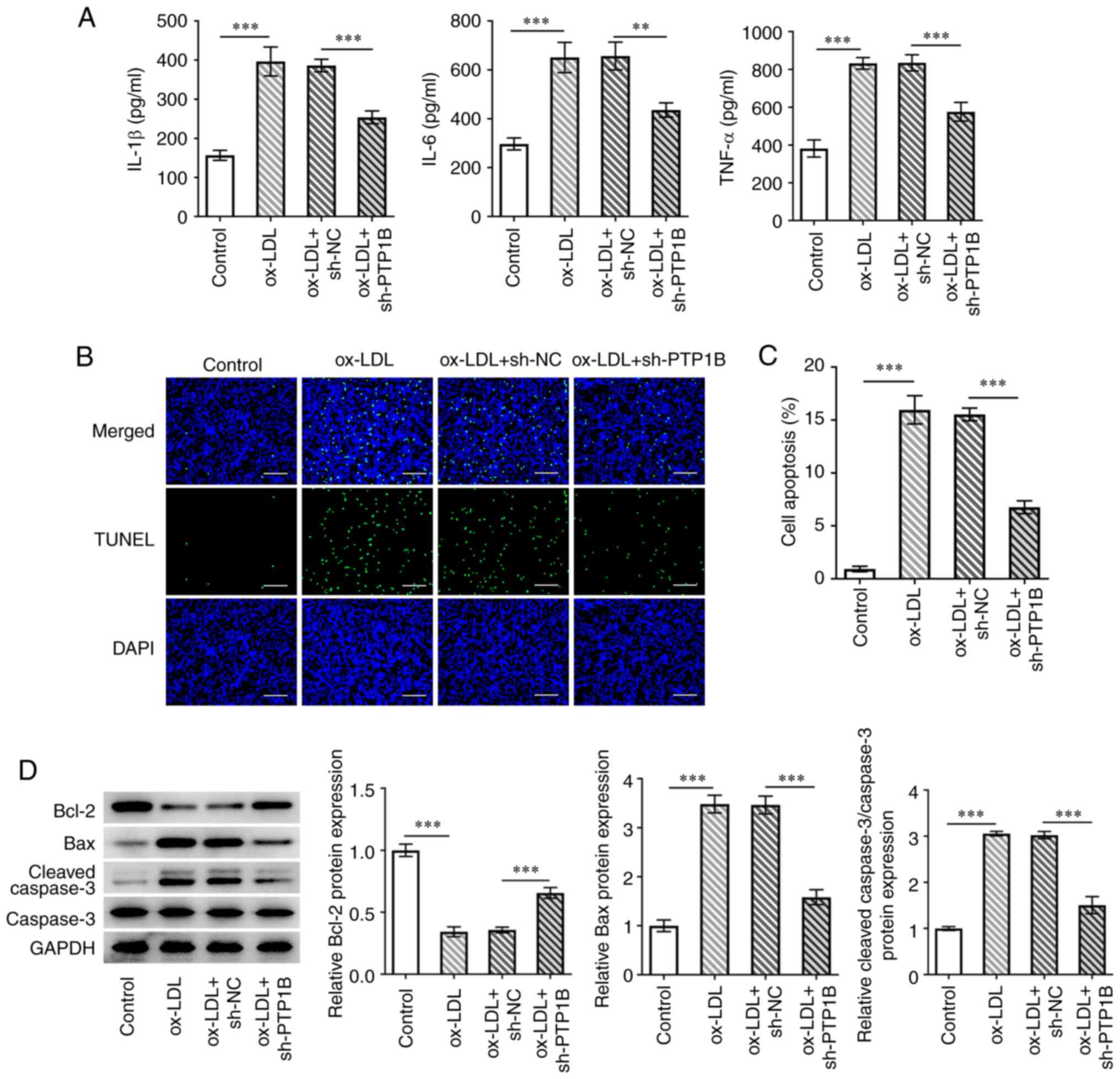
To evaluate the effects of PTP1B silencing on
inflammatory injury in HUVECs in response to ox-LDL, parameters of
inflammation and cell apoptosis were measured. The results
demonstrated that ox-LDL treatment significantly enhanced IL-6,
IL-1β and TNF-α levels compared with those in the control group
(Fig. 2A). However, sh-PTP1B
transfection counteracted these effects of ox-LDL on the three
inflammatory factors aforementioned (Fig. 2A). Furthermore, cell apoptosis was
found to be significantly increased by ox-LDL, which was reversed
following the knockdown of PTP1B expression (Fig. 2B and C). Western blotting results demonstrated
a significant reduction in the Bcl-2 protein expression levels and
a significant increase in Bax and cleaved caspase-3 protein
expression levels in ox-LDL-treated cells compared with those in
the control group (Fig. 2D).
However, the effects of ox-LDL on the expression of these
aforementioned proteins associated with apoptosis were reversed by
PTP1B knockdown (Fig. 2D).
PTP1B knockdown suppresses
ox-LDL-induced oxidative stress in HUVECs and restores
tubule-formation ability
Subsequently, the role of PTP1B in oxidative stress
in HUVECs induced by ox-LDL was explored. The results demonstrated
that ox-LDL treatment significantly increased MDA levels whilst
significantly decreasing SOD and GSH-Px activity compared with
those in the control group (Fig.
3A). By contrast, PTP1B knockdown significantly reversed the
increase in MDA levels whilst also reversing the reduction in SOD
and GSH-Px activity following ox-LDL treatment (Fig. 3A). The effects of PTP1B on the
tubule-formation ability of HUVECs were investigated. Ox-LDL
significantly reduced the number of tubules compared with that in
the control group, which was also significantly reversed by PTP1B
knockdown (Fig. 3C and D). Furthermore, western blotting
demonstrated that ox-LDL significantly reduced VEGFA expression in
HUVECs compared with that in control cells, while a significant
increase in VEGFA protein expression was observed in ox-LDL-treated
HUVECs following PTP1B knockdown compared with that in cells
treated with ox-LDL alone (Fig.
3D).
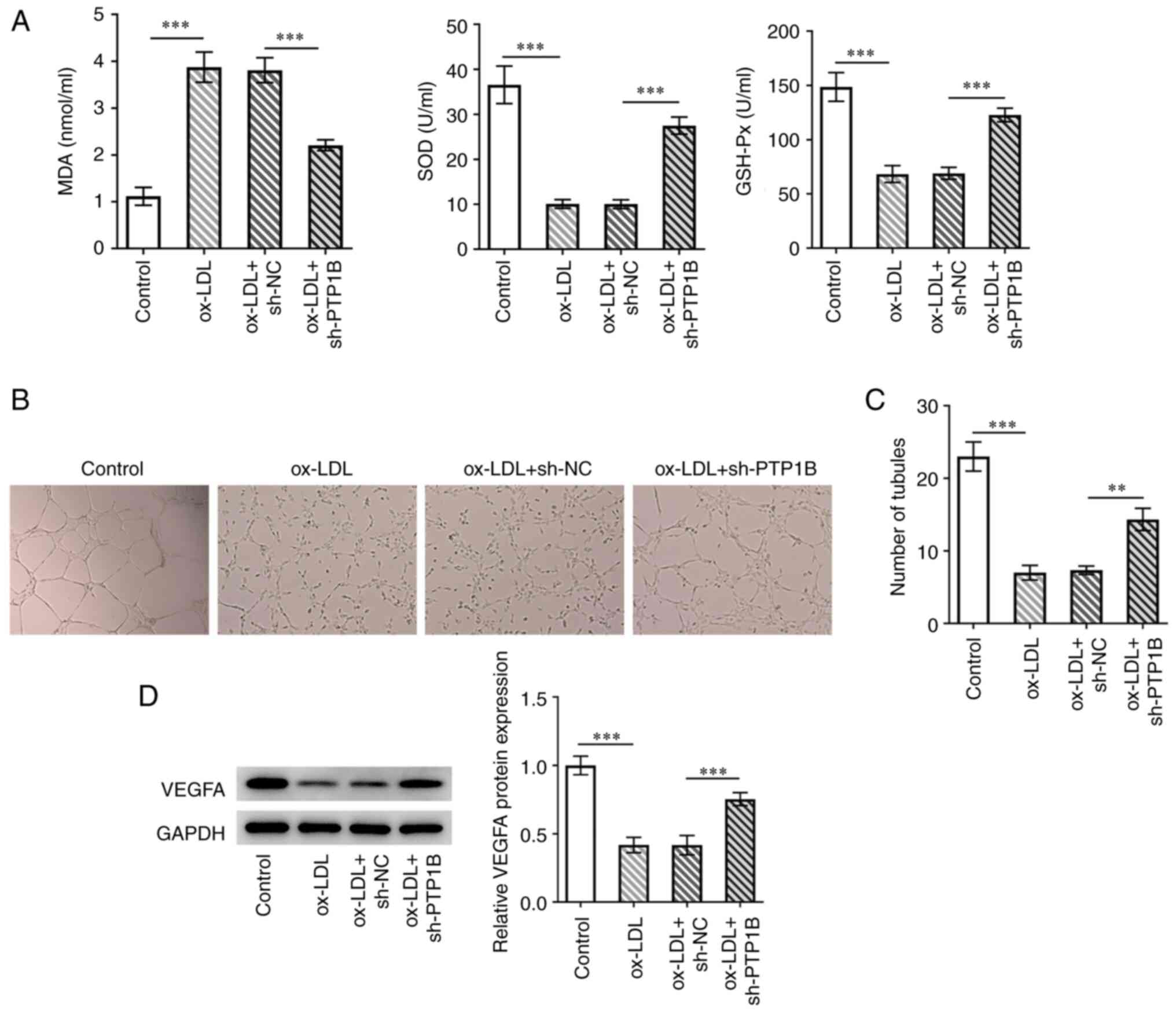 | Figure 3Knockdown of PTP1B expression
alleviates ox-LDL-induced oxidative stress in HUVECs to restore
tubule-formation ability. (A) Levels of MDA, SOD and GSH-Px were
evaluated using corresponding kits. (B) Tubule-formation ability
was explored using endothelial tubule formation assay and (C)
quantified. Magnification, x40. (D) VEGFA protein expression was
measured using western blotting. Results represent the mean ± SD.
**P<0.01 and ***P<0.001. MDA,
malondialdehyde; SOD, superoxide dismutase; GSH-Px, glutathione
peroxidase; PTP1B, protein tyrosine phosphatase 1B; ox-LDL,
oxidized low-density lipoprotein; sh, short-hairpin; VEGFA,
vascular endothelial growth factor A; NC, negative control. |
KLF2 negatively regulates PTP1B
transcription
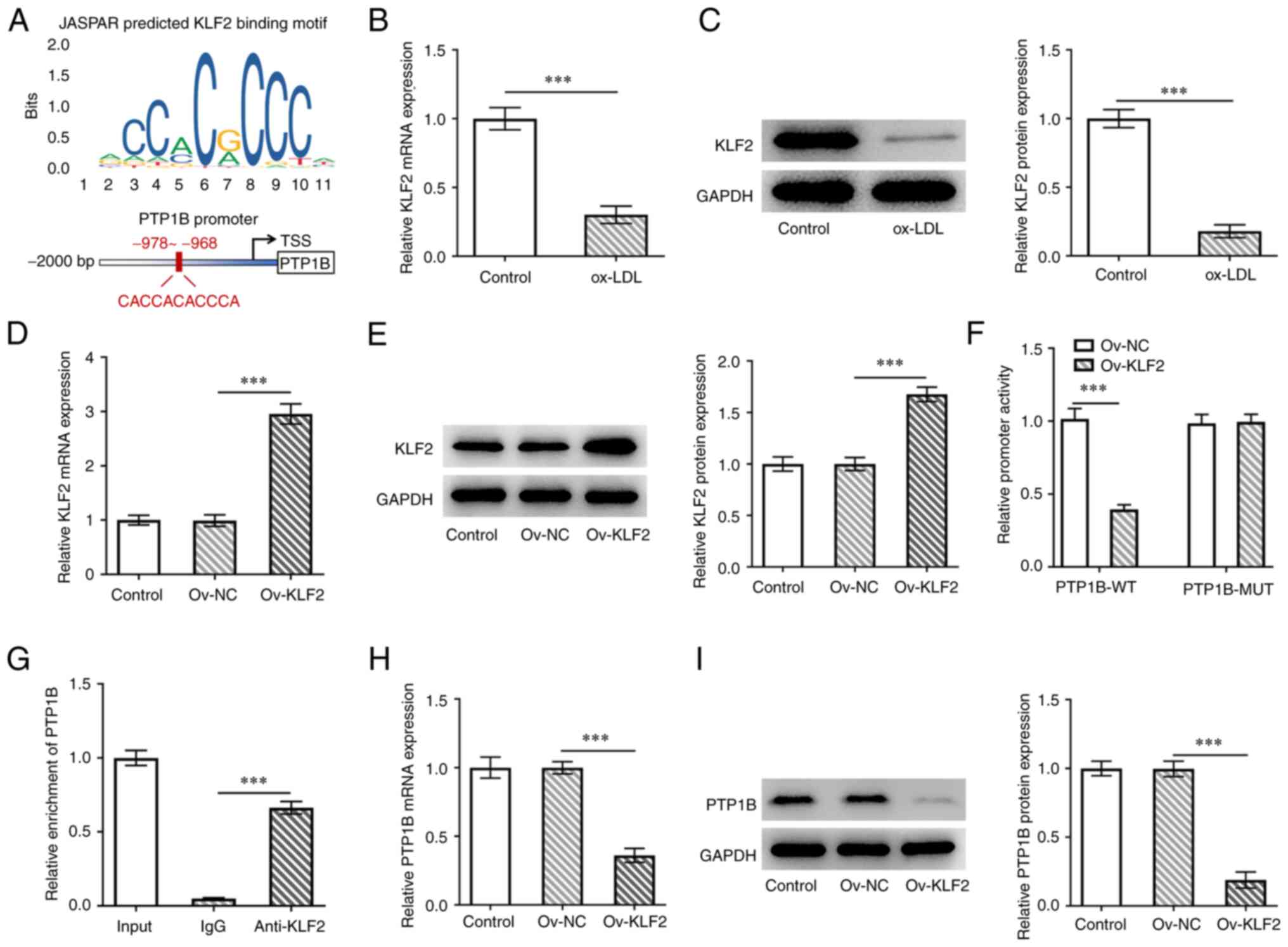
Using the JASPAR database, the transcription factor
KLF2, was predicted to bind to the PTP1B promoter (Fig. 4A). KLF2 mRNA and protein expression
levels were both found to be significantly reduced in HUVECs
following treatment with ox-LDL (Fig.
4B and C). To explore the
effects of KLF2 on PTP1B in HUVECs, KLF2 was overexpressed and
transfection efficiency was verified, as evidenced by the
significantly increased KLF2 expression in cells transfected with
the Ov-KLF2 plasmid compared with that in cells transfected with
the Ov-NC plasmid (Fig. 4D and
E). It was demonstrated that the
luciferase activity of the wild-type PTP1B promoter was
significantly inhibited by KLF2 overexpression, whereas the mutant
PTP1B promoter group displayed no changes in the luciferase
activity (Fig. 4F). ChIP was
performed to further verify the potential binding of KLF2 on the
PTP1B promoter. The results demonstrated that the PTP1B DNA
sequence was significantly enriched in the KLF2 group compared with
that in the IgG group (Fig. 4G).
Furthermore, KLF2 overexpression significantly inhibited the
expression of both PTP1B mRNA and protein compared with that in
cells transfected with the Ov-NC plasmid (Fig. 4H and I). This suggested a negative regulatory
mechanism exerted by KLF2 against PTP1B expression in HUVECs.
KLF2 knockdown reverses the protective
effects of PTP1B silencing on ox-LDL-induced HUVECs by regulating
the AMPK/SIRT1 signaling pathway
To investigate the role of KLF2 in ox-LDL-treated
HUVECs with PTP1B expression knocked down, sh-KLF2 was transfected
into HUVECs and KLF2 expression was prominently decreased after
transfection of sh-KLF2#1/2 plasmids, which verified the
transfection efficiency using RT-qPCR and western blotting
(Fig. 5A and B). Cells transfected with sh-KLF2#2
exhibited lower expression levels of KLF2 compared with those in
the sh-KLF2#1 group (Fig. 5A and
B). Therefore, sh-KLF2#2 was used
for subsequent experiments. Compared with those in the
sh-PTP1B-only group, co-transfection with sh-PTP1B and sh-KLF2
slightly reduced cell viability whilst significantly increasing LDH
activity (Fig. 5C and D), indicating a reversal of the
protective effects initially exerted by PTP1B knockdown. In
addition, IL-6, IL-1β and TNF-α levels in ox-LDL-induced HUVECs
were significantly elevated by the knockdown of KLF2 and PTP1B
compared with those in cells with only PTP1B expression knocked
down (Fig. 5E). KLF2 knockdown
also significantly reversed the protective effects mediated by
sh-PTP1B transfection against cell apoptosis (Fig. 5F), in addition to the expression
levels of Bcl-2, Bax and cleaved caspase-3 (Fig. 6B). Furthermore, KLF2 knockdown
significantly increased MDA levels whilst significantly decreasing
SOD and GSH-Px activity compared with those in cells transfected
with sh-PTP1B only (Fig. 5G).
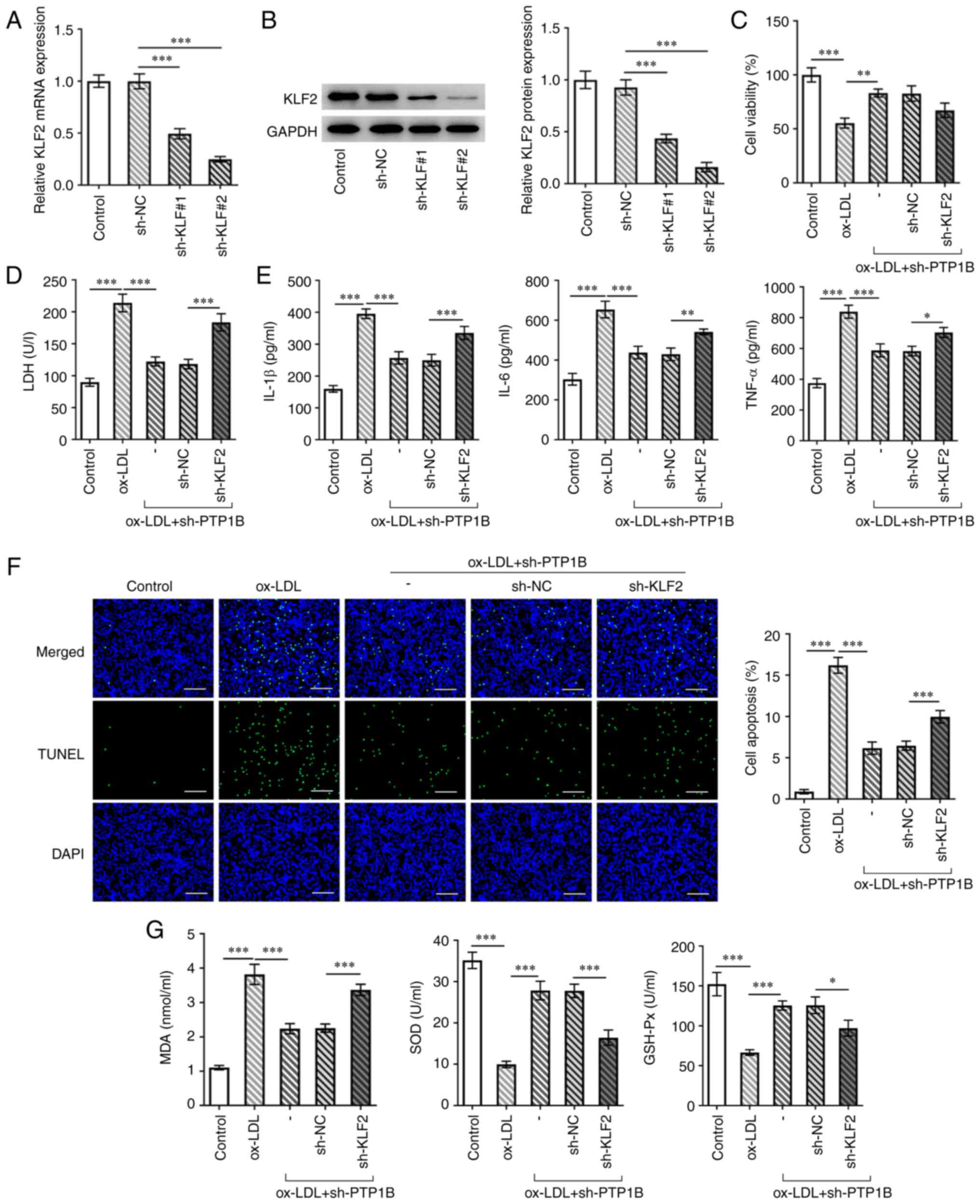 | Figure 5Knocking down KLF2 expression
reverses the effect of PTP1B silencing on cell viability,
inflammation, apoptosis and oxidative stress in ox-LDL-induced
HUVECs. (A) mRNA and (B) protein expression of KLF2 in HUVECs
transfected with or without sh-KLF2 were measured using reverse
transcription-quantitative PCR and western blotting. (C) Cell
viability was measured using Cell Counting Kit-8 assay. (D) LDH
levels were analyzed using a corresponding kit. (E) The levels of
IL-6, IL-1β and TNF-α were measured using ELISA. (F) Cell apoptosis
was measured using TUNEL assay. Scale bar, 50 µm. (G) Levels of
MDA, SOD and GSH-Px were assessed using corresponding kits. Results
represent the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001. KLF2,
Kruppel-like factor 2; PTP1B, protein tyrosine phosphatase 1B;
ox-LDL, oxidized low-density lipoprotein; Ov, overexpression
plasmid; NC, negative control; sh, short hairpin; LDH, lactate
dehydrogenase; MDA, malondialdehyde; SOD, superoxide dismutase;
GSH-Px, glutathione peroxidase. |
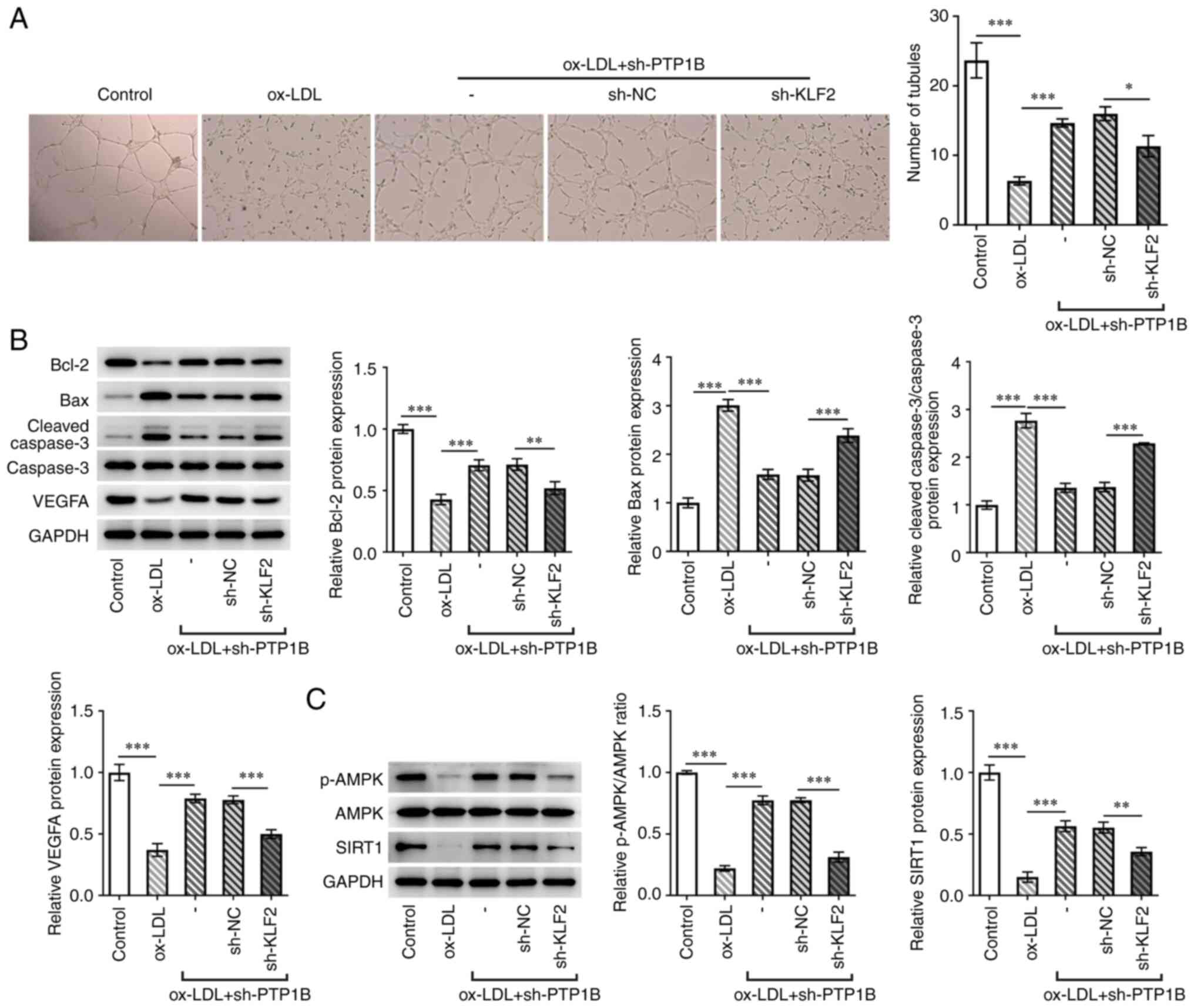 | Figure 6Silencing of KLF2 expression reverses
the protective effects of PTP1B knockdown on ox-LDL-induced HUVECs
by regulating the AMPK/SIRT1 pathway. (A) Tubule-formation capacity
was measured by endothelial tube formation assay. Magnification,
x40. (B) Protein expression levels of Bcl-2, Bax, cleaved
caspase-3, caspase-3 and VEGFA were measured using western blotting
assay. (C) Protein levels of p-AMPK, AMPK and SIRT1 were measured
using western blotting. Results represent the mean ± SD.
*P<0.05, **P<0.01 and
***P<0.001. KLF2, Kruppel-like factor 2; PTP1B,
protein tyrosine phosphatase 1B; ox-LDL, oxidized low-density
lipoprotein; NC, negative control; sh, short hairpin; VEGFA,
vascular endothelial growth factor A; p-, phosphorylated; AMPK,
5'AMP-activated protein kinase; SIRT1, sirtuin 1. |
The number of tubules formed and VEGFA expression
were also significantly reduced in the sh-KLF2 + sh-PTP1B compared
with those in the sh-PTP1B-only group (Fig. 6A and B). The results of the western blotting
assay demonstrated that ox-LDL significantly reduced the levels of
AMPK phosphorylation and SIRT1 expression (Fig. 6C). However, sh-PTP1B transfection
significantly reversed these effects of ox-LDL on the AMPK/SIRT1
signaling pathway (Fig. 6C).
Furthermore, subsequent knockdown of both KLF2 and PTP1B
significantly counteracted these regulatory effects of sh-PTP1B on
AMPK phosphorylation and SIRT1 expression (Fig. 6C).
Discussion
In the present study, the role of PTP1B in
ox-LDL-induced HUVECs was investigated. The results demonstrated
that PTP1B knockdown significantly restored cell viability,
inhibited inflammatory cell injury whilst also restoring tubule
formation ability. Furthermore, the results demonstrated that PTP1B
expression was negatively regulated by KLF2, which may be
associated with the AMPK/SIRT1 signaling pathway.
Atherosclerosis is a chronic inflammatory disease
that is caused by abnormal responses of the blood vessel wall to a
number of noxious stimuli (28-30).
Vascular EC injury is considered to be a common pathological cause
of cardiovascular diseases (31).
HUVECs have been widely acknowledged to be a useful model for
research on the human vascular endothelium (32-35).
Although this model does not represent all EC types in an organism,
it is nevertheless a viable model for studying the main molecular
pathways and properties underlying endothelial functions (36). Ox-LDL can accelerate EC
inflammation, apoptosis and endothelial-mesenchymal transition,
which serves a key role in mediating the early stages of lesion
formation during atherosclerosis (37). Ox-LDL also triggers lipid
metabolism disorders, leading to EC injury and death (38). In the present study, ox-LDL
stimulation was used to establish an in vitro
atherosclerosis model, where a dose of ox-LDL (100 µg/ml) was
selected based on previous studies (23-25).
The results demonstrated that HUVECs treated with ox-LDL exhibited
reduced cell viability, increased inflammatory factor levels,
elevated apoptosis rates and reduced tubule formation capabilities,
all of which are consistent with previous reports (23,24).
PTP1B has been previously reported to serve a role in
atherosclerosis and contribute to the pathophysiology of ECs
(19,39). The present study demonstrated that
PTP1B expression was upregulated in HUVECs after treatment with
ox-LDL. Following PTP1B knockdown, HUVEC viability and tubule
formation ability were significantly restored, whereas LDH
activity, inflammatory factor levels and cell apoptosis were
suppressed in cells stimulated with ox-LDL. Furthermore, the
oxidative stress levels of the cells were also decreased following
PTP1B knockdown. These findings were in consistency with previous
reports, which showed that the upregulation of the combination of
the cAMP response element-binding protein and lysine
methyltransferase 5A inhibited the hyperglycemia-induced
inflammatory factor levels by regulating PTP1B expression in
vascular endothelial cells (40).
These results suggested that PTP1B knockdown may protect HUVECs
against inflammatory injury and dysfunction induced by ox-LDL.
Transcription factors can regulate the expression
levels of target genes by repressing or activating transcription
(41,42). Using the JASPAR database, the
present study predicted a binding site of KLF2 on the PTP1B
promoter. The results demonstrated that KLF2 mRNA and protein
expression levels were decreased in ox-LDL-treated cells compared
with those in the control cells. To verify the binding between KLF2
and the PTP1B promoter, dual-luciferase reporter and ChIP assays
were performed. The results demonstrated an interaction between
KLF2 and the PTP1B promoter. KLFs belong to the zinc finger family
of transcription factors that serve key roles in biological
processes, including cell proliferation, inflammation and
differentiation (43). KLF2 has
been previously demonstrated to serve a role in the regulation of
inflammatory activation in endothelial cells (44). A previous study reported that IFN
regulatory factor 2-binding protein 2 can protect against
ox-LDL-induced endothelial inflammation and epithelial-mesenchymal
transition by activating KLF2 expression (45). In addition, Li et al
(46) reported that synoviolin 1
overexpression inhibited the ox-LDL-induced apoptosis of
endothelial cells, which was positively regulated by KLF2. Another
previous study demonstrated that ox-LDL could reduce the levels of
downstream regulators, such as myocyte enhancer factor 2C, where
ERK5 ameliorated ox-LDL-induced EC death, inflammation and
dysfunction by inhibiting the ERK5/myocyte enhancer factor 2C/KLF2
signaling pathway in an ox-LDL-induced primary human umbilical vein
endothelial cell model (47). In
agreement with these previous studies, the present study showed
that KLF2 knockdown reversed the effects of PTP1B silencing on
ox-LDL-induced HUVECs by inhibiting cell viability and endothelial
function, whilst promoting inflammation, oxidative stress and
apoptosis.
In conclusion, the present study demonstrated that
PTP1B may serve a regulatory role in inflammation and endothelial
dysfunction in ox-LDL-induced HUVECs. The results also suggested
that PTP1B expression is negatively regulated by KLF2, which may be
dependent on the AMPK/SIRT1 signaling pathway. The present study
provided a potential novel therapeutic target for the treatment of
endothelial dysfunction that occurs during atherosclerosis.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ and QG designed the study. YZ, QG and ZW
performed the experiments. YZ and ZW analyzed the data. All authors
read and approved the final manuscript. YZ and OG confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Davies PF, Zilberberg J and Helmke BP:
Spatial microstimuli in endothelial mechanosignaling. Circ Res.
92:359–370. 2003.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Feaver RE, Gelfand BD and Blackman BR:
Human haemodynamic frequency harmonics regulate the inflammatory
phenotype of vascular endothelial cells. Nat Commun.
4(1525)2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Giannotta M, Trani M and Dejana E:
VE-cadherin and endothelial adherens junctions: Active guardians of
vascular integrity. Dev Cell. 26:441–454. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Gimbrone MA: The Gordon Wilson lecture.
Understanding vascular endothelium: A pilgrim's progress.
Endothelial dysfunction, biomechanical forces and the pathobiology
of atherosclerosis. Trans Am Clin Climatol Assoc. 121:115–127.
2010.PubMed/NCBI
|
|
5
|
Garcia-Cardeña G, Comander J, Anderson KR,
Blackman BR and Gimbrone MA Jr: Biomechanical activation of
vascular endothelium as a determinant of its functional phenotype.
Proc Natl Acad Sci USA. 98:4478–4485. 2001.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son
DJ, Visvader JE and Jo H: Discovery of novel mechanosensitive genes
in vivo using mouse carotid artery endothelium exposed to disturbed
flow. Blood. 116:e66–e73. 2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Marchio P, Guerra-Ojeda S, Vila JM,
Aldasoro M, Victor VM and Mauricio MD: Targeting Early
atherosclerosis: A focus on oxidative stress and inflammation. Oxid
Med Cell Longev. 2019(8563845)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tarbell JM, Shi ZD, Dunn J and Jo H: Fluid
mechanics, arterial disease, and gene expression. Annu Rev Fluid
Mech. 46:591–614. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chatterjee S and Fisher AB:
Mechanotransduction in the endothelium: Role of membrane proteins
and reactive oxygen species in sensing, transduction, and
transmission of the signal with altered blood flow. Antioxid Redox
Signal. 20:899–913. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chiu JJ and Chien S: Effects of disturbed
flow on vascular endothelium: Pathophysiological basis and clinical
perspectives. Physiol Rev. 91:327–387. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Su G, Sun G, Liu H, Shu L, Zhang J, Guo L,
Huang C and Xu J: Niacin suppresses progression of atherosclerosis
by inhibiting vascular inflammation and apoptosis of vascular
smooth muscle cells. Med Sci Monit. 21:4081–4089. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Onat D, Brillon D, Colombo PC and Schmidt
AM: Human vascular endothelial cells: A model system for studying
vascular inflammation in diabetes and atherosclerosis. Curr Diab
Rep. 11:193–202. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chen PY, Qin L, Baeyens N, Li G, Afolabi
T, Budatha M, Tellides G, Schwartz MA and Simons M:
Endothelial-to-mesenchymal transition drives atherosclerosis
progression. J Clin Invest. 125:4514–4528. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Yip SC, Saha S and Chernoff J: PTP1B: A
double agent in metabolism and oncogenesis. Trends Biochem Sci.
35:442–449. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Penafuerte C, Feldhammer M, Mills JR,
Vinette V, Pike KA, Hall A, Migon E, Karsenty G, Pelletier J,
Zogopoulos G and Tremblay ML: Downregulation of PTP1B and TC-PTP
phosphatases potentiate dendritic cell-based immunotherapy through
IL-12/IFNγ signaling. Oncoimmunology. 6(e1321185)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Xu Q, Wu N, Li X, Guo C, Li C, Jiang B,
Wang H and Shi D: Inhibition of PTP1B blocks pancreatic cancer
progression by targeting the PKM2/AMPK/mTOC1 pathway. Cell Death
Dis. 10(874)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
da Silva JF, Alves JV, Bolsonni JA, Costa
RM, Rios FJ, Camargo LL, Montezano AC, Touyz RM and Tostes RC:
Protein tyrosine phosphatase type 1B (PTP1B) contributes to
atherosclerotic processes by mechanisms that involve NADPH-oxidase
and calcium influx. FASEB J. 34:1. 2020.
|
|
18
|
Thompson D, Morrice N, Grant L, Le Sommer
S, Lees EK, Mody N, Wilson HM and Delibegovic M: Pharmacological
inhibition of protein tyrosine phosphatase 1B protects against
atherosclerotic plaque formation in the LDLR-/- mouse
model of atherosclerosis. Clin Sci (Lond). 131:2489–2501.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Thompson D, Morrice N, Grant L, Le Sommer
S, Ziegler K, Whitfield P, Mody N, Wilson HM and Delibegović M:
Myeloid protein tyrosine phosphatase 1B (PTP1B) deficiency protects
against atherosclerotic plaque formation in the ApoE-/-
mouse model of atherosclerosis with alterations in IL10/AMPKα
pathway. Mol Metab. 6:845–853. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Maupoint J, Besnier M, Gomez E, Bouhzam N,
Henry JP, Boyer O, Nicol L, Mulder P, Martinet J and Richard V:
Selective vascular endothelial protection reduces cardiac
dysfunction in chronic heart failure. Circ Heart Fail.
9(e002895)2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ali MI, Ketsawatsomkron P, Belin de
Chantemele EJ, Mintz JD, Muta K, Salet C, Black SM, Tremblay ML,
Fulton DJ, Marrero MB and Stepp DW: Deletion of protein tyrosine
phosphatase 1b improves peripheral insulin resistance and vascular
function in obese, leptin-resistant mice via reduced oxidant tone.
Circ Res. 105:1013–1022. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Herren DJ, Norman JB, Anderson R, Tremblay
ML, Huby AC and Belin de Chantemèle EJ: Deletion of protein
tyrosine phosphatase 1B (PTP1B) enhances endothelial cyclooxygenase
2 expression and protects mice from type 1 diabetes-induced
endothelial dysfunction. PLoS One. 10(e0126866)2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhou H, Jiang F and Leng Y: Propofol
ameliorates ox-LDL-induced endothelial damage through enhancing
autophagy via PI3K/Akt/m-TOR pathway: A novel therapeutic strategy
in atherosclerosis. Front Mol Biosci. 8(695336)2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhang Z, Pan X, Yang S, Ma A, Wang K, Wang
Y, Li T and Liu S: miR-155 promotes ox-LDL-induced autophagy in
human umbilical vein endothelial cells. Mediators Inflamm.
2017(9174801)2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Shiotsugu S, Okinaga T, Habu M, Yoshiga D,
Yoshioka I, Nishihara T and Ariyoshi W: The biological effects of
interleukin-17A on adhesion molecules expression and foam cell
formation in atherosclerotic lesions. J Interferon Cytokine Res.
39:694–702. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang XH, Shu J, Jiang CM, Zhuang LL, Yang
WX, Zhang HW, Wang LL, Li L, Chen XQ, Jin R and Zhou GP: Mechanisms
and roles by which IRF-3 mediates the regulation of ORMDL3
transcription in respiratory syncytial virus infection. Int J
Biochem Cell Biol. 87:8–17. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Fava C and Montagnana M: Atherosclerosis
is an inflammatory disease which lacks a common anti-inflammatory
therapy: How human genetics can help to this issue. A narrative
review. Front Pharmacol. 9(55)2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Galkina E and Ley K: Immune and
inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol.
27:165–197. 2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tousoulis D, Oikonomou E, Economou EK,
Crea F and Kaski JC: Inflammatory cytokines in atherosclerosis:
Current therapeutic approaches. Eur Heart J. 37:1723–1732.
2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Sun HJ, Hou B, Wang X, Zhu XX, Li KX and
Qiu LY: Endothelial dysfunction and cardiometabolic diseases: Role
of long non-coding RNAs. Life Sci. 167:6–11. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Pan JX: LncRNA H19 promotes
atherosclerosis by regulating MAPK and NF-kB signaling pathway. Eur
Rev Med Pharmacol Sci. 21:322–328. 2017.PubMed/NCBI
|
|
33
|
Watanabe T and Sato K: Roles of the
kisspeptin/GPR54 system in pathomechanisms of atherosclerosis. Nutr
Metab Cardiovasc Dis. 30:889–895. 2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhu Z, Li J and Zhang X: Salidroside
protects against ox-LDL-induced endothelial injury by enhancing
autophagy mediated by SIRT1-FoxO1 pathway. BMC Complement Altern
Med. 19(111)2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Gao W, Liu H, Yuan J, Wu C, Huang D, Ma Y,
Zhu J, Ma L, Guo J, Shi H, et al: Exosomes derived from mature
dendritic cells increase endothelial inflammation and
atherosclerosis via membrane TNF-α mediated NF-κB pathway. J Cell
Mol Med. 20:2318–2327. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Baudin B, Bruneel A, Bosselut N and
Vaubourdolle M: A protocol for isolation and culture of human
umbilical vein endothelial cells. Nat Protoc. 2:481–485.
2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yang H, Mohamed AS and Zhou SH: Oxidized
low density lipoprotein, stem cells, and atherosclerosis. Lipids
Health Dis. 11(85)2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hurtado-Roca Y, Bueno H, Fernandez-Ortiz
A, Ordovas JM, Ibañez B, Fuster V, Rodriguez-Artalejo F and
Laclaustra M: Oxidized LDL is associated with metabolic syndrome
traits independently of central obesity and insulin resistance.
Diabetes. 66:474–482. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Legeay S, Fautrat P, Norman JB, Antonova
G, Kennard S, Bruder-Nascimento T, Patel VS, Faure S and Belin de
Chantemèle EJ: Selective deficiency in endothelial PTP1B protects
from diabetes and endoplasmic reticulum stress-associated
endothelial dysfunction via preventing endothelial cell apoptosis.
Biomed Pharmacother. 127(110200)2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Huang T, Li X, Wang F, Lu L, Hou W, Zhu M
and Miao C: The CREB/KMT5A complex regulates PTP1B to modulate high
glucose-induced endothelial inflammatory factor levels in diabetic
nephropathy. Cell Death Dis. 12(333)2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Garcia-Alonso L, Iorio F, Matchan A,
Fonseca N, Jaaks P, Peat G, Pignatelli M, Falcone F, Benes CH,
Dunham I, et al: Transcription factor activities enhance markers of
drug sensitivity in cancer. Cancer Res. 78:769–780. 2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bushweller JH: Targeting transcription
factors in cancer-from undruggable to reality. Nat Rev Cancer.
19:611–624. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Jha P and Das H: KLF2 in regulation of
NF-κB-mediated immune cell function and inflammation. Int J Mol
Sci. 18(2383)2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhou Z, Tang AT, Wong WY, Bamezai S,
Goddard LM, Shenkar R, Zhou S, Yang J, Wright AC, Foley M, et al:
Cerebral cavernous malformations arise from endothelial gain of
MEKK3-KLF2/4 signalling. Nature. 532:122–126. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Jiang Y and Shen Q: IRF2BP2 prevents
ox-LDL-induced inflammation and EMT in endothelial cells via
regulation of KLF2. Exp Ther Med. 21(481)2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Li Q, Xuan W, Jia Z, Li H, Li M, Liang X
and Su D: HRD1 prevents atherosclerosis-mediated endothelial cell
apoptosis by promoting LOX-1 degradation. Cell Cycle. 19:1466–1477.
2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Patel R, Varghese JF, Singh RP and Yadav
UCS: Induction of endothelial dysfunction by oxidized low-density
lipoproteins via downregulation of Erk-5/Mef2c/KLF2 signaling:
Amelioration by fisetin. Biochimie. 163:152–162. 2019.PubMed/NCBI View Article : Google Scholar
|